

The results of a trial of metastatic colorectal cancer patients randomized to capecitabine plus oxaliplatin plus bevacizumab as a standard triweekly cycle or a dose-dense biweekly cycle schedule are reported. The biweekly combination at the doses studied cannot be recommended.
Keywords: Capecitabine, Clinical trial, Bevacizumab, Progression-free survival, Metastatic colorectal cancer, Toxicity, XELOX
Abstract
Background.
Capecitabine administered for 7 days biweekly with oxaliplatin (XELOX) biweekly has been reported to have activity and safety profiles similar to those of standard capecitabine given for 14 days triweekly. Multiple studies have shown that the addition of bevacizumab to 5-fluorouracil–based chemotherapy is active and well tolerated.
Methods.
Patients with metastatic colorectal cancer (mCRC) were randomized to XELOX plus bevacizumab using a standard triweekly cycle (Q3W) or a dose-dense biweekly cycle (Q2W) schedule. The primary endpoint was the progression-free survival (PFS) interval. This trial is registered on ClinicalTrials.gov (identifier, NCT00159432).
Results.
In total, 435 U.S. patients were randomized. The median PFS intervals were 9.6 months in the Q3W group and 9.1 months in the Q2W group. The median overall survival times were 28.4 months and 22.1 months and the median times to treatment failure were 5.5 months and 3.4 months, respectively. Overall, gastrointestinal disorders were the most common (93%) adverse event (AE). Grade 3 or 4 AEs occurred in 75% and 81% of patients in the Q3W and Q2W groups, respectively. Treatment discontinuation as a result of diarrhea (5% versus 10%) and hand–foot syndrome (2% versus 9%) was less common in the Q3W group than in the Q2W group, respectively.
Conclusions.
Based on these results, the first-line treatment of U.S. patients with mCRC using a biweekly combination of XELOX and bevacizumab at the doses studied cannot be recommended. XELOX Q3W remains the preferred schedule for the management of mCRC.
Introduction
Colorectal cancer (CRC) is a common malignancy usually treated with a chemotherapy regimen containing 5-fluorouracil (5-FU) [1, 2]. Low-dose, continuous infusion 5-FU produces a higher response rate than with other 5-FU regimens [3–6], but issues associated with continuous infusion remain. Capecitabine is an orally administered fluoropyrimidine carbamate that is absorbed intact from the gastrointestinal (GI) tract and becomes metabolically activated to 5-FU within the tumor [7]. Capecitabine, using a triweekly (Q3W) schedule, has demonstrated equivalency to 5-FU as monotherapy and in combination with oxaliplatin (XELOX) versus oxaliplatin, leucovorin, and 5-FU (FOLFOX-4) with or without bevacizumab for the treatment of metastatic CRC (mCRC) [8–11]. These results, in conjunction with the results of other randomized clinical trials, led to the inclusion of XELOX with or without bevacizumab Q3W as a standard treatment option for CRC in the National Comprehensive Cancer Network guidelines. An alternative dose-dense regimen of oxaliplatin at 85 mg/m2 on day 1 and capecitabine at 2,500–4,000 mg/m2/d on days 1–7 every 2 weeks (Q2W) was developed and found to be active with an acceptable safety profile for patients with advanced CRC [12]. For recurrent CRC or mCRC, capecitabine (1,700 mg/m2/d) on days 1–15 and oxaliplatin (130 mg/m2) on day 1 with bevacizumab (7.5 mg/kg) Q3W was shown to provide good efficacy and tolerability [13]. A phase II randomized trial using oxaliplatin (85 mg/m2) on day 1 and the dose-dense schedule of capecitabine at 3,500 mg/m2/d on days 1–7 Q2W was shown to result in a significantly longer progression-free survival (PFS) interval than with the Q3W schedule, with comparable safety and a 34% greater delivered dose intensity [14]. Consequently, it was reasonable to investigate the comparative efficacy of a dose-dense XELOX with bevacizumab schedule in a larger randomized trial.
The purpose of this trial was to determine whether or not dose-dense XELOX with bevacizumab Q2W is superior to standard XELOX with bevacizumab Q3W in terms of PFS outcomes in patients with mCRC or unresectable, locally advanced CRC.
Materials and Methods
Participants
Male and female outpatients aged ≥18 years were eligible if they had metastatic or unresectable, locally advanced, histologically confirmed adenocarcinoma of the colon or rectum and had not previously received systemic treatment for metastatic disease. Patients may have received prior neoadjuvant therapy for surgically resected stage II or III colon or rectal cancer if ≥12 months had elapsed since completion of the treatment and the appearance of metastatic disease. Patients were required to have at least one target lesion according to the Response Evaluation Criteria in Solid Tumors (RECIST), version 1.0 [15]. Other inclusion criteria were an Eastern Cooperative Oncology Group (ECOG) performance status score ≤2 and adequate hematologic, renal, and hepatic function.
Exclusion criteria included the presence of locally advanced rectal cancer eligible for concurrent chemoradiotherapy, previous cytotoxic chemotherapy for mCRC, surgical treatment of hepatic metastatic disease within 8 weeks, organ allograft, clinically significant cardiac disease, concomitant use of anticoagulants or nonsteroidal anti-inflammatory drugs other than aspirin ≤81 mg/day, central nervous system disorders, known peripheral neuropathy, moderate or severe renal impairment, and pregnancy or lactation. Written informed consent was provided by each patient. The study was conducted in compliance with the principles of the Declaration of Helsinki and Guidelines for Good Clinical Practice. The protocol and any modifications, any accompanying materials provided to the patient, and appropriate consent procedures were reviewed and approved by each study site's institutional review board. The study is registered on ClinicalTrials.gov (identifier, NCT00159432).
Trial Design and Interventions
XELOX-A-DVS (dense versus standard) was a prospective, multicenter, randomized, open-label, phase II study of 435 patients with mCRC who met standard eligibility criteria, but who had not received first-line chemotherapy. Patients continued treatment until disease progression, death, discontinuation of study treatment (maximum 72 weeks), toxicity unresponsive to dose modification or supportive measures, or physician or patient choice. Patients were randomized to receive either capecitabine (850 mg/m2) twice daily (BID) on days 1–14 and oxaliplatin (130 mg/m2) on day 1 plus bevacizumab (7.5 mg/kg) on day 1 every 21 days (XELOX Q3W or standard) or capecitabine (1,500 mg/m2) BID on days 1–7 and oxaliplatin (85 mg/m2) on day 1 plus bevacizumab (5 mg/kg) on day 1 (Q2W or dose dense).
The calculated total daily dose of capecitabine was rounded to the nearest 500 mg. The dose of capecitabine could be increased once at any time after the first two cycles provided that the patient had only experienced toxicity that was grade ≤1. The increase was equal to the addition of one 500-mg tablet per total daily dose in both treatment arms. Doses were modified based on toxicity and grade according to predefined criteria. For capecitabine, a novel dose-reduction strategy was used such that the total daily dose was reduced by one or two 500-mg tablets, versus a standard reduction of 25% (supplemental online Appendix A). If a toxicity was considered to be solely a result of one drug, the doses of the other drugs did not require modification. If a delay related to capecitabine was required, the other study drugs were delayed as well until capecitabine could be restarted. If capecitabine had to be discontinued permanently because of toxicity, the patient was taken off study treatment.
Surgical resection of metastatic disease was permitted based on local medical and surgical judgment. Patients who were candidates for complete surgical resection were to receive ≥12 weeks of study treatment prior to surgery and up to an additional 24 weeks following recovery from surgery.
Diarrhea was managed per protocol using American Society of Clinical Oncology guidelines for cancer treatment–related diarrhea [16].
Patients who continued to respond or experienced disease stabilization at 72 weeks could continue study treatment at the discretion of the investigator. Patients who achieved a complete radiographic response without surgery could continue to receive up to an additional 24 weeks of study treatment.
Randomization and Masking
This multicenter trial was conducted from July 2005 to April 2009 at 98 study centers across the U.S. Treatment allocation was assigned by central randomization, and patients were assigned to one of the two treatment arms in a 1:1 ratio using the permuted block randomization method. Randomization was not stratified by any other variable. This was an open-label study.
Endpoints and Assessments
The primary endpoint of the study was the PFS interval (defined as the time to disease progression or death) and was assessed by individual investigators at each site based on the RECIST, version 1.0 [15]. Secondary endpoints included the time to progression (TTP), time to treatment failure (TTF), objective response rate (ORR) based on RECIST, version 1.0 [15], overall survival (OS) time, and safety. TTF was defined as premature withdrawal because of adverse events (AEs); progressive disease or insufficient therapeutic response; death; failure to return; or refusal of treatment, unwillingness to cooperate, or withdrawn consent.
Tumor assessments were made using computed tomography, magnetic resonance imaging, or x-ray within 21 days prior to treatment start and every 6 weeks through cycle 24 in both groups. Evaluation of target and nontarget lesions and determination of response (complete, partial, progressive disease, stable disease) were in accordance with RECIST, version 1.0 [15]. Patients who discontinued study treatment for a reason other than progressive disease were to continue to have tumor assessments until the time of progressive disease.
Safety
AEs were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE), version 3.0. The causality relationship of study regimen to an AE was assessed by the investigator. All AEs occurring up to 28 days after the last intake of study medication were reported. A formal interim analysis for safety was planned when 33% of the events across the two treatment arms had occurred; that is, at 101 events (disease progression or death) or at ∼51 events per group.
Statistical Analysis
For a fixed-sample study with a 24-month accrual and 4-year study duration, ≥356 evaluable patients (178 patients per treatment arm) would provide 80% power to detect a 28% lower risk for progression (i.e., a median PFS time of 10.5 months in XELOX plus bevacizumab Q3W group versus 14.5 months in XELOX Q2W group) at a 5% level of significance (two-sided log-rank test). The required number of events (disease progression or death) in the two treatment groups combined was 302.
Analyses of the primary and secondary efficacy endpoints were based on the intent-to-treat (ITT) population. The ITT population included all randomized patients whether or not they received study treatment. Patients excluded from the study for a protocol violation following randomization were included in the per-protocol population (Fig. 1). This included patients who did not receive capecitabine, oxaliplatin, and bevacizumab for at least two (Q3W arm) or three (Q2W arm) cycles for reasons other than disease progression or death and patients who received the required number of cycles but received <50% of the planned dose of at least one of the three drugs during that time. Analyses of the per-protocol population were intended primarily as sensitivity analyses for the primary efficacy endpoint.
Figure 1.

Patient disposition.
Abbreviations: ITT, intent to treat; Q2W, every 2 weeks; Q3W, every 3 weeks; XELOX, capecitabine plus oxaliplatin.
The primary efficacy endpoint, the PFS interval, was summarized using the Kaplan–Meier method and compared between the treatment arms using the two-sided log-rank test. The point estimate and associated confidence interval (CI) for the hazard ratio (HR) and the median PFS time as appropriate were summarized using Cox regression analysis (with treatment as the covariate) and the Kaplan–Meier estimation method, respectively. Multivariate Cox regression analysis was performed to evaluate associations between PFS, OS, and time to first grade 2 or 3 hand–foot syndrome (HFS) event and baseline characteristics, including sex, age (<60 years versus ≥60 years), number of metastatic sites (one or fewer versus more than one), the presence of liver metastases, baseline alkaline phosphatase level (at or below the upper limit of normal [ULN] versus above the ULN), baseline ECOG performance status score (<2 versus ≥2), and stage of first diagnosis (locoregional versus metastatic). Folic acid level (≤15 mg/mL versus >15 mg/mL) was also included as an additional factor in the Cox regression analysis in time to first grade 2 or 3 HFS event.
Results
Patient Disposition and Demographics
In total, 435 patients were randomized (Q3W, n = 217; Q2W, n = 218) and constituted the ITT population. The safety population (n = 419) consisted of patients who received at least one dose of study medication (Q3W, n = 208; Q2W, n = 211). Eight (4%) patients in the Q3W group and two (1%) patients in the Q2W group completed the planned 72 weeks of treatment. Patient disposition in the trial is illustrated in Figure 1. Patients in the two groups were well matched. Patient demographics and disease characteristics are outlined in Table 1.
Table 1.
Baseline demographics and disease characteristics (intent to treat population)
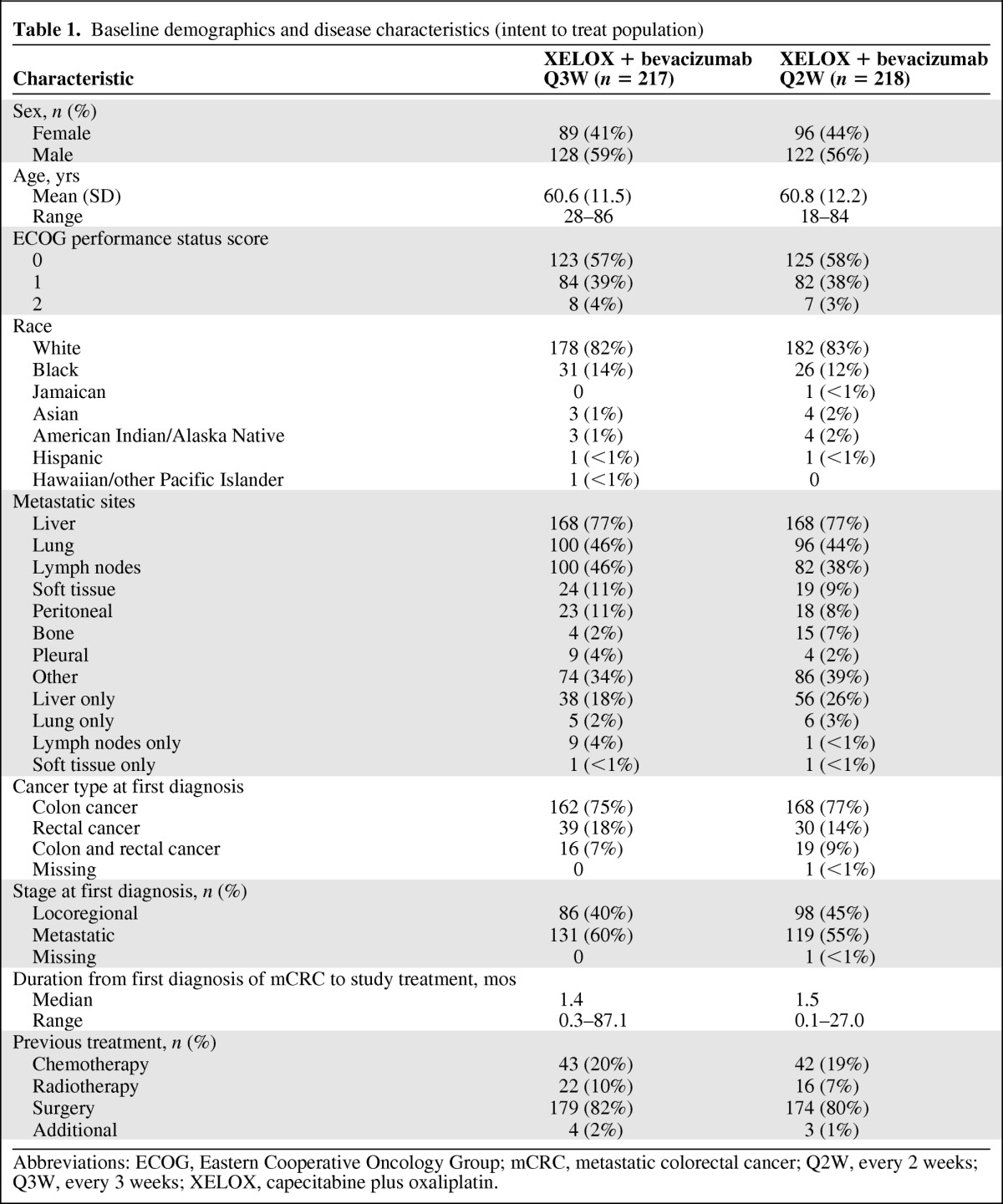
Abbreviations: ECOG, Eastern Cooperative Oncology Group; mCRC, metastatic colorectal cancer; Q2W, every 2 weeks; Q3W, every 3 weeks; XELOX, capecitabine plus oxaliplatin.
Primary Endpoint: PFS Interval
The observed median PFS intervals (ITT population) were 9.6 months and 9.1 months (HR, 0.89; 95% CI, 0.67–1.17; p = .39), favoring the control regimen (Q3W) (Fig. 2A). The median PFS intervals (ITT population) using an on-treatment approach (which included tumor assessments and death events that occurred only up to and including 28 days after the last intake of study medication) were 10.2 months and 11.1 months (HR, 0.94; 95% CI, 0.66–1.34; p = 0.74), favoring the dose-dense regimen (Q2W). The per-protocol analysis produced similar results, favoring the Q3W regimen: 9.8 months versus 9.4 months (HR, 0.94; 95% CI, 0.69–1.28; p = .70). Multivariate Cox regression analyses based on the full model showed the following baseline characteristics to be significant predictors of a longer PFS time in both groups: one or fewer metastatic sites (p < .01) and an ECOG performance status score ≤1 (p ≤ .01).
Figure 2.

Kaplan–Meier plots of progression-free survival (A) and overall survival (B) outcomes in the ITT population.
Abbreviations: ITT, intent to treat; Q2W, every 2 weeks; Q3W, every 3 weeks; XELOX, capecitabine plus oxaliplatin.
Secondary Efficacy Endpoints
Secondary time-dependent and tumor response efficacy endpoints were consistent with the primary finding of the trial. The observed median TTP (ITT population) were 10.5 months and 9.4 months (HR, 0.90; 95% CI, 0.66–1.21; p = .48), favoring the control regimen (Q3W). The TTF (ITT population) was significantly longer in the control arm (Q3W), with an observed median of 5.5 months, compared with 3.4 months for the Q2W group (HR, 0.74; 95% CI, 0.60–0.92; p < .01). A total of 166 (77%) patients in the Q3W group and 168 (77%) patients in the Q2W group experienced treatment failure. AEs (Q3W, 30%; Q2W, 45%) and progressive disease (insufficient therapeutic response) (Q3W, 30%; Q2W, 17%) were the most common reasons for treatment failure.
The ORR was higher in the Q3W group. The overall clinical response rates (complete and partial responses) were 33% in the Q3W group and 23% in the Q2W group (between-group difference, 9.8%; 95% CI, 0.9%–18.7%). Stable disease was observed in 88 of 217 (41%) patients in the Q3W group and 74 of 218 (34%) patients in the Q2W group. Progressive disease was observed in 7% and 11% of patients in the Q3W and Q2W groups, respectively.
Ninety-three (43%) patients in the Q3W group and 106 (49%) patients in the Q2W group died during the study. Most patients died as a result of their cancer. The HR for the OS outcome was 0.85 (95% CI, 0.64–1.12; p = .25), with median OS times of 28.4 months and 22.1 months for the Q3W and Q2W groups, respectively (Fig. 2B). One hundred twenty-four of the 208 (60%) patients in the Q3W group and 144 of the 211 (68%) patients in the Q2W group received further postprotocol treatment with at least one drug. The drugs most commonly used are shown in Table 2. Results of the multivariate Cox regression analyses showed that key predictors of a longer OS duration in both groups were one or fewer metastatic sites (p < .0001), age <60 years (p < .05), an ECOG performance status score ≤1 (p < .05), and an alkaline phosphatase level ≤ ULN (p < .05).
Table 2.
Summary of additional postprotocol drug therapy
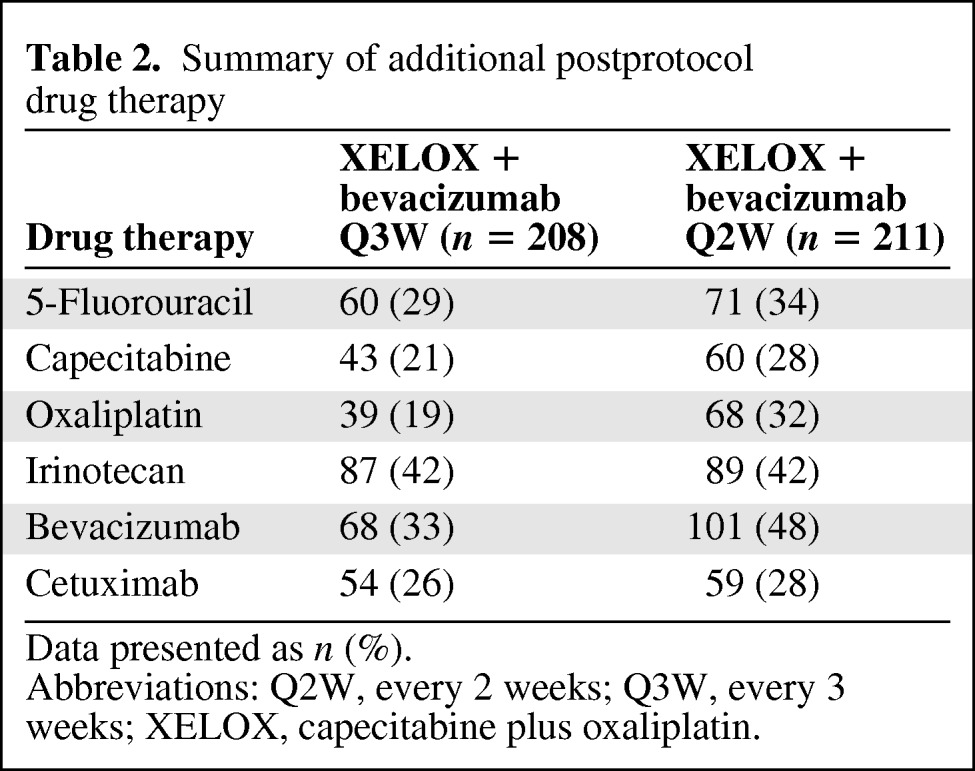
Data presented as n (%).
Abbreviations: Q2W, every 2 weeks; Q3W, every 3 weeks; XELOX, capecitabine plus oxaliplatin.
Safety
Treatment exposure was based on an analysis of the predefined safety population. Patients received a median of six treatment cycles in both groups (Table 3). The median percentages of delivered versus planned doses for capecitabine (91.8% versus 92.3%), oxaliplatin (99.7% versus 99.8%), and bevacizumab (100.5% versus 100.9%) were comparable across study arms. The mean (standard deviation [SD]) daily capecitabine doses were 3,065.2 mg (452.5 mg) and 5,366.9 mg (930.8 mg) for the Q3W and Q2W groups, respectively. The mean (SD) total capecitabine doses were 335,260 mg (276,060 mg) in the Q3W group and 295,580 mg (267,760 mg) in the Q2W group. Dose reduction for AE management was required in 46% and 42% of patients at median treatment times of 2.2 and 1.4 months for the Q3W and Q2W groups, respectively.
Table 3.
Exposure to capecitabine
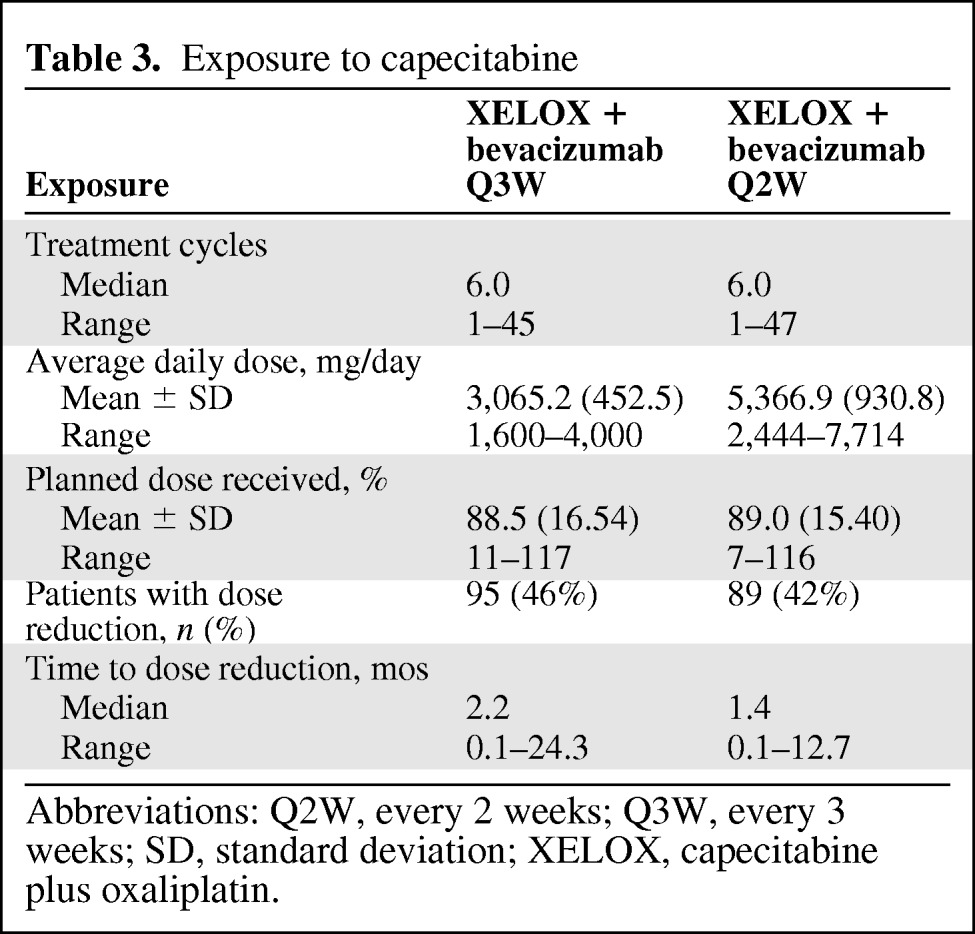
Abbreviations: Q2W, every 2 weeks; Q3W, every 3 weeks; SD, standard deviation; XELOX, capecitabine plus oxaliplatin.
Of the AEs in the Q3W group, 67% were treatment related; in the Q2W group, 66% were treatment related (Table 4). One hundred fifty-five (75%) patients in the Q3W group and 171 (81%) patients in the Q2W group had at least one AE assessed by the investigator as NCI CTCAE grade 3 or 4 (Table 4). Nineteen percent of the AEs in each group were assessed as NCI grade 3 or 4, of which 304 (70%) in the Q3W group and 310 (72%) in the Q2W group were treatment related. Seventy-nine (58%) treatment-related AEs were assessed as serious in the Q3W group and 97 (66%) were assessed as serious in the Q2W group.
Table 4.
Adverse events (AEs) with an incidence ≥5%
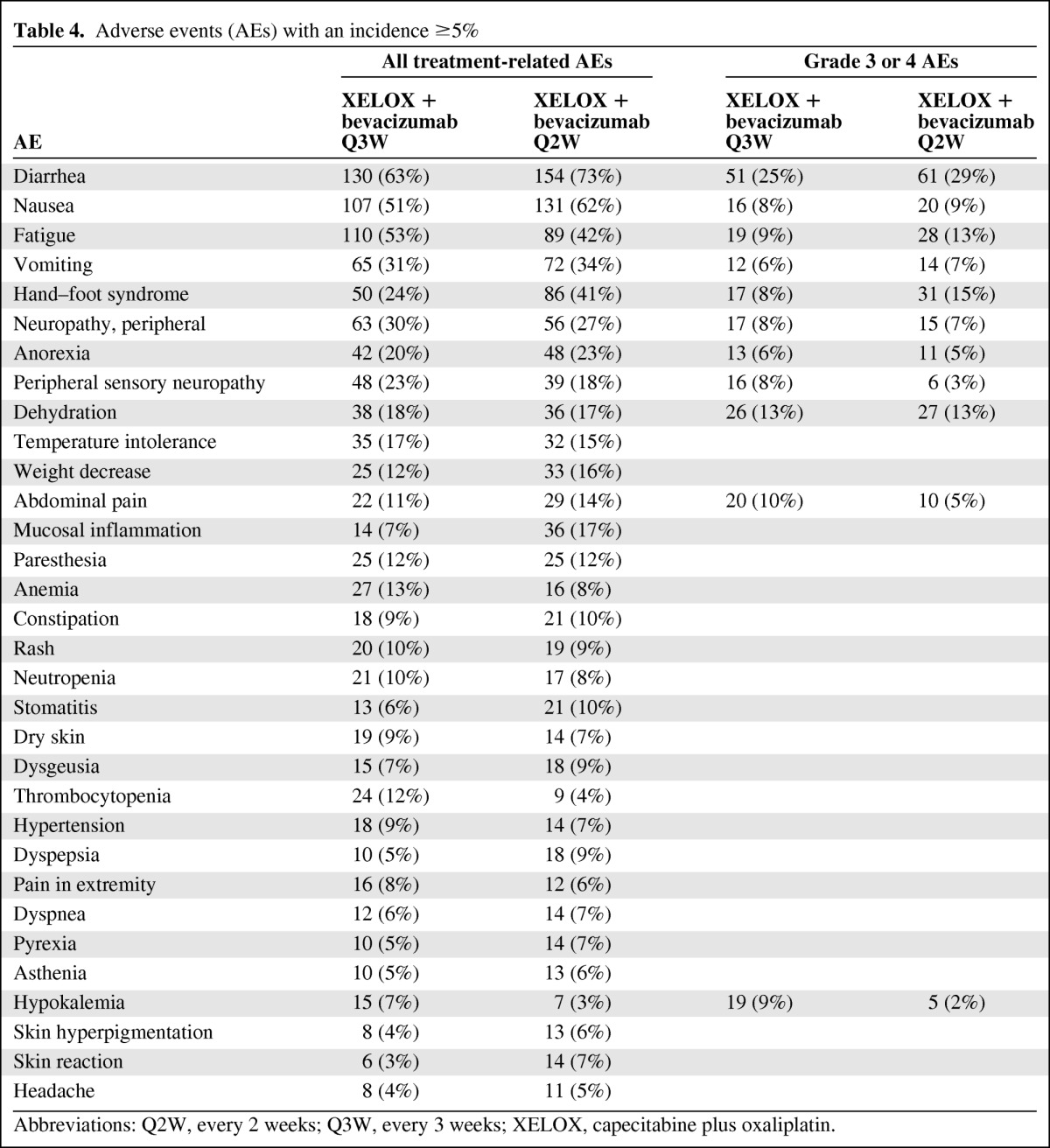
Abbreviations: Q2W, every 2 weeks; Q3W, every 3 weeks; XELOX, capecitabine plus oxaliplatin.
Diarrhea was the most common grade 3 or 4 AE, occurring in 25% of patients in the Q3W group and 29% of patients in the Q2W group. The onset of grade 3 or 4 diarrhea was numerically faster in the Q2W group (HR, 0.75; 95% CI, 0.52–1.09) (Fig. 3A). The median time to grade 2 or 3 HFS was not reached in the Q3W group, but it was 9.1 months for the Q2W group (HR, 0.35; 95% CI, 0.23–0.53) (Fig. 3B). Multivariate Cox analysis showed that male gender was a significant predictor of earlier occurrence of grade 2 or 3 HFS (p < .001), whereas the difference in the absence of liver metastases (p = .06) did not reach statistical significance.
Figure 3.

Kaplan–Meier plots for time to onset of grade 3 or 4 diarrhea (A) and grade 2 or 3 hand–foot syndrome (B).
Abbreviations: Q2W, every 2 weeks; Q3W, every 3 weeks; XELOX, capecitabine plus oxaliplatin.
Grade 3 or 4 AEs that occurred with a frequency difference ≥5% in the Q3W and Q2W groups, respectively, were abdominal pain (10% versus 5%), hypokalemia (9% versus 2%), fatigue (9% versus 13%), HFS (8% versus 15%), and peripheral sensory neuropathy (8% versus 3%).
Sixty-six (32%) patients in the Q3W group and 104 (49%) patients in the Q2W group discontinued study treatment because of one or more AE. Overall, the most common AEs leading to discontinuation of study treatment were (Q3W versus Q2W) diarrhea (5% versus 10%) and HFS (2% versus 9%). Rates of treatment discontinuations resulting from oxaliplatin-related neurotoxicity were comparable between the standard and dose-dense regimens, 18% versus 15%, respectively. Reintroduction of oxaliplatin treatment was uncommon, occurring in 3% and 2% of patients, respectively.
Discussion
This phase II clinical trial was conducted to determine if a regimen of intermittent XELOX with bevacizumab given as a dose-dense, 14-day (Q2W) cycle was superior to the standard regimen given as a 21-day (Q3W) cycle in terms of the PFS outcomes of patients with metastatic or unresectable, locally advanced CRC. At the doses used in this study, the Q2W cycle was not superior to the standard Q3W cycle in terms of the PFS outcome. Furthermore, the results of other secondary efficacy endpoints were generally consistent with the results of the PFS evaluation. There were no significant differences between treatment groups in the median TTP or median OS time. However, the TTF was significantly shorter with the Q2W schedule than with the Q3W schedule. At the same time, although the safety profiles of the Q3W and Q2W groups were generally similar, patients in the Q2W group were more likely to experience HFS and to discontinue treatment as a result of diarrhea or HFS. In fact, discontinuation of treatment because of AEs occurred in 30% of patients in the Q3W group and 47% of patients in the Q2W group.
The results of this phase II trial in mCRC patients differ from the results observed in a phase II trial of patients with metastatic breast cancer when given a dose-dense regimen of capecitabine. Using a seven-on–seven-off schedule of capecitabine (2,000 mg BID) in combination with bevacizumab (10 mg/kg) Q2W, a partial response was observed in seven of 23 evaluable patients, stable disease for >6 months was observed in six patients, and stable disease for <6 months was observed in seven patients, with progressive disease in three patients [17]. As in the present study, grade 2 or 3 HFS was common.
The results of this trial suggest that, although the dose-dense Q2W regimen is active, it causes unacceptable toxicity, which leads to a large percentage of patients experiencing treatment failure before disease progression. This is likely because of the high starting dose of capecitabine and the inability of the dose modification strategy used in this trial to prevent the recurrence of unacceptable toxicity. The mean daily doses of capecitabine were 3,065.2 mg/day in the Q3W group and 5,366.9 mg/day in the Q2W group. However, the cumulative capecitabine exposure was less in the dose-dense arm. In addition, the mean percentage of the planned dose received was 89.5% in the Q3W group and 89.0% in the Q2W group, with a reduction in the capecitabine dose in 46% and 42% of patients, respectively.
We considered other factors that may have contributed to the high incidence of toxicity in the Q2W group and the inability to confirm treatment effects observed by Scheithauer et al. [14] Several factors were evaluated. Regional differences in patient tolerability of fluoropyrimidine treatment, including 5-FU, UFT, S-1, and capecitabine, have been reported. U.S. patients have higher rates of grade 3 or 4 toxicity, and particularly GI AEs, than non-U.S. patients [18]. The explanation for these observed differences is unclear, for example, ethnicity and pharmacogenomics, dietary folate intake, cultural differences in perception, and AE reporting. Ethnic and cultural differences were unlikely in this trial because 83% of the participants were white, and the study was conducted solely in the U.S. Baseline folate levels were collected during the study in a limited number of patients; however, the results are inconclusive because of the small number of patients. The inability to deliver a dose intensity and safety profile similar to those of Scheithauer et al. [14] may be explained in part by the known regional differences in fluoropyrimidine tolerability.
Another difference between the trials was the inclusion of bevacizumab in the current trial. Cassidy et al. [9] reported slightly higher rates of diarrhea, vomiting, and HFS in patients receiving bevacizumab in combination with FOLFOX or XELOX versus placebo. Another possible explanation is that the commercial availability of all drugs used in this regimen may have increased the potential for patient dropout in the settings of unwanted toxicity or inconvenience.
Conclusion
This U.S.-based phase II study failed to confirm earlier reports in European patients [12, 14] of activity and tolerability of dose-dense Q2W XELOX and bevacizumab. However, our results are comparable with those reported in a large, international, randomized, controlled trial [9, 10] and further demonstrate the activity and tolerability of XELOX and bevacizumab given as a 21-day cycle in a U.S. population.
Based on the lack of benefit in terms of the PFS outcome as well as a shorter TTF and higher incidences of diarrhea and HFS, the first-line treatment of patients with mCRC using a 14-day combination of XELOX and bevacizumab at the doses studied in this trial cannot be recommended. Standard 21-day XELOX and bevacizumab remains the reference standard for this regimen.
See www.TheOncologist.com for supplemental material available online.
Supplementary Material
Acknowledgments
This research was supported by Roche Laboratories, Inc. Editorial support was provided by Elizabeth Yepez of Insight Medical Communications, funded by Roche Laboratories, Inc. Edward McKenna and Sylvia Hu are employees of Roche-Genentech USA and therefore also disclose stock holdings. Herbert Hurwitz received a grant to cover the expenses of consultancy and travel in undertaking this trial. Ambrose Kwok received compensation from Everest Clinical Research Services, Inc., for his work performing statistical analysis. Thomas Cartwright and Yehuda Z. Patt declare consultancy fees from Roche-Genentech USA. The Kimmel Cancer Center, employer of Edith P. Mitchell, received funding in the form of a grant to cover the expense of undertaking this trial.
Footnotes
- (C/A)
- Consulting/advisory relationship
- (RF)
- Research funding
- (E)
- Employment
- (H)
- Honoraria received
- (OI)
- Ownership interests
- (IP)
- Intellectual property rights/inventor/patent holder
- (SAB)
- Scientific advisory board
Author Contributions
Conception/Design: Herbert Hurwitz, Edith P. Mitchell, Thomas Cartwright, Ambrose Kwok, Sylvia Hu, Edward McKenna, Yehuda Z. Patt
Collection and/or assembly of data: Herbert Hurwitz, Edith P. Mitchell, Thomas Cartwright, Yehuda Z. Patt
Data analysis and interpretation: Herbert Hurwitz, Edith P. Mitchell, Thomas Cartwright, Ambrose Kwok, Sylvia Hu, Edward McKenna, Yehuda Z. Patt
Manuscript writing: Herbert Hurwitz, Edith P. Mitchell, Thomas Cartwright, Edward McKenna
Final approval of manuscript: Herbert Hurwitz, Edith P. Mitchell, Thomas Cartwright, Ambrose Kwok, Sylvia Hu, Edward McKenna, Yehuda Z. Patt
References
- 1.National Comprehensive Cancer Network. Colon Cancer. v. 2.2010. [accessed May 14, 2010]. Available at http://www.nccn.org/professionals/physician_gls/PDF/colon.pdf.
- 2.National Comprehensive Cancer Network. Rectal Cancer. v. 2.2010. [accessed May 14, 2010]. Available at http://www.nccn.org/professionals/physician_gls/PDF/rectal.pdf.
- 3.Lokich JJ, Ahlgren JD, Gullo JJ, et al. A prospective randomized comparison of continuous infusion fluorouracil with a conventional bolus schedule in metastatic colorectal carcinoma: A Mid-Atlantic Oncology Program study. J Clin Oncol. 1989;7:425–432. doi: 10.1200/JCO.1989.7.4.425. [DOI] [PubMed] [Google Scholar]
- 4.Leichman CG, Leichman L, Spears CP, et al. Prolonged continuous infusion of fluorouracil with weekly bolus leucovorin: A phase II study in patients with disseminated colorectal cancer. J Natl Cancer Inst. 1993;85:41–44. doi: 10.1093/jnci/85.1.41. [DOI] [PubMed] [Google Scholar]
- 5.Hansen RM. 5-Fluorouracil by protracted venous infusion: A review of recent clinical studies. Cancer Invest. 1991;9:637–642. doi: 10.3109/07357909109039875. [DOI] [PubMed] [Google Scholar]
- 6.Mori A, Bertoglio S, Guglielmi A, et al. Activity of continuous-infusion 5-fluorouracil in patients with advanced colorectal cancer clinically resistant to bolus 5-fluorouracil. Cancer Chemother Pharmacol. 1993;33:179–180. doi: 10.1007/BF00685339. [DOI] [PubMed] [Google Scholar]
- 7.Miwa M, Ura M, Nishida M, et al. Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5-fluorouracil selectively in tumours by enzymes concentrated in human liver and cancer tissue. Eur J Cancer. 1998;34:1274–1281. doi: 10.1016/s0959-8049(98)00058-6. [DOI] [PubMed] [Google Scholar]
- 8.Cassidy J, Tabernero J, Twelves C, et al. XELOX (capecitabine plus oxaliplatin): Active first-line therapy for patients with metastatic colorectal cancer. J Clin Oncol. 2004;22:2084–2091. doi: 10.1200/JCO.2004.11.069. [DOI] [PubMed] [Google Scholar]
- 9.Cassidy J, Clarke S, Díaz-Rubio E, et al. XELOX vs FOLFOX-4 as first-line therapy for metastatic colorectal cancer: NO16966 updated results. Br J Cancer. 2011;105:58–64. doi: 10.1038/bjc.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saltz LB, Clarke S, Díaz-Rubio E, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: A randomized phase III study. J Clin Oncol. 2008;26:2013–2019. doi: 10.1200/JCO.2007.14.9930. [DOI] [PubMed] [Google Scholar]
- 11.Arkenau HT, Arnold D, Cassidy J, et al. Efficacy of oxaliplatin plus capecitabine or infusional fluorouracil/leucovorin in patients with metastatic colorectal cancer: A pooled analysis of randomized trials. J Clin Oncol. 2008;26:5910–5917. doi: 10.1200/JCO.2008.16.7759. [DOI] [PubMed] [Google Scholar]
- 12.Scheithauer W, Kornek GV, Raderer M, et al. Intermittent weekly high-dose capecitabine in combination with oxaliplatin: A phase I/II study in first-line treatment of patients with advanced colorectal cancer. Ann Oncol. 2002;13:1583–1589. doi: 10.1093/annonc/dkf281. [DOI] [PubMed] [Google Scholar]
- 13.Hochster HS, Hart LL, Ramanathan RK, et al. Safety and efficacy of oxaliplatin and fluoropyrimidine regimens with or without bevacizumab as first-line treatment of metastatic colorectal cancer: Results of the TREE Study. J Clin Oncol. 2008;26:3523–3529. doi: 10.1200/JCO.2007.15.4138. [DOI] [PubMed] [Google Scholar]
- 14.Scheithauer W, Kornek GV, Raderer M, et al. Randomized multicenter phase II trial of two different schedules of capecitabine plus oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol. 2003;21:1307–1312. doi: 10.1200/JCO.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 15.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 16.Benson AB, 3rd, Ajani JA, Catalano RB, et al. Recommended guidelines for the treatment of cancer treatment-induced diarrhea. J Clin Oncol. 2004;22:2918–2926. doi: 10.1200/JCO.2004.04.132. [DOI] [PubMed] [Google Scholar]
- 17.Traina TA, Theodoulou M, Dugan U, et al. A novel capecitabine dosing schedule combined with bevacizumab is safe and effective in patients with metastatic breast cancer. Cancer Res. 2009;69(suppl):395S. [Google Scholar]
- 18.Haller DG, Cassidy J, Clarke SJ, et al. Potential regional differences for the tolerability profiles of fluoropyrimidines. J Clin Oncol. 2008;26:2118–2123. doi: 10.1200/JCO.2007.15.2090. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.