

Abstract
This article presents the case of an adult patient with tuberous sclerosis complex who presented with large right benign and left malignant Leydig cell tumors. The tumors were examined to determine if they showed the classic hallmarks of TSC1/TSC2 involvement.
Keywords: Tuberous sclerosis, Leydig cell tumor, TSC1
Introduction
Leydig cell tumors (LCT) are the most common form of adult sex cord stromal tumors, but they account for less than 5% of all testicular tumors. There are few known genetic or environmental risk factors for sex cord stromal tumors. Cryptorchidism is a contributory cause. Two cases of adult Leydig cell tumors in patients with mutations in fumarate hydratase have been reported [1]. Two previous reports identified the occurrence of Leydig cell tumors in the neurocutaneous disorder tuberous sclerosis complex (TSC) [2, 3]. We recently encountered an adult patient with TSC who also presented with large right benign and left malignant Leydig cell tumors. TSC is associated with the development of a wide variety of tumors in multiple organ systems, including the brain, eyes, heart, lungs, kidneys, thyroid, and pancreas. It is due to mutations in either of two genes, TSC1 and TSC2 [4, 5, 6].
The protein products of the TSC1 and TSC2 genes form a complex that serves a critical function in the regulation of mammalian target of rapamycin (mTOR) complex 1 (mTORC1) [7, 8]. Both TSC1 and TSC2 proteins are required for the TSC1/TSC2 complex to function properly in this regulation. Complete loss of either TSC1 or TSC2 typically occurs in TSC-related tumors through a two-hit mechanism in which germline mutation in either gene is complemented by second hit loss of the remaining allele, which can be detected by analysis of heterozygosity in the tumor. In the absence of functional TSC1/TSC2 complex, mTORC1 activation leads to downstream kinase activation and phosphorylation of ribosomal protein S6, which serves as a convenient marker in immunohistochemistry studies.
In the light of previous reports of Leydig cell tumors in TSC, we examined whether the LCT tumors developing in this individual showed these classic hallmarks of TSC1/TSC2 involvement, similar to other tumors in TSC.
Case Report
A male infant was diagnosed with tuberous sclerosis complex (TSC) at 1 year of age when he presented with onset of infantile spasms. Wood's lamp examination demonstrated hypopigmented macules on his trunk but no other features indicative of TSC. At age 9 years, he had severe learning difficulties and limited speech capacity. Physical examination showed facial angiofibromas and periungal fibromas, typical of TSC. A computed tomography scan of the brain identified subependymal nodules, also consistent with TSC. Renal ultrasound showed no abnormalities.
His mother and brother were also affected with TSC. Genetic testing of family members identified a TSC1 exon 17 mutation c.2074C>T (R692X).
The patient presented at age 27 with right groin swelling. Ultrasound examination showed a 1-cm cystic area at the superior pole of the normally situated right testis and a left undescended testicle, with an ill-defined circular lesion consistent with testicular carcinoma. A left orchidectomy was carried out; histopathology confirmed a well-circumscribed 1.5-cm mass with pathology consistent with a Leydig cell tumor. There was virtually no staining for lipofuscin pigment and strong staining for inhibin and vimentin. No tumor spread into the tunica albuginea or rete testis was identified. Follow-up orchidectomy on the normally situated right testis was performed 12 months later because of concern that the cystic area also represented a malignancy. Pathology showed a well-circumscribed area of Leydig cell hyperplasia, but no evidence of Leydig cell tumor.
DNA was prepared from paraffin sections of tumor and normal tissue from the patient using the Gentra Puregene kit (Qiagen, Gaithersburg, MD). TSC1 exon 17 was amplified and sequenced by Sanger methodology. Tumor sections were examined by immunohistochemistry for tuberin and pS6-S235/236 (Cell Signaling Technology, Beverly, MA) expression.
Results
Both tumor and normal tissue were heterozygous for the TSC1 germline mutation exon 17 c.2074C>T R692X (Fig. 1A) known to be present in this patient. This indicates that there was retention of the wild-type TSC1 allele in the tumor and no loss of heterozygosity. Immunohistochemistry staining indicated that tuberin (TSC2 gene product) was expressed at relatively high levels in the tumor, whereas phosphorylated S6 (at residues S235/236) was not seen (Fig. 1B). Complete loss of TSC1 typically leads to markedly reduced levels of TSC2 (tuberin) expression, as TSC2 is not stable in the absence of TSC1. Thus, these three studies were all consistent in indicating that there was not complete loss of TSC1 in this tumor, with expression of TSC2 and absence of mTORC1 activation. These findings are all against the hypothesis that this LCT had developed through the two-hit mechanism predicted by the classic tumor suppressor gene model of tumor development in TSC.
Figure 1.
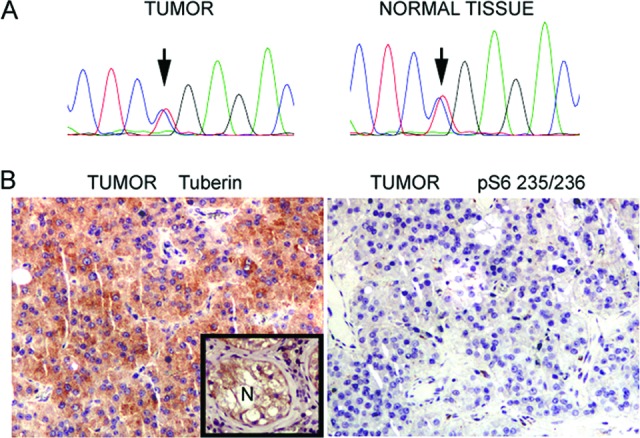
Histological and molecular studies on a Leydig cell tumor from a patient with tuberous sclerosis complex. (A): Sanger sequencing trace from tumor and normal tissue shows the TSC1 germline mutation (exon 17 c.2074C>T), heterozygous in both samples. (B): Immunostaining of the tumor: tuberin with inset positive control (normal tissue, left) and pS6–S235/236 (right).
Discussion
The molecular basis of Leydig cell tumors is poorly understood. Activating mutations in the luteinizing hormone receptor are a cause of childhood Leydig cell hyperplasia and adenoma, by driving Leydig cell growth and causing familial male-limited gonadotropin-independent precocious puberty due to excess testosterone production [9]. Fumarate hydratase mutations, which cause hereditary leiomyomatosis and renal cell cancer (HLRCC) syndrome, have been reported in association with adult Leydig cell tumors at least one case, and possibly in a second case [1].
Our patient with a TSC1 R692X mutation with bilateral Leydig cell abnormalities stimulated us to examine the possible role of the TSC genes in LCT development [9]. The results suggest that the TSC1-TSC2 protein complex is expressed at normal levels and mTORC1 signalling is not overactive in the tumor tissue in this case. The tumor likely developed independent of his TSC1 gene mutation and is therefore unlikely to be directly related to TSC. The two other cases of TSC and LCT in the literature may represent simple coincidence and/or reporting bias. In our case, the testicular tumors developed bilaterally, suggesting the possibility of TSC1 genetic predisposition. The right testis, which was normally situated in the scrotal sac, developed LC hyperplasia; the undescended left testis developed LCT. It appears likely that the lack of descent of the left testis contributed to testicular tumor development, and this may have been the primary cause in this patient. Of note, no family members had any evidence of cutaneous leiomyomas, arguing against concurrent HLRCC in this patient.
No other cases of LCT have been documented in patients with TSC in an epidemiological study of the Northern Ireland population with 73 known affected individuals, other than the case described here. Extracranial and extrarenal malignancies in that cohort occurred in less than 4% of cases and are therefore rare; they included endometrial, ovarian, and colorectal cancers. Routine imaging of the brain and renal tracts is of course likely to provide a more accurate reflection of cancers in these organs, but the fact that no male extrarenal genitourinary cancers were identified on long-term follow-up suggests that TSC is not a major cause of LCTs [10]. Further study is required to explore in greater detail the possibility that other genes predispose patients to LCT, as well as to characterize the somatic genetic events that occur in the adult form of these tumors.
Conclusions
Genetic and immunohistochemistry analyses indicate that the Leydig cell tumors in this patient with TSC occurred independent of his TSC1 gene mutation and are unlikely to be directly related to TSC. Genes predisposing patients to adult Leydig cell tumors are currently unknown but may include FH. The somatic genetic events that drive Leydig cell tumor development are also unknown.
Acknowledgments
This study was supported by the National Institutes of Health (2R37NS031535 and 1P01CA120964).
Footnotes
- (C/A)
- Consulting/advisory relationship
- (RF)
- Research funding
- (E)
- Employment
- (H)
- Honoraria received
- (OI)
- Ownership interests
- (IP)
- Intellectual property rights/inventor/patent holder
- (SAB)
- Scientific advisory board
Author Contributions
Conception/Design: David J. Kwiatkowski, Patrick J. Morrison
Provision of study material or patients: Charles W. Shepherd, Deirdre E. Donnelly, Rachel Hardy, Rosemary Clarke, David J. Kwiatkowski, Patrick J. Morrison
Collection and/or assembly of data: Izabela A. Malinowska, David J. Kwiatkowski, Patrick J. Morrison
Data analysis and interpretation: Izabela A. Malinowska, David J. Kwiatkowski, Patrick J. Morrison
Manuscript writing: Izabela A. Malinowska, Charles W. Shepherd, Deirdre E. Donnelly, Rachel Hardy, Rosemary Clarke, David J. Kwiatkowski, Patrick J. Morrison
Final approval of manuscript: Izabela A. Malinowska, Charles W. Shepherd, Deirdre E. Donnelly, Rachel Hardy, Rosemary Clarke, David J. Kwiatkowski, Patrick J. Morrison
References
- 1.Carvajal-Carmona LG, Alam NF, Pollard PJ, et al. Adult leydig cell tumors of the testis caused by germline fumarate hydratase mutations. J Clin Endocrinol Metab. 2006;91:3071–3075. doi: 10.1210/jc.2006-0183. [DOI] [PubMed] [Google Scholar]
- 2.Martin RW, 3rd, Rady P, Arany I, Tyring SK. Benign Leydig cell tumor of the testis associated with human papillomavirus type 33 presenting with the sign of Leser-Trélat. J Urol. 1993;150:1246–1250. doi: 10.1016/s0022-5347(17)35745-2. [DOI] [PubMed] [Google Scholar]
- 3.Perez-Atayde AR, Nunez AE, Carroll WL, et al. Large-cell calcifying sertoli cell tumor of the testis. An ultrastructural, immunocytochemical, and biochemical study. Cancer. 1983;51:2287–2292. doi: 10.1002/1097-0142(19830615)51:12<2287::aid-cncr2820511220>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 4.Morrison PJ, Donnelly DE, Atkinson AB, et al. Advances in the genetics of familial renal cancer. The Oncologist. 2010;15:532–538. doi: 10.1634/theoncologist.2010-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Millar S, Bradley L, Donnelly DE, et al. Familial pediatric endocrine tumors. The Oncologist. 2011;16:1388–1396. doi: 10.1634/theoncologist.2011-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kwiatkowski DJ, Whittemore V, Thiele E, editors. Tuberous Sclerosis Complex: Genes, Clinical Features, and Therapeutics. New York: Wiley; 2010. [Google Scholar]
- 7.Goto J, Talos DM, Klein PM, et al. Regulable neural progenitor-specific TSC1 loss yields giant cells with organellar dysfunction in a model of tuberous sclerosis complex. Proc Natl Acad Sci U S A. 2011;108:E1070–1079. doi: 10.1073/pnas.1106454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kwiatkowski DJ, Manning BD. Tuberous sclerosis: A gap at the crossroads of multiple signaling pathways. Hum Mol Genet. 2005;14:R251–258. doi: 10.1093/hmg/ddi260. [DOI] [PubMed] [Google Scholar]
- 9.Boot AM, Lumbroso S, Verhoef-Post M, et al. Mutation analysis of the LH receptor gene in Leydig cell adenoma and hyperplasia and functional and biochemical studies of activating mutations of the LH receptor gene. J Clin Endocrinol Metab. 2011;96:E1197–1205. doi: 10.1210/jc.2010-3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Devlin L, Crawford H, Shepherd C, et al. Tuberous sclerosis complex; clinical features, diagnosis and prevalence within Northern Ireland. Dev Med Child Neurol. 2006;48:495–499. doi: 10.1017/S0012162206001058. [DOI] [PubMed] [Google Scholar]