

Abstract
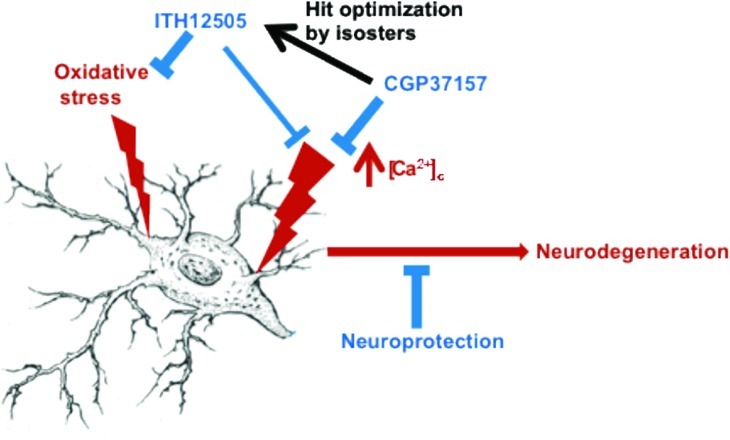
Benzothiazepine CGP37157 is widely used as tool to explore the role of mitochondria in cell Ca2+ handling, by its blocking effect of the mitochondria Na+/Ca2+ exchanger. Recently, CGP37157 has shown to exhibit neuroprotective properties. In the trend to improve its neuroprotection profile, we have synthesized ITH12505, an isosteric analogue having a methyl instead of chlorine at C2′ of the phenyl ring. ITH12505 has exerted neuroprotective properties similar to CGP37157 in chromaffin cells and hippocampal slices stressed with veratridine. Also, both compounds afforded neuroprotection in hippocampal slices stressed with glutamate. However, while ITH12505 elicited protection in SH-SY5Y cells stressed with oligomycin A/rotenone, CGP37157 was ineffective. In hippocampal slices subjected to oxygen/glucose deprivation plus reoxygenation, ITH12505 offered protection at 3–30 μM, while CGP37157 only protected at 30 μM. Both compounds caused blockade of Ca2+ channels in high K+-depolarized SH-SY5Y cells. An in vitro experiment for assaying central nervous system penetration (PAMPA-BBB; parallel artificial membrane permeability assay for blood-brain barrier) revealed that both compounds could cross the blood–brain barrier, thus reaching their biological targets in the central nervous system. In conclusion, by causing a mild isosteric replacement in the benzothiazepine CGP37157, we have obtained ITH12505, with improved neuroprotective properties. These findings may inspire the design and synthesis of new benzothiazepines targeting mitochondrial Na+/Ca2+ exchanger and L-type voltage-dependent Ca2+ channels, having antioxidant properties.
Keywords: Alzheimer’s disease, cell calcium, CGP37157, mitochondrial sodium calcium exchanger, neuroprotection, oxidative stress
In excitable cells, Ca2+ homeostasis has been implicated in a huge number of cellular mechanisms, including neurogenesis, secretion, differentiation, circuit formation, or neuronal plasticity.1 However, when the fine regulation of cytosolic Ca2+ concentrations ([Ca2+]c) is disrupted, a plethora of events leading to cell death are triggered.2 The observation that not only high Ca2+ overload3 but also Ca2+ antagonists,4 reducing [Ca2+]c, can cause neuronal death, has raised the hypothesis that Ca2+ ions behave as both cell survival supporters and cell death promoters, depending on both amplitude and temporal extension of the cell Ca2+ signal, together with other physiological conditions.5 Hence, there has to be a critical set point where Ca2+-derived cytoprotective signals might turn into cytotoxic.6
In this framework, the relationship between Ca2+ dysregulation and neuronal death has been well-documented in several neurodegenerative diseases, such as Parkinson's disease,7 cerebral ischemia,8 and amyotrophic lateral sclerosis.9 As far as Alzheimeŕs disease (AD) is concerned, several contributions have related altered [Ca2+]c to physiopathological events characterizing AD.10 For instance, Aβ destabilizes Ca2+ homeostasis,11 rendering neurons vulnerable to neurofibrillary degeneration12 and impairing both proliferation and neuronal differentiation of cultured human neural progenitor cells.13 Murray and co-workers analyzed brain tissue from AD patients, showing that a sustained [Ca2+] increase is associated with neurofibrillary tangle-bearing neurons.14 More recently, Dreses-Werringloer et al. found a new genetic factor associated with the development of AD: the P86L polymorphism of the new Ca2+ channel CALHM1.15 The expression of this mutated form, P86L-CALHM1, promotes accumulation of Aβ and alters membrane permeability to Ca2+, resulting in dysregulation of Ca2+ homeostasis.
Despite these findings, AD therapeutics has scarcely focused on the clinical development of cell Ca2+-regulating agents,16 except for NMDA-sensitive glutamate receptor blockers,17 even when new biological targets modulating [Ca2+]c, such as the above-mentioned CALHM-1 channel, have been related to AD. Nevertheless, the search of optimal medicines to counteract the progression of AD,18 by means of targeting well-known physiopathological markers, that is, Aβ19 or aggregated tau (τ) protein,20 as well as the so-called cholinergic hypothesis,21 has been disappointing.22
During the past decade, our research group has been studying new voltage-dependent Ca2+ channel (VDCC) ligands as new [Ca2+] regulators for the treatment of AD.23−25 Our current goal aims on the study of new Ca2+-regulating biological targets implicated in AD, and the search of new drugs for their pharmacological regulation. In this regard, we recently discovered that CALHM1 generates transient elevations of both [Ca2+]c and mitochondrial Ca2+ concentrations ([Ca2+]m); however, its P86L mutated form also causes cell Ca2+ entry, but generates a plateau of [Ca2+]m that was prolonged in time.26 Furthermore, we have described the neuroprotective properties of the mitochondrial Na+/Ca2+ exchanger (mNCX) blocker CGP37157 to rescue cultured cells27 and hippocampal slices28 from the toxic stimulus exerted by veratridine, a model of cell death by Na+ and Ca2+ overload.
Indeed, the scientific community devoted to the research on neurodegenerative diseases has not paid attention to the mNCX until recently, due to its emerging role in the mitochondrial/cytosolic [Ca2+] trafficking29 and its implication in the development of neurological diseases.30 Existence of a Na+/Ca2+ exchange in mitochondria was first reported by Carafoli.31 The further discovery of the benzothiazepine derivative CGP37157 as a mNCX blocker constituted a breakthrough for the functional characterization of the mNCX32 (Figure 1). However, CGP37157 is not only a mNCX blocker, at submicromolar concentrations,33 but also it has been described as blocker of both VDCCs34 and plasmalemal NCX, at micromolar concentrations,35 as well. Pei and co-workers have communicated the poor water solubility and short half-life of CGP37157.36 In that paper, authors describe the efforts to synthesize CGP37157 derivatives, with improved pharmacokinetic profile and mNCX blocking activity, for their use as potential drugs for Type-II diabetes mellitus. Unfortunately, none of the new compounds improved that mNCX blockade elicited by CGP37157, while enhancement of the pharmacokinetic profile was not experimentally demonstrated.
Figure 1.

Chemical structures of the mNCX blocker CGP37157 and its isosteric analogue ITH12505.
Focusing on the search of new mNCX ligands for the treatment of AD, we appreciated from the work of Pei and co-workers that the compound numbered as 1c was only 4.5-fold less potent (IC50 = 6.3 μM) than CGP37157 (IC50 = 1.4 μM)36 to block mNCX, and can be considered as the closest isosteric derivative of CGP37157, where the chlorine atom at the pendant phenyl ring has been replaced by a methyl group. Replacement of isosters and bioisosters is a common strategy in medicinal chemistry for the optimization of hit compounds, modulating both pharmacodynamic and pharmacokinetic properties.37 In this context, we believed that this compound deserved a deeper pharmacological study as a new potential neuroprotective drug, similarly to what was described for CGP37157 by our research group, hypothesizing that this isosteric replacement will not be able to dramatically change the pharmacodynamic properties of CGP37157, while improving its pharmacokinetic profile.
Results and Discussion
Thus, in this work, we present the pharmacological characterization of the 2′-methyl isosteric analogue of CGP37157, compound 1c from the work of Pei et al.,36 that we have synthesized and incorporated into our chemical library as compound ITH12505 (Figure 1). We report here the comparative properties of compounds CGP37157 and ITH12505 in eliciting neuroprotection and blocking Ca2+ uptake into K+-depolarized cells. ITH12505 has shown similar neuroprotective properties to CGP37157 against Ca2+overload-elicited excitoxicity and presented additional protective effect in oxidative stress models targeting mitochondrial dysfunction models.
ITH12505 Protected against the Cytotoxic Effects of Veratridine in Chromaffin Cells
We first evaluated whether ITH12505 was able to reproduce the neuroprotective profile of CGP37157 in our earliest neuroprotection evaluation experimental model, that is, bovine chromaffin cells stressed with veratridine.38 We have used bovine adrenal medulla chromaffin cells, a paraneuronal cell type that, as neurons, possesses Na+ and Ca2+ channels, as well as K+ channels. Furthermore, mitochondrial Ca2+ fluxes, including the use of CGP37157, have been thoroughly studied in these cells.39 Veratridine was selected as toxic stimulus because it elicits cell death through Na+ and Ca2+ overload40 so, mitochondria are able to regulate cell Na+ and Ca2+ signals through mNCX recruitment. In fact, the mNCX blocker CGP37157 protected bovine chromaffin cells against cytotoxicity induced by veratridine with an EC50 of 10 μM.27 We selected 30 μM veratridine as toxic stimulus based on our previous study where this concentration afforded a reliable neuronal damage.27 Veratridine augmented the release of LDH with respect to nontreated cells by 31%. This release was reduced by increasing concentrations of ITH12505, though it was only statistically significant at 30 μM. Thus, in the presence of 30 μM of ITH12505, veratridine-elicited LDH release was reduced 13%, meaning a 59% protection. Protection elicited by CGP37157 was tested at 20 μM in each experiment for comparative purposes; at this concentration CGP37157 afforded 85% protection (Figure 2). Hence, ITH12505 protected chromaffin cells stressed with veratridine with a slightly higher EC50 than that found with CGP37157.
Figure 2.

Protection by compound ITH12505 against the cytotoxic effects of veratridine (Ver). Data are mean ± SEM of triplicates of at least three independent experiments: ###, p < 0.001 respect to basal; ***, p < 0.001, with respect to Ver alone.
Effects of CGP37157 and ITH12505 on the Neurotoxicity Elicited by Rotenone/Oligomycin A (O/R) in SH-SY5Y Cells
We have recently reported how cytoprotective effects of CGP37157 are exclusively found in Na+/Ca2+ overload cell death models,27 as it was unable to rescue chromaffin cells subjected to a toxic stimulus related to the mitochondrial disruption-derived oxidative stress, for example, blockade of the mitochondrial respiratory chain by combining 10 μM oligomycin A and 30 μM rotenone. Rotenone and oligomycin A (O/R) block complexes I and V, respectively, of the mitochondrial electron transport chain, thereby causing free radical generation and blockade of ATP synthesis.41 Therefore, exposure of SH-SY5Y neuroblastoma or chromaffin cells to O/R constitutes a good model of oxidative stress, having its origin in mitochondria. Recently, mitochondrial complex I blockade by rotenone has been considered a very reproducible in vitro model of hypoxia occurred in physiopatological events related to cerebral ischemia.42 CGP37157 not only failed against the O/R exposure, but in fact augmented cell-damaging effects of O/R in chromaffin cells.27 Herein, SH-SY5Y cells were incubated with CGP37157 or ITH12505 before the addition of O/R, and coincubated with compounds plus O/R for an additional 24 h period. Cell viability at the end of this period was evaluated by the MTT method. N-Acetylcysteine (NAC)43 and melatonin were used as reference compounds. In the range of concentrations evaluated, CGP37157 did not protect SH-SY5Y cells against O/R (Figure 3b), while ITH12505 slightly protected against the loss of cell viability exerted by O/R with statistically significant values (p < 0.01) (Figure 3a). At 0.3 μM, ITH12505 afforded 40% protection, a figure similar to that of melatonin and NAC.
Figure 3.

Protection by ITH12505 (a), but not with CGP37157 (b), against the cytotoxic effects of O/R in neuroblastoma cells. Basal (control) group was considered as 100% of viability and represents cell viability of cells incubated only with cell culture medium. Data are the mean ± SEM of triplicates of five different cell batches: ###p < 0.001, comparing control and O/R-lesioned cells; ** p < 0.01 and * p < 0.05, comparing to O/R-lesioned cells in the absence of drug.
Moreover, in per se toxicity experiments, ITH12505, at much higher concentrations, up to 30 μM, did not affect to this neuronal model (Figure 4a). By contrast, CGP37157, exposed at 30 μM, generated a loss of cell viability comparable to that found for the toxic cocktail O/R (Figure 4b).
Figure 4.

Effect of ITH12505 (a), and of CGP37157 (b), on the SH-SY5Y neuroblastoma cell viability, in absence of toxic stimulus. Basal (control) group was considered as 100% of viability and represents cell viability of cells incubated only with cell culture medium. An O/R group was included for comparative purposes, as a positive control of the expected damage. Data are the mean ± SEM of triplicates of six different cell batches: *** p < 0.001, comparing to control cells in the absence of drugs or O/R.
The neuroprotective activity of ITH12505 in this in vitro model against O/R prompted us to study its antioxidant properties in a more physiological and complex model of neurodegeneration. Should the antioxidant activity of ITH12505 be confirmed, together with the maintenance of the protective profile against cell Ca2+ dysregulation of CGP37157, we would have found a very interesting neuroprotective benzothiazepine, as it is capable to protect neurons against the two main physiological events causing cell death, that is, Ca2+ overload and oxidative stress.
Effects of Compounds ITH12505 and CGP37157 on Rat Hippocampal Slices Stressed with Veratridine
We have reported that CGP37157 protected rat hippocampal slices subjected to veratridine exposure, in a concentration-dependent manner, with a maximal protection at 30 μM.28 Similarly, after a stabilization period of 30 min at 34 °C, slices were preincubated with ITH12505 at concentrations of 3, 10, or 30 μM for 30 min at 37 °C; thereafter, slices continued in the presence of ITH12505 plus veratridine 30 μM for an additional 3.5 h period. Measured by the method of the MTT reduction, veratridine caused a 41% diminution of viability; this neuronal lesion was prevented by increasing concentrations of compound ITH12505, in a concentration-dependent manner, with a maximal protection at 30 μM (35% protection). This protection was comparable to that of CGP37157 at 30 μM, used as reference (Figure 5).28
Figure 5.

ITH12505 protected hippocampal slices against the neurotoxic effects of veratridine (Ver). Data are mean ± SEM of quadruplicates of five independent experiments: ###p < 0.001 respect to control; **p < 0.01, ***p < 0.001 respect to Veratridine group.
Effects of Compounds ITH12505 and CGP37157 on Glutamate-Lesioned Rat Hippocampal Slices
Among all the experimental models of neuronal death based on Ca2+ dysregulation, glutamate receptor-mediated Ca2+ overload appears to be the most relevant from a pathogenic point of view, since it is closely related to various neurodegenerative disorders and stroke.44 In fact, the only noncholinergic drug of those five approved by drug regulatory agencies for the treatment of AD is memantine (MEM), a NMDA-sensitive glutamate receptor blocker.17 Moreover, recent observations have confirmed the influence of mitochondria-mediated cell Ca2+ regulation on glutamate-induced excitoxicity.45 Thus, stimulated by the finding that compound ITH12505 could be an optimized drug for the treatment of neurodegenerative diseases from the head compound CGP37157, by both its cell Ca2+ regulatory and its antioxidant properties, we evaluated the neuroprotective feature of ITH12505, as well as of CGP37157, in rat hippocampal slices subjected to glutamate (Glu) excitotoxicity.
Following the experimental protocol indicated for the preparation of the rat hippocampal slices, after a stabilization period of 30 min at 34 °C, slices were coincubated with glutamate at 1 mM and ITH12505 at concentrations of 3, 10, or 30 μM for 4 h at 37 °C. Glutamate caused a 32% reduction of cell viability in nontreated tissues, analogously to the observed in previous reports from our group and others;46 this neuronal lesion was prevented by increasing concentrations of compound ITH12505, in a concentration-dependent manner, with a maximal protection at 30 μM (Figure 6a). At this concentration, MTT was reduced by 85% compared to control, having 52% more viability than that found in glutamate slices. This protection was comparable to that of memantine (10 μM), used as reference compound.47 Under the same experimental conditions, CGP37157 afforded a full protection of hippocampal slices subjected to glutamate excitotoxicity, and counteracted the glutamate-induced loss of viability in a concentration-dependent manner, with a maximal protection at 30 μM, which caused a near full protection (98% reduction of MTT compared to control cells) (Figure 6b). Again, CGP37157 exerted a higher neuroprotection, but ITH12505 showed a decent behavior as neuroprotectant against glutamate excitotoxicity. In summary, the chemical modification of CGP37157 producing the methyl isosteric analogue ITH12505 resulted in maintenance of the protection against Ca2+ overload-induced neurotoxicity, though losing some of activity.
Figure 6.

ITH12505 (a) and CGP37157 (b) protected hippocampal slices against the neurotoxic effects of glutamate (Glu). Data are mean ± SEM of quadruplicates of at least five independent experiments: ###p < 0.001 respect to control; * p < 0.05, **p < 0.01, ***p < 0.001 respect to glutamate group.
Effects of Compounds ITH12505 and CGP37157 on Oxygen and Glucose Deprivation-Lesioned Rat Hippocampal Slices
Stimulated by the neuroprotective properties of compound ITH12505 against the model of oxidative stress defined by SH-SY5Y neuroblastoma cells subjected O/R exposure (Figure 3a), and rat hippocampal slices to glutamate excitotoxicity (Figure 6), we considered of interest to test whether compound ITH12505 exerted neuroprotection in a experimental model more closely related to brain ischemia, for example, the rat hippocampal slice damaged with oxygen and glucose deprivation (OGD) followed by reoxygenation. The functional impairment of mitochondria, promoted by the absence of the oxygen supply, is improved when reoxygenation triggers a massive production of reactive oxygen species raised by the OGD-induced overwork of the NADPH oxidase (NOX) enzyme. Thus, 15 min slice exposure to OGD followed by 2 h reoxygenation produced 26% decrease of cell viability with respect to basal slices, as previously described by our group and others.48,49 We found that compound ITH12505 was able to increase cell viability in a concentration-dependent manner (3–30 μM), with a maximal protection of 47% at 30 μM. At 10 μM, neuroprotection afforded by ITH12505 was 44%; this protection was comparable to that of the reference drug galantamine,50 which protected by 41% at 15 μM (Figure 7a). Figure 7b shows the effect of CGP37157 on the loss of viability evoked by OGD. OGD reduced by 21% the cell viability of control slices, and this loss of cell viability was counteracted by CGP37157 at the concentration of 30 μM, as cell viability was 93% with respect to control, meaning a 67% reduction of OGD-induced loss of viability. CGP37157 was unable to protect against OGD plus reoxygenation at lower concentrations (Figure 7). Therefore, we appreciated that ITH12505 presents a better neuroprotection profile against OGD plus reoxygenation than CGP37157, which only significantly protected hippocampal slices at 30 μM. In a previous paper, other authors described similar data for CGP37157; by using organotypic hippocampal slices cultures stressed with OGD, CGP37157 showed a gentle reduction of OGD-induced loss of viability at the highest concentration tested, 10 μM, after a 24 h period of preincubation with the drug.51
Figure 7.

Protection by ITH12505 (a) and CGP37157 (b) against neurotoxicity elicited by oxygen and glucose deprivation (OGD) followed by reoxygenation (reox) in rat hippocampal slices. MTT reduction in control slices was taken as 100% of tissue viability. Data are mean ± SEM of quadruplicates of at least five independent experiments: ##p < 0.01, ###p < 0.001 comparing basal respect to OGD; * p < 0.05, ***p < 0.001 in comparison to OGD.
Collecting our results on O/R exposure in SH-SY5Y cells and those from OGD-damaged hippocampal slices, we can state that ITH12505 had a better protection profile against oxidative stress, compared with its parent compound CGP37157. Considering that oxidative stress is an important factor in the pathogenesis of various neurodegenerative diseases52 and that several VDCC blockers possess antioxidant activity,53 multitarget benzothiazepine compounds having the capacity of mitigating enhanced Ca2+ cycling and exhibiting antioxidant effects may exhibit better and more efficient neuroprotective properties. One could postulate that antioxidant activity of ITH12505 is basically related to its methyl group at the phenyl ring, presumably sensitive to oxidation to carboxylic acid, but we discard the antioxidant effects of ITH12505 are due to this radical scavenging feature, because neither ITH12505 nor CGP37157 showed an appreciable radical capture capacity in the oxygen radical absorbance capacity assay (ORAC)54 (data not shown).
Effects of Compounds ITH12505 and CGP37157 on VDCC-Mediated Ca2+ Entry
In previous studies, we have reported the cytoprotective feature of CGP37157 in several models of cell death induced by Ca2+ overload.27,28 This effect can be partially related to its direct blockade of VDCCs, as previously described.34 Actually, first evidence of a mNCX blockade was found in VDCC blockers, such as diltiazem, prenylamine, fendiline, nifedipine, or verapamil.55,56
The altered Ca2+ influx through VDCCs, preferentially via L-type (Cav1.1–1.4), has been reported to be a key mechanism of neuronal death occurring in several physiopathological events,3 such as neurodegenerative processes. Taking into account that compound ITH12505 has kept most of the neuroprotective activity against cell Ca2+ deregulation-derived excitoxicity found in the parental compound CGP37157, the VDCC blocking activity by ITH12505 has been studied, using the Ca2+-sensitive fluorescent dye Fluo-4/AM on SH-SY5Y cells, stimulating Ca2+ entry through VDCCs by a high K+ concentration. Fluo-4/AM-loaded cells were incubated with different concentrations of ITH12505, from 0.3 μM to 100 μM for 10 min and then a concentrated solution of KCl was administered, to get a final extracellular K+ concentration of 70 mM. The Ca2+-antagonist nimodipine (Nimo) was used as reference.3 The same experiment was carried out with CGP37157. Compound ITH12505 blocked K+-induced Ca2+ entry in a concentration-dependent manner, with a maximum at 100 μM, where Ca2+ entry was reduced to 12% (Figure 8a). Nimodipine, at the concentration of 10 μM, attenuated Ca2+ entry up to 42%. As previously described in chromaffin cells,27 CGP37157 was also able to block Ca2+ entry in SH-SY5Y neuroblastoma cells. This activity was similar to that found for ITH12505 (Figure 8b); maximal blockade was found at 100 μM, where Ca2+ entry was reduced to 13%. Figure 8c shows a comparative plot of the concentration–responses curves for the blockade elicited by both compounds. Traces represent nonlinear regression percentages of blockade with respect to control versus concentration. In this graph, CGP37157 exhibited an EC50 to block Ca2+ entry of 6.12 μM, while compound ITH12505 had an EC50 of 6.80 μM. Hence, the VDCCs blocking activity of both CGP37157 and ITH12505 is quite similar in SH-SY5Y cells, with no statistically significant differences between them from 0.3 to 100 μM. Furthermore, at the concentrations displaying neuroprotective activity, ITH12505 and CGP37157 have also targeted VDCCs. On the one hand, this second target limits the validity of ITH12505 and CGP37157 to clearly implicate the mNCX as the only target to justify their neuroprotective properties. On the other, by contrast, the simultaneous blockade of the mNCX and the L-type VDCC could be convenient to increase their efficacy in re-establishing the Ca2+ balance that is known to be perturbed in most brain disorders leading to neuronal loss, namely, neurodegenerative diseases, stroke, and spinal cord/brain trauma. In this context, it is noticeable that CGP37157 reduces the severity of ischemic damage occurring during middle cerebral artery occlusion in the rat57 and reproduces the mitochondrial Ca2+ imbalance seen in AD patients.58
Figure 8.

Blockade by ITH12505 (a) and CGP37157 (b) on the [Ca2+]c increase induced by high K+ in SH-SY5Y neuroblastoma cells. Panel (c) shows nonlinear regressions of increments of blockade versus concentration for ITH12505 (black circles, continuous line) and CGP37157 (white circles, dotted line). Data are mean ± SEM of at least four independent experiments: **p < 0.01, ***p < 0.001 respect to control.
The dual action over both VDCCs and mNCX makes our hypothesis fulfill the queries of the multitarget strategy about the search of new drugs for the treatment of AD. This theory establishes that one drug, possessing moderate activities on two or more targets, will be more effective than highly potent, selective drugs acting on a single target.59 Examples of this approach are widely found in AD therapeutics.25,60 On this assumption, we have found that subeffective concentrations of neuroprotective compounds have additive or even synergistic neuroprotective effects.61 In high contrast with this approach is the single-target design of highly potent compounds that target, for instance, Aβ aggregation or γ-secretase inhibition; in phase-II clinical trials, these compounds have shown relevant adverse events without improving cognition.62,63
In Vitro Blood–Brain Barrier Permeation Assay
In today’s drug discovery research, screening for the blood–brain barrier (BBB) penetration is of great importance. One major problem for successful CNS drugs is to cross the BBB and reach their therapeutic targets. On the other hand, for drugs acting in peripheral tissues, penetration into the central nervous system (CNS) might cause unwanted side effects. In the last years, several in silico/in vitro methods have been used to predict the BBB permeation of investigational drugs. Among them, the parallel artificial membrane permeation assays (PAMPA) have the advantage of predicting passive blood–brain barrier permeation with high success, high throughput, and reproducibility.
To evaluate the brain penetration of ITH12505 and CGP37157 we used the PAMPA-BBB method described by Di et al.64 and subsequently optimized by Rodríguez-Franco and co-workers for molecules with limited water-solubility.60,65 In the last years, this later method was successfully applied by some of us to different classes of compounds.24,66−69 The in vitro permeability (Pe) values of ITH12505 and CGP37157 through a lipid extract of porcine brain were determined by using PBS/ethanol (70:30) (Table 1). In the same assay, 15 commercial drugs of known CNS penetration were also tested and their experimental values were compared to reported values, giving a good lineal correlation. Both tested compounds showed good permeability values, higher than that reported for verapamil (Pe = 16 × 10–6 cm s–1) that is generally used as a high permeability standard.64 Consequently, it is expected both compounds could penetrate into the CNS and reach their cerebral targets.
Table 1. Brain Penetration of ITH12505 and CGP37157 Measured by PAMPA-BBB (Pe = 10–6 cm s–1)a.

Data are the mean ± SD of three independent experiments.
Permeability values (Pe) are expressed as ×10–6 cm s–1, and PBS/EtOH (70:30) was used as solvent.
Our study suggests that even minimal changes in the functional groups of the benzothiazepine CGP37157 could modify its neuroprotective profile. Therefore, we predict that further modeling may augment the scope of neuroprotection afforded by benzothiazepine derivatives, and it could also lead to greater potency and improved pharmacokinetic properties. It must be stressed that CGP37157 presents poorer water solubility and a short half-life.36 We hypothesized that the isosteric replacement of the chlorine by a methyl group at C2′ could lead to a higher blood–brain barrier cross, as increasing lipophilicity is expected in ITH12505. However, CGP37157 showed a slight better cross of the blood–brain barrier than ITH12505 in the in vitro PAMPA assay. Otherwise, both compounds present an optimal result, with Pe values above 20 × 106 cm s–1, higher than those found for well-known CNS drugs, such as the Ca2+ antagonist verapamil (Pe = 16 × 10–6 cm s–1).
Conclusions
In conclusion, on the basis of the findings presented herein, we are designing and synthesizing new benzothiazepine derivatives having a multitarget neuroprotective profile, by acting at least on two of the numerous ion channels and transporters controlling the delicate Ca2+ balance of neurons, namely, the mNCX and the L-type (α1D) VDCC. We have found that benzothiazepine CGP37157 and its isosteric derivative ITH12505 exhibit neuroprotective properties in various stress models of cytotoxicity. We have confirmed that CGP37157 shows interesting neuroprotective properties against Ca2+ overload-induced neurotoxicity, and described that ITH12505 represents an improve in its neuroprotective profile, mainly in oxidative stress-inducing stimuli, and maintaining partially the activity against Ca2+ overload toxic stimuli. This feature can be interesting taking into account the rising interest in the oxidative stress-induced mechanisms leading to AD.52 There are a huge amount of contributions highlighting such a relationship. For instance, elevated biomarkers of oxidative stress have already been found in the light-moderate stage of the disease.70 Also, links between oxidative stress and alterations of the cytoskeleton, thereby correlating with tau-triggered neurodegeneration, have been reported.71 Moreover, it seems that ITH12505 presents a wider therapeutic range, as it did not show toxicity in SH-SY5Y at micromolar concentrations, unlike CGP37157, which exerted a signficative loss of cell viability at 30 μM.
The mechanism of the neuroprotective action of compounds ITH12505 and CGP37157 is still poorly understood. Inasmuch as both compounds exerted a blocking effect on Ca2+ entry (Figure 8), likely occurring through L-type (α1D, CaV 1.3) VDCCs expressed by SH-SY5Y cells,72 an action on Ca2+ homeostatic mechanisms could be the underlying mechanism for neuroprotection. Since CGP37157 was first shown to target the mNCX,32 this has been the object of numerous studies to figure the Ca2+ handling by mitochondria and its effect on cell function.39,45
More recently, the mNCX has been considered an interesting target to develop novel compounds for neuroprotection.30 In this context, blockade of the mNCX by ITH12505 and CGP37157 could be a contributing factor in their protection effect. Thus, futile mitochondrial Ca2+ cycling (mCC) causes energy dissipation, what is particularly active in neurons overstimulated by glutamate.73 Under this frame, mitigation of the rate of mitochondrial Ca2+ efflux into the cytosol by ITH12505 and CGP37157 could slow down the rate of futile mCC, and reduce the level of [Ca2+]c that, as stated in the introduction, is critical to trigger survival, as well as cell death signaling pathways.6
Methods
Reagents
N-Acetylcysteine (NAC), dimethyl sulfoxide (DMSO), glutamic acid, melatonin, memantine, nimodipine, rotenone, and oligomycin A were purchased from Sigma Aldrich (Madrid, Spain). CGP37157 was purchased from Tocris House (Bristol, U.K.). Fluo-4/AM was purchased from Molecular Probes (Invitrogen, Barcelona, Spain).
Synthesis of 7-Chloro-5-o-tolyl-1,2,3,5-tetrahydro-4,1-benzothiazepiin-2-one (ITH12505)
The isosteric analogue of CGP37157 where chlorine at position orto of the phenyl ring (C2′) has been replaced by a methyl group (Figure 1), namely, ITH12505, was synthesized according to the experimental procedure described,36 obtaining ITH12505 with a 58% yield, showing analytical and spectral data according to the literature.36
Culture of Chromaffin Cells
Adrenal glands were obtained from the city slaughterhouse, under the supervision of the local veterinary service. Bovine adrenal medullar chromaffin cells were isolated as described previously, with some modifications.74 Cells were suspended in Dulbecco's modified Eagle's medium (DMEM, Invitrogen, Madrid, Spain), supplemented with 5% fetal bovine serum, 50 IU mL–1 penicillin, and 50 μg mL–1 streptomycin. Cells were preplated for 30 min and proliferation inhibitors (10 μM cytosine arabinoside, 10 μM fluorodeoxyuridine, and 10 μM leucine methyl ester) were added to the medium to prevent excessive growth of fibroblasts that could mask cell death measurements. Total cell number was determined as described previously. For cell death studies, cells were plated at a density of 5 × 105 cells per well on 24-well dishes. Cultures were maintained in an incubator at 37 °C in a humidified atmosphere with 5% CO2. Cell treatments were performed in serum-free DMEM, as serum interferes with LDH measurements.
Culture of SH-SY5Y Neuroblastoma Cells
SH-SY5Y cells were maintained in a 1:1 mixture of F-12 nutrient mixture (Ham12) (Sigma-Aldrich, Madrid, Spain) and Eagle’s minimum essential medium (EMEM) supplemented with 15 nonessential aminoacids, 1 mM sodium pyruvate, 10% heat-inactivated fetal bovine serum (FBS), 100 IU mL–1 penicillin, and 100 μg mL–1 streptomycin (reagents from Invitrogen, Madrid, Spain). Cultures were seeded into flasks containing supplemented medium and maintained at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. For assays, SH-SY5Y cells were subcultured in 48 well plates at a seeding density of 1 × 105 cells per well. Cells were treated with the drugs before confluence in EMEM with 1% FBS. All cells were used at a low passage number (<13).
Cell Incubation with Compounds Solutions
To prepare stock solutions of the various reagents, they were dissolved in DMSO at the concentration of 10–2 M. All solutions were stored in aliquots at −20 °C. Once defrosted for a given experiment, the aliquot was discarded. The final concentrations of DMSO used (always <0.1%) did not cause cell toxicity.
LDH Assay
Samples of incubation media were collected at the end of the 24 h cell incubation period with veratridine alone or with a given compound, in order to estimate extracellular LDH, an indication of cell death.75 LDH activity was measured in cells (5 × 105 per well) after treatment with 10% Triton X-100 (intracellular LDH). LDH activity was measured spectrophotometrically at 490 and 620 nm using a microplate reader (iEMS reader MF; Thermo Fisher Scientific, Waltham, MA). Total LDH activity was defined as the sum of intracellular plus extracellular LDH activity. Released LDH was defined as the percentage of extracellular compared with total LDH activity.
MTT Assay
Cell viability, virtually the mitochondrial activity of living cells, was measured by quantitative colorimetric assay with the dye MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide), as described previously.76 SH-SY5Y cells were seeded into 48-well culture. MTT was added to the wells (5 mg mL–1) and allowed to incubate in the dark at 37 °C for 2 h, followed by cell lysis. The tetrazolium ring of MTT can be cleaved by active reductases in order to produce a precipitated formazan derivative. The generated formazan was dissolved by adding 200 μL of DMSO, resulting in a colored solution whose optical density was measured in a colorimetric plate reader at 540 nm (FLUOstar Optima, BMG, Germany). All MTT assays were performed in triplicate. Data were expressed as percentage of MTT reduction, taking the maximum control tissue capability in each individual experiment as 100%. In some part of the text, data are discussed as percentage of protection afforded by a given treatment; for instance, a 50% decrease of MTT reduction means 50% cell death; hence, a decrease of 25% cell death by a given treatment means 50% neuroprotection.
Neuronal Viability Experiments in Rat Hippocampal Slices Stressed with Veratridine or Glutamate
Experiments were performed in hippocampal slices prepared from brains of 2-month-old Sprague–Dawley rats (275–325 g weight), following the European Union Council Directive issued for these purposes, and were approved by the Ethics Committee of the Facultad de Medicina, Universidad Autónoma de Madrid, Spain. All efforts were made to minimize the number of animals and their suffering. We followed the protocol described by Egea and co-workers77 with slight modifications.78 Rats were quickly decapitated under sodium pentobarbital anesthesia (60 mg kg–1, ip). Forebrains were rapidly removed from the skull and placed into ice-cold Krebs-bicarbonate dissection buffer (pH 7.4) containing the following: NaCl 120 mM, KCl 2 mM, CaCl2 0.5 mM, NaHCO3 26 mM, MgSO4 10 mM, KH2PO4 1.18 mM, glucose 11 mM, and sucrose 200 mM. Hippocampi were quickly dissected, and slices (350 μm thick) were rapidly prepared using a McIlwain tissue chopper, separated in Krebs buffer at 4 °C, and allowed to recover for 45 min in Krebs-bicarbonate buffer at 34 °C. Experiments were performed at 37 °C. A control and neurotoxicity group was included in all experiments. To perform the experiments, we followed the protocols shown on top of the figures and briefly described in their legends. Hippocampal slices were collected immediately after the neurotoxic compound exposure period and were incubated with MTT (0.5 mg mL–1) in Krebs-bicarbonate solution for 30 min at 37 °C. Hippocampal slice viability was determined by the ability of cells to reduce MTT.76 Formazan production was measured as described above.
Viability Experiments in Rat Hippocampal Slices Subjected to Oxygen and Glucose Deprivation Plus Reoxygenation (OGD)
Following the general experimental procedure for the isolation of hippocampal slices, after the 45 min period of stabilization at 34 °C, slices submitted to oxygen and glucose deprivation (OGD) plus reoxygenation were incubated in a glucose-free Krebs solution, equilibrated with a 95% N2 and 5% CO2 gas mixture. Glucose was replaced by 2-deoxyglucose. After this OGD period, slices were returned to an oxygenated normal Krebs solution containing glucose (reoxygenation period). Experiments were performed at 37 °C. A basal and toxicant group was included in all experiments. To perform the experiments, we followed the protocols shown on top of the figures and briefly described in their legends. Hippocampal slice viability was determined as mentioned above.
Measurement of Cytosolic Ca2+ Concentrations, [Ca2+]c
SH-SY5Y neuroblastoma cells were grown at confluence in 96-well black dishes. Cells were loaded with 4 μM Fluo-4/AM for 45 min at 37 °C in EMEM. Cells were then washed with Krebs-Hepes solution and kept at room temperature for 10 min before the beginning of the experiment. Compounds were incubated 5 min before application of K+ (70 mM) to enhance VDCC opening and [Ca2+]c increase. At the end of the experiment, Triton X-100 (5%) and 1 mM MnCl2 were applied to record maximal and basal fluorescence, respectively. Fluorescence was measured in a fluorescence microplate reader (FLUOstar Optima, BMG, Germany). Wavelengths of excitation and emission were 485 and 520 nm, respectively. Data were represented as percentage of fluorescence increase with respect to control, according to the formula ΔF/ ΔFcontrol ×100, where ΔF is the percent increase of fluorescence with respect to the Fmax–Fmin interval.
In Vitro Blood–Brain Barrier Permeation Assay by PAMPA Method
Prediction of the brain penetration was evaluated using a parallel artificial membrane permeation assay (PAMPA), in a similar manner as described previously.24,60,64−66 Commercial drugs, phosphate buffered saline solution at pH 7.4 (PBS), and dodecane were purchased from Sigma, Aldrich, Acros, and Fluka. Millex filter units (PVDF membrane, diameter 25 mm, pore size 0.45 μm) were acquired from Millipore. The porcine brain lipid (PBL) was obtained from Avanti Polar Lipids. The donor microplate was a 96-well filter plate (PVDF membrane, pore size 0.45 μm) and the acceptor microplate was an indented 96-well plate, both from Millipore. The acceptor 96-well microplate was filled with 180 μL of PBS/ethanol (70:30) and the filter surface of the donor microplate was impregnated with 4 μL of porcine brain lipid (PBL) in dodecane (20 mg mL–1). Compounds were dissolved in PBS/ethanol (70:30) at 100 μg mL–1, filtered through a Millex filter, and then added to the donor wells (180 μL). The donor filter plate was carefully put on the acceptor plate to form a sandwich, which was left undisturbed for 240 min at 25 °C. After incubation, the donor plate is carefully removed and the concentration of compounds in the acceptor wells was determined by UV spectroscopy. Every sample is analyzed at five wavelengths, in four wells and at least in three independent runs, and the results are given as the mean ± standard deviation. In each experiment, 15 quality control standards of known BBB permeability were included to validate the analysis set.
Data Analysis
Data are presented as mean ± standard error of the mean (SEM). Comparisons between experimental and control groups were performed by one-way ANOVA followed by Newman-Keuls post hoc test. Differences were considered to be statistically significant when p ≤ 0.05. All statistical procedures were carried out using GraphPad Prism software version 5.0 for an IBM compatible computer.
Acknowledgments
We thank the continued support of Fundación de Investigación Biomédica, Hospital Universitario de la Princesa, and Fundación Teófilo Hernando.
Glossary
Abbreviations
- Aβ
amyloid beta peptide
- AD
Alzheimer's disease
- CGP
- CNS
central nervous system
- LDH
lactate dehydrogenase
- MEM
memantine
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- mNCX
mitochondrial Na+/Ca2+ exchanger
- NAC
N-acetylcysteine
- NMDA
N-methyl-d-aspartate
- O/R
rotenone plus oligomycin A
- τ
tau protein
- Ver
veratridine
- VDCC
voltage-dependent Ca2+ channels
Author Contributions
C.d.l.R., A.G.G., and M.V. oversaw and designed both chemical synthesis and pharmacology, M.G.L. oversaw experiments in hippocampal slices, M.I.R.-F. oversaw and designed PAMPA and ORAC experiments, J.E. designed and performed experiments in hippocampal slices, A.M.G.-D.D. designed experiments in chromaffin cells, L.G.-L. conducted experiments in SH-SY5Y cells and in hippocampal slices, R.L. designed and performed measurements of cytosolic Ca2+, F.J.M.-S. performed the chemical synthesis, C.M. conducted experiments in chromaffin cells, L.M. conducted PAMPA experiments, and C.P. performed ORAC experiments.
C.d.l.R., R.L., and L.G.-L. thank IS Carlos III for research contract under Miguel Servet Program. This work was supported by the following grants from Spanish Institutions to C.d.l.R.: Programa Miguel Servet (CP10/00531, IS Carlos III); to R.L.: Programa Miguel Servet (CP11/00165); A.G.G.: (1) Fundación CIEN, IS Carlos III, No. PI016/09; (2) MICINN, SAF2010–21795; (3) IS Carlos III, RD 06/0026 RETICS, RENEVAS; (4) Grant NDE 07/09, Agencia Lain Entralgo, Comunidad de Madrid; M.G.L.: (1) Spanish Ministry of Science and Innovation ref: SAF2009-12150 and (2) Spanish Ministry of Health (Instituto de Salud Carlos III) RETICS-RD06/0026; and M.I.R.-F.: Spanish Ministry of Science and Innovation (SAF2006-01249 and SAF2009-13015-C02-01).
The authors declare no competing financial interest.
References
- Wang J. M.; Sun C. (2010) Calcium and neurogenesis in Alzheimer’s disease. Front. Neurosci. 4, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson M. P. (2007) Calcium and neurodegeneration. Aging Cell 6, 337–350. [DOI] [PubMed] [Google Scholar]
- Cano-Abad M. F.; Villarroya M.; Garcia A. G.; Gabilan N. H.; Lopez M. G. (2001) Calcium entry through L-type calcium channels causes mitochondrial disruption and chromaffin cell death. J. Biol. Chem. 276, 39695–39704. [DOI] [PubMed] [Google Scholar]
- Koh J. Y.; Cotman C. W. (1992) Programmed cell death: its possible contribution to neurotoxicity mediated by calcium channel antagonists. Brain Res. 587, 233–240. [DOI] [PubMed] [Google Scholar]
- Turner C. P.; Connell J.; Blackstone K.; Ringler S. L. (2007) Loss of calcium and increased apoptosis within the same neuron. Brain Res. 1128, 50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike T.; Martin D. P.; Johnson E. M. Jr. (1989) Role of Ca2+ channels in the ability of membrane depolarization to prevent neuronal death induced by trophic-factor deprivation: evidence that levels of internal Ca2+ determine nerve growth factor dependence of sympathetic ganglion cells. Proc. Natl. Acad. Sci. U.S.A. 86, 6421–6425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z.; Wood N. W. (2009) Cell death pathways in Parkinson’s disease: role of mitochondria. Antioxid. Redox Signal. 11, 2135–2149. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Lipton P. (1999) Cytosolic Ca2+ changes during in vitro ischemia in rat hippocampal slices: major roles for glutamate and Na+-dependent Ca2+ release from mitochondria. J. Neurosci. 19, 3307–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosskreutz J.; Van Den Bosch L.; Keller B. U. (2010) Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium 47, 165–174. [DOI] [PubMed] [Google Scholar]
- Khachaturian Z. S. (1994) Calcium hypothesis of Alzheimer’s disease and brain aging. Ann. N.Y. Acad. Sci. 747, 1–11. [DOI] [PubMed] [Google Scholar]
- Resende R.; Pereira C.; Agostinho P.; Vieira A. P.; Malva J. O.; Oliveira C. R. (2007) Susceptibility of hippocampal neurons to Abeta peptide toxicity is associated with perturbation of Ca2+ homeostasis. Brain Res. 1143, 11–21. [DOI] [PubMed] [Google Scholar]
- Mattson M. P. (1994) Calcium and neuronal injury in Alzheimer’s disease. Contributions of beta-amyloid precursor protein mismetabolism, free radicals, and metabolic compromise. Ann. N.Y. Acad. Sci. 747, 50–76. [PubMed] [Google Scholar]
- Haughey N. J.; Nath A.; Chan S. L.; Borchard A. C.; Rao M. S.; Mattson M. P. (2002) Disruption of neurogenesis by amyloid beta-peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer’s disease. J. Neurochem. 83, 1509–1524. [DOI] [PubMed] [Google Scholar]
- Murray F. E.; Landsberg J. P.; Williams R. J.; Esiri M. M.; Watt F. (1992) Elemental analysis of neurofibrillary tangles in Alzheimer’s disease using proton-induced X-ray analysis. Ciba Found. Symp. 169, 201–210, discussion 210–206. [DOI] [PubMed] [Google Scholar]
- Dreses-Werringloer U.; Lambert J. C.; Vingtdeux V.; Zhao H.; Vais H.; Siebert A.; Jain A.; Koppel J.; Rovelet-Lecrux A.; Hannequin D.; Pasquier F.; Galimberti D.; Scarpini E.; Mann D.; Lendon C.; Campion D.; Amouyel P.; Davies P.; Foskett J. K.; Campagne F.; Marambaud P. (2008) A polymorphism in CALHM1 influences Ca2+ homeostasis, Abeta levels, and Alzheimer’s disease risk. Cell 133, 1149–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollefson G. D. (1990) Short-term effects of the calcium channel blocker nimodipine (Bay-e-9736) in the management of primary degenerative dementia. Biol. Psychiatry 27, 1133–1142. [DOI] [PubMed] [Google Scholar]
- Fillit H. M.; Doody R. S.; Binaso K.; Crooks G. M.; Ferris S. H.; Farlow M. R.; Leifer B.; Mills C.; Minkoff N.; Orland B.; Reichman W. E.; Salloway S. (2006) Recommendations for best practices in the treatment of Alzheimer’s disease in managed care. Am. J. Geriatr. Pharmacother. 4(Suppl A), S9–S24, quiz S25–S28. [DOI] [PubMed] [Google Scholar]
- van Marum R. J. (2008) Current and future therapy in Alzheimer’s disease. Fundam. Clin. Pharmacol. 22, 265–274. [DOI] [PubMed] [Google Scholar]
- Rauk A. (2009) The chemistry of Alzheimer’s disease. Chem. Soc. Rev. 38, 2698–2715. [DOI] [PubMed] [Google Scholar]
- Bulic B.; Pickhardt M.; Mandelkow E. M.; Mandelkow E. (2010) Tau protein and tau aggregation inhibitors. Neuropharmacology 59, 276–289. [DOI] [PubMed] [Google Scholar]
- Geula C.; Mesulam M. M. (1995) Cholinesterases and the pathology of Alzheimer disease. Alzheimer Dis. Assoc. Disord. 9(Suppl 2), 23–28. [DOI] [PubMed] [Google Scholar]
- Mangialasche F.; Solomon A.; Winblad B.; Mecocci P.; Kivipelto M. (2010) Alzheimer’s disease: clinical trials and drug development. Lancet Neurol. 9, 702–716. [DOI] [PubMed] [Google Scholar]
- Leon R.; de Los Rios C.; Marco-Contelles J.; Lopez M. G.; Garcia A. G.; Villarroya M. (2008) Synthesis of 6-amino-1,4-dihydropyridines that prevent calcium overload and neuronal death. Eur. J. Med. Chem. 43, 668–674. [DOI] [PubMed] [Google Scholar]
- Marco-Contelles J.; Leon R.; de los Rios C.; Samadi A.; Bartolini M.; Andrisano V.; Huertas O.; Barril X.; Luque F. J.; Rodriguez-Franco M. I.; Lopez B.; Lopez M. G.; Garcia A. G.; Carreiras Mdo C.; Villarroya M. (2009) Tacripyrines, the first tacrine-dihydropyridine hybrids, as multitarget-directed ligands for the treatment of Alzheimer’s disease. J. Med. Chem. 52, 2724–2732. [DOI] [PubMed] [Google Scholar]
- Egea J.; De Los Rios C. (2011) 1,8-Naphthyridine derivatives as cholinesterases inhibitors and cell Ca2+ regulators, a multitarget strategy for Alzheimer’s disease. Curr. Top. Med. Chem. 11, 2807–2823. [DOI] [PubMed] [Google Scholar]
- Moreno-Ortega A. J.; Ruiz-Nuno A.; Garcia A. G.; Cano-Abad M. F. (2010) Mitochondria sense with different kinetics the calcium entering into HeLa cells through calcium channels CALHM1 and mutated P86L-CALHM1. Biochem. Biophys. Res. Commun. 391, 722–726. [DOI] [PubMed] [Google Scholar]
- Nicolau S. M.; de Diego A. M.; Cortes L.; Egea J.; Gonzalez J. C.; Mosquera M.; Lopez M. G.; Hernandez-Guijo J. M.; Garcia A. G. (2009) Mitochondrial Na+/Ca2+-exchanger blocker CGP37157 protects against chromaffin cell death elicited by veratridine. J. Pharmacol. Exp. Ther. 330, 844–854. [DOI] [PubMed] [Google Scholar]
- Nicolau S. M.; Egea J.; Lopez M. G.; Garcia A. G. (2010) Mitochondrial Na+/Ca2+ exchanger, a new target for neuroprotection in rat hippocampal slices. Biochem. Biophys. Res. Commun. 400, 140–144. [DOI] [PubMed] [Google Scholar]
- Hernandez-SanMiguel E.; Vay L.; Santo-Domingo J.; Lobaton C. D.; Moreno A.; Montero M.; Alvarez J. (2006) The mitochondrial Na+/Ca2+ exchanger plays a key role in the control of cytosolic Ca2+ oscillations. Cell Calcium 40, 53–61. [DOI] [PubMed] [Google Scholar]
- Castaldo P.; Cataldi M.; Magi S.; Lariccia V.; Arcangeli S.; Amoroso S. (2009) Role of the mitochondrial sodium/calcium exchanger in neuronal physiology and in the pathogenesis of neurological diseases. Prog. Neurobiol. 87, 58–79. [DOI] [PubMed] [Google Scholar]
- Carafoli E.; Tiozzo R.; Lugli G.; Crovetti F.; Kratzing C. (1974) The release of calcium from heart mitochondria by sodium. J. Mol. Cell Cardiol. 6, 361–371. [DOI] [PubMed] [Google Scholar]
- Chiesi M.; Schwaller R.; Eichenberger K. (1988) Structural dependency of the inhibitory action of benzodiazepines and related compounds on the mitochondrial Na+-Ca2+ exchanger. Biochem. Pharmacol. 37, 4399–4403. [DOI] [PubMed] [Google Scholar]
- Cox D. A.; Conforti L.; Sperelakis N.; Matlib M. A. (1993) Selectivity of inhibition of Na(+)-Ca2+ exchange of heart mitochondria by benzothiazepine CGP-37157. J. Cardiovasc. Pharmacol. 21, 595–599. [DOI] [PubMed] [Google Scholar]
- Baron K. T.; Thayer S. A. (1997) CGP37157 modulates mitochondrial Ca2+ homeostasis in cultured rat dorsal root ganglion neurons. Eur. J. Pharmacol. 340, 295–300. [DOI] [PubMed] [Google Scholar]
- Czyz A.; Kiedrowski L. (2003) Inhibition of plasmalemmal Na(+)/Ca(2+) exchange by mitochondrial Na(+)/Ca(2+) exchange inhibitor 7-chloro-5-(2-chlorophenyl)-1,5-dihydro-4,1-benzothiazepin-2(3H)-one (CGP-37157) in cerebellar granule cells. Biochem. Pharmacol. 66, 2409–2411. [DOI] [PubMed] [Google Scholar]
- Pei Y.; Lilly M. J.; Owen D. J.; D’Souza L. J.; Tang X. Q.; Yu J.; Nazarbaghi R.; Hunter A.; Anderson C. M.; Glasco S.; Ede N. J.; James I. W.; Maitra U.; Chandrasekaran S.; Moos W. H.; Ghosh S. S. (2003) Efficient syntheses of benzothiazepines as antagonists for the mitochondrial sodium-calcium exchanger: potential therapeutics for type II diabetes. J. Org. Chem. 68, 92–103. [DOI] [PubMed] [Google Scholar]
- Meanwell N. A. (2011) Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem. 54, 2529–2591. [DOI] [PubMed] [Google Scholar]
- Maroto R.; De la Fuente M. T.; Artalejo A. R.; Abad F.; Lopez M. G.; Garcia-Sancho J.; Garcia A. G. (1994) Effects of Ca2+ channel antagonists on chromaffin cell death and cytosolic Ca2+ oscillations induced by veratridine. Eur. J. Pharmacol. 270, 331–339. [DOI] [PubMed] [Google Scholar]
- Garcia A. G.; Garcia-De-Diego A. M.; Gandia L.; Borges R.; Garcia-Sancho J. (2006) Calcium signaling and exocytosis in adrenal chromaffin cells. Physiol. Rev. 86, 1093–1131. [DOI] [PubMed] [Google Scholar]
- Novalbos J.; Abad-Santos F.; Zapater P.; Cano-Abad M. F.; Moradiellos J.; Sanchez-Garcia P.; Garcia A. G. (1999) Effects of dotarizine and flunarizine on chromaffin cell viability and cytosolic Ca2+. Eur. J. Pharmacol. 366, 309–317. [DOI] [PubMed] [Google Scholar]
- Egea J.; Rosa A. O.; Cuadrado A.; Garcia A. G.; Lopez M. G. (2007) Nicotinic receptor activation by epibatidine induces heme oxygenase-1 and protects chromaffin cells against oxidative stress. J. Neurochem. 102, 1842–1852. [DOI] [PubMed] [Google Scholar]
- Galkin A.; Abramov A. Y.; Frakich N.; Duchen M. R.; Moncada S. (2009) Lack of oxygen deactivates mitochondrial complex I: implications for ischemic injury?. J. Biol. Chem. 284, 36055–36061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd S.; Dean O.; Copolov D. L.; Malhi G. S.; Berk M. (2008) N-acetylcysteine for antioxidant therapy: pharmacology and clinical utility. Expert Opin. Biol. Ther. 8, 1955–1962. [DOI] [PubMed] [Google Scholar]
- Choi D. W. (1988) Glutamate neurotoxicity and diseases of the nervous system. Neuron 1, 623–634. [DOI] [PubMed] [Google Scholar]
- Abramov A. Y.; Duchen M. R. (2010) Impaired mitochondrial bioenergetics determines glutamate-induced delayed calcium deregulation in neurons. Biochim. Biophys. Acta 1800, 297–304. [DOI] [PubMed] [Google Scholar]
- Molz S.; Dal-Cim T.; Budni J.; Martin-de-Saavedra M. D.; Egea J.; Romero A.; del Barrio L.; Rodrigues A. L.; Lopez M. G.; Tasca C. I. (2011) Neuroprotective effect of guanosine against glutamate-induced cell death in rat hippocampal slices is mediated by the phosphatidylinositol-3 kinase/Akt/ glycogen synthase kinase 3beta pathway activation and inducible nitric oxide synthase inhibition. J. Neurosci. Res. 89, 1400–1408. [DOI] [PubMed] [Google Scholar]
- Volbracht C.; van Beek J.; Zhu C.; Blomgren K.; Leist M. (2006) Neuroprotective properties of memantine in different in vitro and in vivo models of excitotoxicity. Eur. J. Neurosci. 23, 2611–2622. [DOI] [PubMed] [Google Scholar]
- Siqueira I. R.; Cimarosti H.; Fochesatto C.; Nunes D. S.; Salbego C.; Elisabetsky E.; Netto C. A. (2004) Neuroprotective effects of Ptychopetalum olacoides Bentham (Olacaceae) on oxygen and glucose deprivation induced damage in rat hippocampal slices. Life Sci. 75, 1897–1906. [DOI] [PubMed] [Google Scholar]
- Mozes E.; Hunya A.; Posa A.; Penke B.; Datki Z. (2012) A novel method for the rapid determination of beta-amyloid toxicity on acute hippocampal slices using MTT and LDH assays. Brain Res. Bull. 87, 521–525. [DOI] [PubMed] [Google Scholar]
- Sobrado M.; Roda J. M.; Lopez M. G.; Egea J.; Garcia A. G. (2004) Galantamine and memantine produce different degrees of neuroprotection in rat hippocampal slices subjected to oxygen-glucose deprivation. Neurosci. Lett. 365, 132–136. [DOI] [PubMed] [Google Scholar]
- Martinez-Sanchez M.; Striggow F.; Schroder U. H.; Kahlert S.; Reymann K. G.; Reiser G. (2004) Na(+) and Ca(2+) homeostasis pathways, cell death and protection after oxygen-glucose-deprivation in organotypic hippocampal slice cultures. Neuroscience 128, 729–740. [DOI] [PubMed] [Google Scholar]
- Bonda D. J.; Wang X.; Perry G.; Nunomura A.; Tabaton M.; Zhu X.; Smith M. A. (2010) Oxidative stress in Alzheimer disease: a possibility for prevention. Neuropharmacology 59, 290–294. [DOI] [PubMed] [Google Scholar]
- Lukic-Panin V.; Kamiya T.; Zhang H.; Hayashi T.; Tsuchiya A.; Sehara Y.; Deguchi K.; Yamashita T.; Abe K. (2007) Prevention of neuronal damage by calcium channel blockers with antioxidative effects after transient focal ischemia in rats. Brain Res. 1176, 143–150. [DOI] [PubMed] [Google Scholar]
- Davalos A.; Gomez-Cordoves C.; Bartolome B. (2004) Extending applicability of the oxygen radical absorbance capacity (ORAC-fluorescein) assay. J. Agric. Food Chem. 52, 48–54. [DOI] [PubMed] [Google Scholar]
- Vaghy P. L.; Johnson J. D.; Matlib M. A.; Wang T.; Schwartz A. (1982) Selective inhibition of Na+-induced Ca2+ release from heart mitochondria by diltiazem and certain other Ca2+ antagonist drugs. J. Biol. Chem. 257, 6000–6002. [PubMed] [Google Scholar]
- Uceda G.; Garcia A. G.; Guantes J. M.; Michelena P.; Montiel C. (1995) Effects of Ca2+ channel antagonist subtypes on mitochondrial Ca2+ transport. Eur. J. Pharmacol. 289, 73–80. [DOI] [PubMed] [Google Scholar]
- Pilitsis J. G.; Diaz F. G.; O’Regan M. H.; Phillis J. W. (2002) Inhibition of mitochondrial Na(+)/Ca(2+) exchange by 7-chloro-5-(2-chlorophenyl)-1,5-dihydro-4,1-benzothiazepin-2(3H)-one attenuates free fatty acid efflux in rat cerebral cortex during ischemia-reperfusion injury. Neurosci. Lett. 321, 1–4. [DOI] [PubMed] [Google Scholar]
- Thiffault C.; Bennett J. P. Jr. (2005) Cyclical mitochondrial deltapsiM fluctuations linked to electron transport, F0F1 ATP-synthase and mitochondrial Na+/Ca+2 exchange are reduced in Alzheimer’s disease cybrids. Mitochondrion 5, 109–119. [DOI] [PubMed] [Google Scholar]
- Espinoza-Fonseca L. M. (2006) The benefits of the multi-target approach in drug design and discovery. Bioorg. Med. Chem. 14, 896–897. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Franco M. I.; Fernandez-Bachiller M. I.; Perez C.; Hernandez-Ledesma B.; Bartolome B. (2006) Novel tacrine-melatonin hybrids as dual-acting drugs for Alzheimer disease, with improved acetylcholinesterase inhibitory and antioxidant properties. J. Med. Chem. 49, 459–462. [DOI] [PubMed] [Google Scholar]
- Romero A.; Egea J.; Garcia A. G.; Lopez M. G. (2010) Synergistic neuroprotective effect of combined low concentrations of galantamine and melatonin against oxidative stress in SH-SY5Y neuroblastoma cells. J. Pineal. Res. 49, 141–148. [DOI] [PubMed] [Google Scholar]
- Ritchie C. W.; Bush A. I.; Mackinnon A.; Macfarlane S.; Mastwyk M.; MacGregor L.; Kiers L.; Cherny R.; Li Q. X.; Tammer A.; Carrington D.; Mavros C.; Volitakis I.; Xilinas M.; Ames D.; Davis S.; Beyreuther K.; Tanzi R. E.; Masters C. L. (2003) Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch. Neurol. 60, 1685–1691. [DOI] [PubMed] [Google Scholar]
- Frolich L.; Ashwood T.; Nilsson J.; Eckerwall G. (2011) Effects of AZD3480 on cognition in patients with mild-to-moderate Alzheimer’s disease: a phase IIb dose-finding study. J. Alzheimer's Dis. 24, 363–374. [DOI] [PubMed] [Google Scholar]
- Di L.; Kerns E. H.; Fan K.; McConnell O. J.; Carter G. T. (2003) High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem. 38, 223–232. [DOI] [PubMed] [Google Scholar]
- Reviriego F.; Rodriguez-Franco M. I.; Navarro P.; Garcia-Espana E.; Liu-Gonzalez M.; Verdejo B.; Domenech A. (2006) The sodium salt of diethyl 1H-pyrazole-3,5-dicarboxylate as an efficient amphiphilic receptor for dopamine and amphetamines. crystal structure and solution studies. J. Am. Chem. Soc. 128, 16458–16459. [DOI] [PubMed] [Google Scholar]
- Fernandez-Bachiller M. I.; Perez C.; Campillo N. E.; Paez J. A.; Gonzalez-Munoz G. C.; Usan P.; Garcia-Palomero E.; Lopez M. G.; Villarroya M.; Garcia A. G.; Martinez A.; Rodriguez-Franco M. I. (2009) Tacrine-melatonin hybrids as multifunctional agents for Alzheimer’s disease, with cholinergic, antioxidant, and neuroprotective properties. ChemMedChem 4, 828–841. [DOI] [PubMed] [Google Scholar]
- Arce M. P.; Rodriguez-Franco M. I.; Gonzalez-Munoz G. C.; Perez C.; Lopez B.; Villarroya M.; Lopez M. G.; Garcia A. G.; Conde S. (2009) Neuroprotective and cholinergic properties of multifunctional glutamic acid derivatives for the treatment of Alzheimer’s disease. J. Med. Chem. 52, 7249–7257. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Munoz G. C.; Arce M. P.; Lopez B.; Perez C.; Romero A.; del Barrio L.; Martin-de-Saavedra M. D.; Egea J.; Leon R.; Villarroya M.; Lopez M. G.; Garcia A. G.; Conde S.; Rodriguez-Franco M. I. (2011) N-acylaminophenothiazines: neuroprotective agents displaying multifunctional activities for a potential treatment of Alzheimer’s disease. Eur. J. Med. Chem. 46, 2224–2235. [DOI] [PubMed] [Google Scholar]
- Fernandez-Bachiller M. I.; Perez C.; Monjas L.; Rademann J.; Rodriguez-Franco M. I. (2012) New tacrine-4-oxo-4H-chromene hybrids as multifunctional agents for the treatment of Alzheimer’s disease, with cholinergic, antioxidant, and beta-amyloid-reducing properties. J. Med. Chem. 55, 1303–1317. [DOI] [PubMed] [Google Scholar]
- Zafrilla P.; Mulero J.; Xandri J. M.; Santo E.; Caravaca G.; Morillas J. M. (2006) Oxidative stress in Alzheimer patients in different stages of the disease. Curr. Med. Chem. 13, 1075–1083. [DOI] [PubMed] [Google Scholar]
- Santa-Maria I.; Smith M. A.; Perry G.; Hernandez F.; Avila J.; Moreno F. J. (2005) Effect of quinones on microtubule polymerization: a link between oxidative stress and cytoskeletal alterations in Alzheimer’s disease. Biochim. Biophys. Acta 1740, 472–480. [DOI] [PubMed] [Google Scholar]
- Reuveny E.; Narahashi T. (1993) Two types of high voltage-activated calcium channels in SH-SY5Y human neuroblastoma cells. Brain Res. 603, 64–73. [DOI] [PubMed] [Google Scholar]
- Wang G. J.; Thayer S. A. (2002) NMDA-induced calcium loads recycle across the mitochondrial inner membrane of hippocampal neurons in culture. J. Neurophysiol. 87, 740–749. [DOI] [PubMed] [Google Scholar]
- Moro M. A.; Lopez M. G.; Gandia L.; Michelena P.; Garcia A. G. (1990) Separation and culture of living adrenaline- and noradrenaline-containing cells from bovine adrenal medullae. Anal. Biochem. 185, 243–248. [DOI] [PubMed] [Google Scholar]
- Koh J. Y.; Choi D. W. (1987) Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J. Neurosci. Methods 20, 83–90. [DOI] [PubMed] [Google Scholar]
- Denizot F.; Lang R. (1986) Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 89, 271–277. [DOI] [PubMed] [Google Scholar]
- Egea J.; Rosa A. O.; Sobrado M.; Gandia L.; Lopez M. G.; Garcia A. G. (2007) Neuroprotection afforded by nicotine against oxygen and glucose deprivation in hippocampal slices is lost in alpha7 nicotinic receptor knockout mice. Neuroscience 145, 866–872. [DOI] [PubMed] [Google Scholar]
- de Los Rios C.; Egea J.; Marco-Contelles J.; Leon R.; Samadi A.; Iriepa I.; Moraleda I.; Galvez E.; Garcia A. G.; Lopez M. G.; Villarroya M.; Romero A. (2010) Synthesis, inhibitory activity of cholinesterases, and neuroprotective profile of novel 1,8-naphthyridine derivatives. J. Med. Chem. 53, 5129–5143. [DOI] [PubMed] [Google Scholar]