

The results of a phase III multicenter, open-label, randomized trial to assess the efficacy and safety of 400 mg imatinib once daily for 12 months versus 36 months in patients with Kit+ (CD117+) gastrointestinal stromal tumors are presented. Thirty-six months of treatment resulted in longer relapse-free and overall survival times.
Keywords: Imatinib, Gleevec, Kit, CD117, Gastrointestinal stromal tumors
Abstract
On January 31, 2012, the U.S. Food and Drug Administration granted regular approval of imatinib mesylate tablets (Gleevec®; Novartis Pharmaceuticals Corporation, East Hanover, NJ) for the adjuvant treatment of adult patients following complete gross resection of Kit+ (CD117+) gastrointestinal stromal tumors (GISTs). The recommended dose of imatinib is 400 mg/day administered daily for 3 years.
Three hundred ninety-seven patients were enrolled in a randomized adjuvant, multicenter, open label, phase III trial comparing 12 months with 36 months of imatinib treatment. Eligible patients had one of the following: tumor diameter >5 cm and mitotic count >5 per 50 high power fields (HPFs); tumor diameter >10 cm and any mitotic count; tumor of any size with mitotic count >10/50 HPFs; or tumor ruptured into the peritoneal cavity. The primary endpoint was the recurrence-free survival (RFS) interval.
The median follow-up for patients without an RFS event was 42 months. There were 84 (42%) RFS events in the 12-month treatment arm and 50 (25%) RFS events in the 36-month treatment arm. Thirty-six months of imatinib treatment led to a significantly longer RFS interval than with 12 months of treatment.
The median follow-up for overall survival (OS) evaluation in patients still living was 48 months. Thirty-six months of imatinib treatment led to a significantly longer OS time than with 12 months of imatinib treatment.
The most common adverse reactions, as noted in previous imatinib studies, were diarrhea, fatigue, nausea, edema, decreased hemoglobin, rash, vomiting, and abdominal pain.
Introduction
On January 31, 2012, the U.S. Food and Drug Administration (FDA) granted regular approval for imatinib mesylate tablets (Gleevec®; Novartis Pharmaceuticals Corporation, East Hanover, NJ) for the adjuvant treatment of adult patients following complete gross resection of Kit+ (CD117+) gastrointestinal stromal tumors (GISTs). Accelerated approval for this indication was granted on December 19, 2008. The initial approval was based on a single phase III randomized, double-blinded, placebo-controlled study that enrolled a total of 713 adult patients who had a histological diagnosis of Kit+ (CD117+) GIST, a resected tumor size ≥3 cm, and a complete gross resection within 14–70 days prior to registration. Imatinib mesylate, 400 mg orally once daily, was administered for 1 year.
The primary objective of the clinical trial was to compare the recurrence-free survival (RFS) outcomes of imatinib- and placebo-treated patients. The overall survival (OS) time was a secondary endpoint [1, 2].
With a median follow-up for the intent-to-treat (ITT) population of 14.0 months, 30 RFS events were observed in the imatinib group and 70 RFS events were observed in the placebo group, as determined by blinded central independent review (hazard ratio [HR], 0.398; 95% confidence interval [CI], 0.259–0.610; two-sided p-value < .0001). The estimated 1-year RFS rates were 98% for imatinib-treated patients and 82% for placebo-treated patients. Based on these results, the trial was terminated and patients still receiving placebo were allowed to cross over to imatinib treatment. At the time of submission, the OS results were immature, with five deaths in the imatinib mesylate group and eight deaths in the placebo group.
The RFS analysis indicated that the greatest effect of imatinib was seen during the 1-year treatment period, with the RFS curves gradually coming together after that time point. This suggested that imatinib might be exerting a cytostatic effect and that a longer duration of treatment might be beneficial.
The current study, a subpart H (accelerated approval) postmarketing requirement (PMR), tested the above hypothesis and compared imatinib treatment for 3 years with imatinib treatment for 1 year in a higher risk population than was included in the initial GIST adjuvant trial. Because follow-up for the initial 1 year versus placebo study was short, submission of additional RFS and OS analyses after 4 years and 5 years of follow-up were also PMRs.
A manuscript and editorial summarizing the submitted study were recently published [3, 4].
Patients and Methods
The current submission is based on data from a study conducted by the Scandinavian Sarcoma Group together with Arbeitsgemeinschaft Internistische Onkologie of Germany, which was a phase III multicenter, open-label, randomized trial to assess the efficacy and safety of 400 mg imatinib once daily for 12 months or 36 months in Kit+ (CD117+) GIST patients aged ≥18 years who had all macroscopic tumor tissue removed at surgery but were estimated to be at high risk for disease recurrence (>50% risk for recurrence within the first 5 years following the diagnosis). High recurrence risk was defined as one or more of the following: largest tumor diameter (measured by a pathologist) >10.0 cm with any mitotic count in the resected tissue specimen, mitotic count >10 mitoses per 50 high power fields (HPFs) with any tumor size, largest tumor diameter >5.0 cm and mitotic count > 5 per 50 HPFs, tumor spillage into the abdominal cavity (rupture could have occurred either before or during surgery), or microscopically infiltrated margins (or suspected positive margins, i.e., R1 resection) [5–8].
Additional inclusion criteria included immunohistochemical documentation of Kit (CD117) within 12 weeks of study entry; an Eastern Cooperative Oncology Group (ECOG) performance status score of 0, 1, or 2; and adequate hematologic, hepatic, and renal function (absolute neutrophil count >1.5 × 109/L, platelet count > 100× 109/L, total bilirubin <1.5× the upper limit of normal (ULN), serum aspartate aminotransferase and alanine aminotransferase <2.5× ULN, and creatinine <1.5× ULN). Female patients of childbearing potential had to have had a negative pregnancy test within 7 days before initiation of study drug. Postmenopausal women had to be amenorrheic for ≥12 months to be considered of nonchildbearing potential. Male and female patients of reproductive potential agreed to employ an effective barrier method of birth control throughout the study and for up to 3 months following discontinuation of study drug. Written, voluntary informed consent was also required.
Exclusion criteria included inoperable GIST, <1 week or >12 weeks from the time of surgery, recurrent GIST, investigational drug treatment within 28 days of study drug dosing, <5 years free of another primary malignancy (except basal cell skin cancer or a cervical carcinoma in situ), grade III or IV cardiac problems as defined by the New York Heart Association criteria (i.e., congestive heart failure or myocardial infarction within 6 months of study), pregnancy or breast-feeding, the presence of severe or uncontrolled medical disease, the concurrent use of warfarin or acetaminophen, the presence of known chronic liver disease, and HIV infection. Also excluded were patients who had received prior chemotherapy for a GIST, patients who had received neoadjuvant imatinib therapy prior to randomization, patients who had received radiotherapy to >25% of the bone marrow, and patients with any significant history of noncompliance to medical regimens or with an inability to grant reliable informed consent.
An independent central pathology review was performed to confirm the diagnosis of GIST and to confirm mitotic index.
The primary objective was to compare the RFS probabilities of GIST patients with a high (>50%) risk for disease recurrence within the first 5 years following surgery who were treated with adjuvant imatinib for either 12 months or 36 months. The time to event was calculated from the date of randomization to the earliest date of recurrence or death from any cause. The recurrence date was defined as the date when the physician first suspected recurrence in the sequence of events that led to a diagnosis of recurring GIST. The secondary objective was to compare overall survival (OS) and GIST-specific survival outcomes. The time to event was calculated from the date of randomization to the date of death for the OS analysis and to the date of death considered to be caused by the GIST for the GIST-specific survival analysis.
Three analysis patient populations were defined. The ITT population comprised 397 randomized patients who signed the informed consent form. The patients were analyzed according to the treatment group to which they were assigned at randomization. The efficacy population included 358 patients (39 patients from the ITT population were excluded, including 24 patients who had overtly metastatic GIST at baseline and 15 patients in whom GIST was not confirmed by central pathology review). The safety population included 392 patients who signed the informed consent form and who received at least one dose of study medication (five patients from the ITT population did not receive study drug). One patient who was randomized to the 12-month arm but (wrongly) treated according to the 36-month regimen was included in the 36-month group for the safety population.
At the time of the supplemental new drug application (sNDA) submission, there were four outstanding PMRs, one related to the submission of the current study and three related to the initial 1-year imatinib adjuvant GIST study. The latter PMRs were to provide additional 4-year and 5-year RFS and OS results of the initial 1-year adjuvant study. Those data are not summarized in this manuscript.
Results
In total, 400 patients were randomized into the study (Sweden, 32 patients; Norway, 27 patients; Finland, 30 patients; Germany, 311 patients) during February 4, 2004 to September 29, 2008. Of these 400 patients, three were randomized despite not having provided informed consent. No data are available for those patients and they were excluded from the ITT, efficacy, and safety populations. Patient demographics, ECOG performance status scores, disease characteristics, and risk factors for the remaining 397 study patients are shown in Table 1.
Table 1.
Patient demographics and disease characteristics (intent-to-treat population)
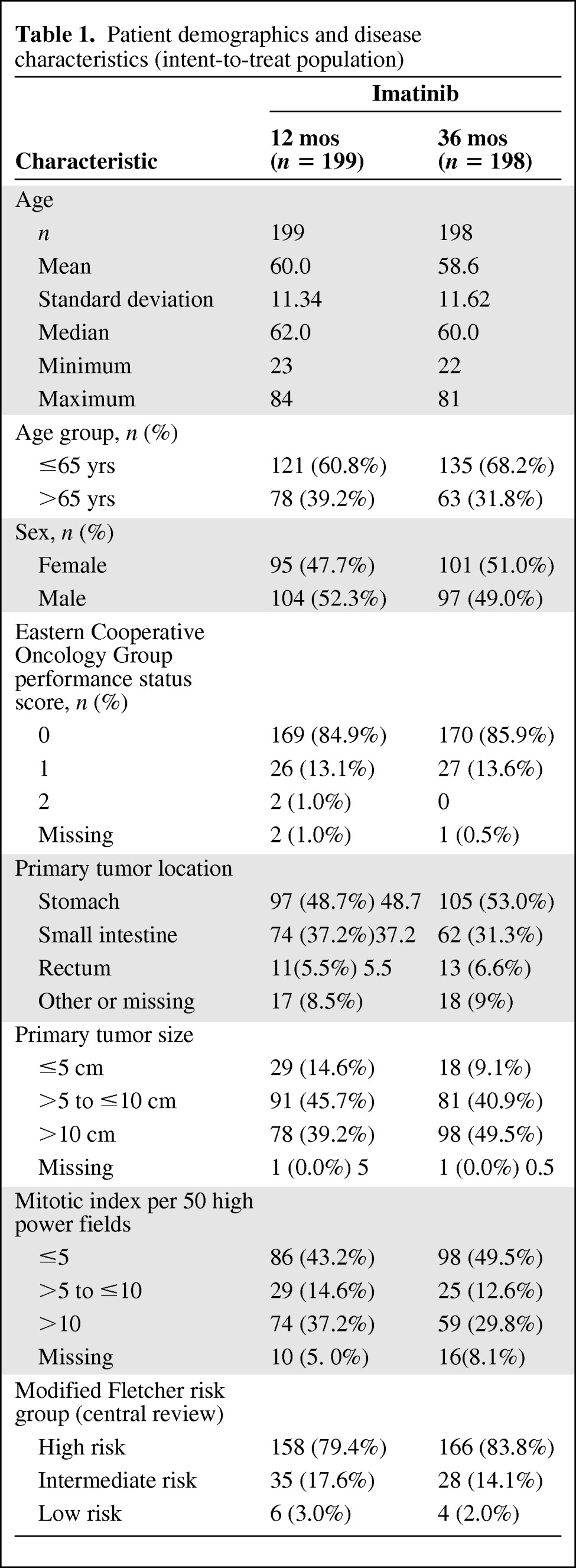
By central review, 93.0% of patients in the 12-month arm and 85.4% of patients in the 36-month arm had GIST that expressed Kit (CD117). In the 12-month arm, the risk was classed as high according to the modified Fletcher criteria in 79.4% of patients and classed as intermediate in 17.6% of patients. In the 36-month arm, the risk was classed as high in 83.8% of patients and intermediate in 14.1% of patients.
The primary efficacy endpoint, the RFS interval, was significantly longer in the 36-month arm than in the 12-month arm (hazard ratio [HR],0.46; 95% confidence interval [CI], 0.32–0.65; p < .0001). Eighty-four RFS events were reported in the 12-month arm versus 50 RFS events in the 36-month arm (Fig. 1). During the first year, when all patients were scheduled to receive imatinib, the rates of recurrence were similar in both the 12-month and 36-month arms. From 12 months to 36 months, a treatment effect in favor of the 36-month arm was observed (HR, 0.22; 95% CI, 0.13–0.37). Following this period, the rate of recurrence was higher in the 36-month arm, although the number of patients at risk decreased leading to less reliable estimates.
Figure 1.

Recurrence-free survival probability in the intent-to-treat population, Kaplan–Meier estimate.
Sensitivity analyses were performed by the FDA to verify the reliability and robustness of RFS effect. These analyses included censoring events occurring in the first year, adjustment of RFS for prognostic factors (primary tumor size, tumor location, mitotic count, age, and gender), backdating an event to a scheduled visit in the event of missing visits, and backdating an event to a nonrecurrence censored observation prior to a missing visit. For all these analyses, the HR was 0.40–0.48.
After the first recurrence, 94 patients in both treatment groups received systemic therapy (Table 2). Of those 94 patients, 81 (86%) received imatinib. The most frequent imatinib starting dose was 400 mg/day in both study arms (94% of the 12-month arm and 74% of the 36-month arm), and imatinib doses were subsequently escalated in only 8% of the 12-month group and 25% of the 36-month group. The median durations of first-line recurrence therapy were 15 months for the 12-month treatment arm (range, 0.1–49 months) and 12 months for the 36 month arm (range, 1.1–36 months). Other treatment options for recurrent disease were surgery and radiation therapy.
Table 2.
Treatment of recurrent GIST (efficacy population)

Abbreviation: GIST, gastrointestinal stromal tumor.
In total, there were 37 deaths observed in the study, with 25 deaths in the 12-month arm and 12 deaths in the 36-month arm. Analysis of the OS probability demonstrated the superiority of the 36-month treatment to the 12-month treatment (HR, 0.45; 95% CI, 0.22–0.89; p = .019, two-sided log rank test). Regarding the GIST-specific survival analysis, 14 deaths classified as GIST related by the investigators were observed in the 12-month arm and seven GIST-related deaths were observed in the 36-month arm. GIST-specific survival estimates indicated a trend toward superiority of the 36-month arm over the 12-month arm (HR, 0.46; 95% CI, 0.19–1.14; p-value = .0872).
Safety
The safety population consisted of 392 patients (194 in the 12-month group and 198 in the 36-month group) who received at least one dose of study drug. The mean (± standard deviation) and median duration of imatinib exposure were 10.8 (±3.2) months and 12.0 months in the 12-month group, respectively, and 28.8 (±11.3) months and 35.9 months in the 36-month group, respectively.
Discontinuation of imatinib therapy because of adverse reactions occurred in 15 patients (8%) and 27 patients (14%) in the 12-month and 36-month treatment arms, respectively. Serious adverse events occurred in 47 patients (24.2%) and 56 patients (28.3%), respectively. Adverse events leading to dose adjustment or interruption occurred in 38 (19.6%) and 58 (29.3%) patients, respectively. Deaths on treatment or within 30 days of the last imatinib dose occurred in two patients, one in each treatment arm. These two deaths were attributed to cardiac failure for one patient and an “unconfirmed meningioma” in a patient with a pre-existing stage IV malignant melanoma.
Almost all patients in each study arm experienced at least one adverse reaction at some time. The most frequently reported adverse reactions were similar to those reported in other clinical studies in other patient populations and include anemia, periorbital edema, diarrhea, nausea, muscle spasms, fatigue, leukopenia, pain, peripheral edema, rash, and hepatic enzyme elevations. No new adverse reactions were reported in the adjuvant GIST treatment setting that had not been previously reported in other patient populations, including patients with unresectable and metastatic GISTs. Edema, gastrointestinal disturbances (nausea, vomiting, abdominal distention, and diarrhea), fatigue, low hemoglobin, and rash were the most frequently reported adverse reactions at the time of discontinuation (Table 3).
Table 3.
Adverse Events (AEs) of CTC grade ≥3 regardless of relationship to study drug reported in imatinib-treated adjuvant gastrointestinal stromal tumor patients

Abbreviations: CTC, Common Toxicity Criteria.
Discussion
Imatinib was granted accelerated approval for the adjuvant treatment of GISTs on December 19, 2008. Approval was based on an American College of Surgeons Oncology Group randomized, placebo-controlled trial [1, 2]. Under the accelerated approval procedure, the FDA may approve a new drug or biologic if adequate and well-controlled trials establish that the product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit. The accelerated approval regulations require the applicant to verify and describe the drug's clinical benefit in additional adequate and well-controlled trials or by continued follow-up of the original study population. Such studies must be conducted with due diligence (21 CFR 314.510 Subpart H and 601.41 Subpart E).
At the time accelerated approval was granted, the median follow-up was 14 months. Although there was a significant difference between imatinib- and placebo-treated patients in terms of the RFS rate, the OS results were immature. Consequently, the FDA required the sponsor to provide further follow-up of RFS and OS outcomes from this study and to also provide results from an ongoing study of 1 year versus 3 years of adjuvant imatinib. The latter study is the basis for this report.
The FDA's authorization to require postmarketing studies (observational epidemiologic studies, animal studies, and laboratory experiments) and postmarketing clinical trials (prospective investigations in which the sponsor or investigator determines the method of assigning treatment or other interventions to one or more human subjects) derives from The Food and Drug Administration Modernization Act of 1997 and the Food and Drug Administration Amendments Act of 2007. In addition, the FDA was provided additional authority to monitor the progress of postmarketing commitments (PMCs) and PMRs by requiring applicants to submit annual status reports. These reports must also include the reasons, if any, for failure to complete the commitment. A timetable for trial completion with milestone dates including, but not limited to, the final protocol submission date, study or clinical trial completion date, and final report submission date must be established and adhered to unless there are compelling reasons to change the dates. The FDA is required to track these PMCs and PMRs and to report on them annually in the Federal Register.
The benefit-to-risk assessment, the basis for an FDA approval decision, of the present application is strongly weighted toward benefit given the observed statistically significant higher RFS and OS probabilities, with HRs of 0.46 and 0.45, respectively. Although, as expected, the adverse event rates and drug discontinuation rates were generally higher with a longer duration of imatinib treatment, the rates of grade 3 and 4 toxicities were only slightly higher. No new adverse reactions were reported in the patient population receiving imatinib for 3 years that had not been previously reported in other patient populations.
At the time of the sNDA submission, additional data were submitted concerning three other outstanding PMRs related to the 1-year imatinib adjuvant GIST study, including the 4-year and 5-year RFS and OS results. Sufficient information was provided to fulfill those commitments.
Agreement was reached with the sponsor on physician labeling. The safety and efficacy results from the current trial and the RFS and OS updates from the previous trial were incorporated into the current label. The dosage and administration section of the label was revised to state that 3 years of imatinib is recommended for adjuvant GIST treatment but that the optimal duration of imatinib therapy is not known.
As part of the approval process, the applicant was asked for, and agreed to, a postmarketing commitment to continue to follow GIST patients in the 1-year versus 3-year imatinib adjuvant trial for ≥5 years from the date that the last patient was randomized. Updated RFS and OS results, including datasets, will be provided as an addendum to the full clinical trial report. The survival data cutoff date will be January 2014, with the final report and dataset submission in March 2015. Because it is an efficacy update and is not related to a specific safety issue, it is a voluntary PMC and not a PMR.
Acknowledgment
The views expressed are the result of independent work and do not necessarily represent the views and findings of the U.S. Food and Drug Administration.
Author Contributions
Conception/Design: Martin H. Cohen, John R. Johnson, Robert Justice, Richard Pazdur
Provision of study material or patients: Martin H. Cohen, John R. Johnson, Robert Justice, Richard Pazdur
Collection and/or assembly of data: Martin H. Cohen, John R. Johnson, Robert Justice, Richard Pazdur
Data analysis and interpretation: Martin H. Cohen, John R. Johnson, Robert Justice, Richard Pazdur
Manuscript writing: Martin H. Cohen, John R. Johnson, Robert Justice, Richard Pazdur
Final approval of manuscript: Martin H. Cohen, John R. Johnson, Robert Justice, Richard Pazdur
References
- 1.Cohen MH, Cortazar P, Justice R, et al. Approval summary: Imatinib mesylate in the adjuvant treatment of malignant gastrointestinal stromal tumors. The Oncologist. 2010;15:300–307. doi: 10.1634/theoncologist.2009-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dematteo RP, Ballman KV, Antonescu CR, et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: A randomised, double-blind, placebo-controlled trial. Lancet. 2009;373:1097–1104. doi: 10.1016/S0140-6736(09)60500-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Joensuu H, Eriksson M, Sundby Hall K, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: A randomized trial. JAMA. 2012;307:1265–1272. doi: 10.1001/jama.2012.347. [DOI] [PubMed] [Google Scholar]
- 4.Blanke CD. Optimal duration of adjuvant therapy for patients with resected gastrointestinal stromal tumors. JAMA. 2012;307:1312–1314. doi: 10.1001/jama.2012.368. [DOI] [PubMed] [Google Scholar]
- 5.Gold JS, Gönen M, Gutiérrez A, et al. Development and validation of a prognostic nomogram for recurrence-free survival after complete surgical resection of localised primary gastrointestinal stromal tumour: A retrospective analysis. Lancet Oncol. 2009;10:1045–1052. doi: 10.1016/S1470-2045(09)70242-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rutkowski P, Bylina E, Wozniak A, et al. Validation of the Joensuu risk criteria for primary resectable gastrointestinal stromal tumour—the impact of tumour rupture on patient outcomes. Eur J Surg Oncol. 2011;37:890–896. doi: 10.1016/j.ejso.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 7.Joensuu H. Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum Pathol. 2008;39:1411–1419. doi: 10.1016/j.humpath.2008.06.025. [DOI] [PubMed] [Google Scholar]
- 8.Hornick JL, Fletcher CD. The role of KIT in the management of patients with gastrointestinal stromal tumors. Hum Pathol. 2007;38:679–687. doi: 10.1016/j.humpath.2007.03.001. [DOI] [PubMed] [Google Scholar]