

Abstract
Kv1.3 channels play an important role in modulating lymphocyte proliferation and apoptosis. We hypothesized that Kv1.3 channels in B lymphocytes might be regulated by rituximab, an antibody to CD20, a drug for treatments of B-cell lymphomas and autoimmune diseases. Using both whole-cell and cell-attached patch-clamp techniques, we found that rituximab inhibited Kv1.3 channels in Daudi human B lymphoma cells by promoting the channel inactivation at a concentration which was much greater than that required for activation of CD20. The effect of rituximab on Kv1.3 channels was abolished after selective blockade of FcγRIIB receptors with anti-FcγRIIB antibody. Western blot experiments showed that Daudi B cells expressed both Kv1.3 channel and the low affinity Fc receptor, FcγRIIB, which could be activated by the Fc region of rituximab. In contrast, normal lymphocytes expressed less Kv1.3 channels with faster inactivation. Confocal microscopy and flow cytometry data showed that rituximab induced apoptosis of Daudi B cells and that the effect was attenuated by blockade of FcγRIIB receptors and partially mimicked by inhibition of Kv1.3 channels. These results suggest that in addition to previously described complement-dependent cytotoxicity, rituximab also induces apoptosis of malignant B lymphocyte by stimulating FcγRIIB receptors and inhibiting Kv1.3 channels.
Keywords: Voltage-dependent potassium channel, Fc receptor, Rituximab, Apoptosis, Patch-clamp technique, Confocal microscopy
1. Introduction
The non-Hodgkin’s lymphomas rank fifth in cancer incidence and sixth in cancer mortality in the United States. Rituximab (Rituxan, IDEC-C2B8), a chimeric mouse anti-human CD20 antibody, has been used for the treatment of non-Hodgkin’s lymphomas [9,25,33]. Several lines of evidence suggest that the clinical outcomes of rituximab are due to depletion of B cells via CD20-associated activation of complement-dependent cytotoxicity [26,33,45], antibody-dependent cellular cytotoxicity [6,21,30], or phagocytosis [7,40]. These events are triggered by rituximab cross-linking of CD20 molecules. The binding affinity of rituximab to CD20 is relatively high; it has been reported that the Kd is less than 6 nM [12,14]. Since rituximab is a chimeric antibody with human IgG1 Fc [19], the Fc region of rituximab also plays a very critical role in mediating its cytotoxicity by ligation of Fc receptors on the surface of nature killer cells [30] and macrophages [22]. The constant serum concentration of rituximab in the patients treated with a regular dose of 375 mg/m2 weekly for 4 weeks can reach 96.8 μg/ml (0.7 μM) [5], which could ligate any Fc receptors [24]. The transient serum concentration could be even higher. Since the low affinity FcγRIIB receptor is highly expressed on the surface of B cells [34] and plays an important role in down-regulating immune responses [17,20,29,37], rituximab may directly target B cells by stimulating this receptor.
The Kv1.3 channel in B lymphocytes plays an important role in promoting proliferation [28] and differentiation [43]. We have previously shown that the Kv1.3 channel is expressed in Daudi B cells and has a unique incomplete inactivation [44]. However, it remains largely unknown how the Kv1.3 channel in B lymphocytes is regulated by the receptors on the surface of B cells and whether the incomplete inactivation represents a unique gating of the Kv1.3 channel in malignant Daudi B cells. Since CD20 can induce B cell apoptosis [36] and Kv1.3 channel is regulated by the death receptor, Fas [39], we originally hypothesized that cross-linking of CD20 with rituximab might result in apoptosis of B cells not only via the pathways described previously [1,10], but also through regulation of Kv1.3 channels. However, as described above, the serum concentration of rituximab can reach levels that could stimulate the low affinity FcγRIIB receptor [5]. Among Fc receptors, the FcγRIIB receptor is the only Fc receptor which down-regulates the functions of immune cells including B lymphocytes [17,20,29,37]. Therefore, it is also possible that rituximab regulates Kv1.3 channels by stimulating the low affinity FcγRIIB receptor. In the present study, we show that rituximab inhibits Kv1.3 channels in Daudi B cells by stimulating the FcγRIIB receptor and induces Daudi B cells to undergo apoptosis partially through activation of FcγRIIB receptors.
2. Materials and methods
2.1. Cell culture and patch-clamp techniques
Daudi B cell culture and patch-clamp experiments were performed as we reported previously [41,44]. Freshly isolated human lymphocytes were provided by Binli Tao (University of Alabama at Birmingham).
2.2. Western blotting
Daudi B cell lysate (20 μg) was loaded and electrophoresed on 7.5% SDS-PAGE gels (Mini-Protean® TGX™ Precast Gels, Bio-Rad) for 60 to 90 min. Gels were blotted onto nitrocellulose membranes for 1 h at 90 V. After 1 h blocking with 5% milk in TBS-T buffer, PVDF membranes were incubated with primary antibody (1:1000 dilution) of either goat anti-human FcγRIIB polyclonal antibody (R&B, systems) or rabbit anti-human Kv1.3 antibody (Alomone Labs) overnight at 4 °C, and then respectively incubated with ReserveAP™ phosphatase-conjugated rabbit anti-goat or goat anti-rabbit IgG secondary antibody (1:5000 dilution, KPL) for 1 h after 3 vigorous washes. Blots were developed by chemiluminescence using ECL Plus Western Blotting Detection System (GE healthcare).
2.3. Confocal microscopy imaging and flow cytometry analysis
To evaluate apoptosis, Daudi B cells were stained with both FITC-conjugated annexin V (AV) and propidium iodide (PI). Confocal microscopy experiments were performed to determine whether and how rituximab induces apoptosis in Daudi B cells. The cell membrane of apoptotic cells was stained with AV because phosphatidylserine, a lipid which has a high binding affinity to AV, is externalized in apoptotic cells. The nuclei of apoptotic cells were stained with PI because the nuclear membrane of apoptotic cells becomes permeable to PI. AV was excited with 488 nm laser and visualized through a 515 nm emission filter, shown in green. PI was excited with 488 nm laser and visualized through a 590 nm emission filter, shown in red. In each set of experiments, images were taken using the same parameter settings. To quantify the number of apoptotic cells, the percentages of apoptotic cells, as determined by AV- and PI-positive cells, were measured on a FACScalibur flow cytometer (Becton Dickinson) within 30 min. AV and PI were excited with 488 nm laser. The emissions were detected through either a 515 nm filter for AV or a 580 nm filter for PI.
2.4. Chemicals and solutions
Most chemicals were obtained from Sigma Chemical Co. Rituximab was provided by Dr. Mansoor N. Saleh (University of Alabama at Birmingham, Birmingham, Alabama, USA). Goat anti-human FcγRIIB antibody was purchased from R&D Systems. NaCl (or KCl) bath solution contained (in mM): 145 NaCl (or 145 KCl), 5 KCl (or 5 NaCl), 1 CaCl2, 1 MgCl2, and 10 N-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), at a pH of 7.4. KCl (or NaCl) pipette solution contained (in mM): 145 KCl (or 145 NaCl), 50 nM free Ca2+ (after titration with 2 mM ethylene glycol-bis(b-aminoethyl ether)-N,N,N′,N′-tetraacetic acid), 1 MgCl2, 2 Na2-ATP (or 2 K2-ATP), and 10 HEPES, at a pH of 7.2. All the concentrations throughout this article are shown as the final concentration.
2.5. Statistical analysis
Paired-t test was used for the comparison between two groups of data from the same patch-clamp recording before and after experimental manipulations. Student’s t test was used for the comparison between two groups of data from two separate patch-clamp recordings. The analysis of variance for multiple comparisons was used for the comparison among multiple groups of data. Data are shown as mean±SD. p<0.05 is considered statistically significant.
3. Results
3.1. The gating and expression of Kv1.3 channels are upregulated in Daudi cells
Our previous report showed that Kv1.3 channel was expressed in Daudi B cells and that the channel could not inactivate completely in response to prolonged depolarization [44]. Consistent with our previous finding, the present study showed that depolarizing voltage-step pulses induced outward currents which did not inactivate completely, but were almost completely blocked with 10 nM MgTX, a selective blocker for Kv1.3 and Kv1.2 channels (Fig. 1A). Since we previously showed that the current was almost abolished with antisense to Kv1.3 channel [44], we concluded that this current resulted from activation of Kv1.3 channels. To determine whether the incomplete inactivation represents the unique gating of Kv1.3 channel in malignant Daudi B cells, the whole-cell recording was also established in normal human lymphocytes. An outward current was also observed in these lymphocytes. Compared to the Kv1.3 currents in Daudi cells, the Kv1.3 currents in normal lymphocytes were much smaller and inactivate completely (Fig. 1B). Therefore, the decay rate of the currents induced by a voltage-step pulse from the holding potential of −60 mV to +60 mV was analyzed and compared between Daudi and normal lymphocytes. The representative Kv1.3 currents induced by a voltage-step pulse from a holding potential of −60 mV to +60 mV in either a Daudi cells or a normal lymphocyte were fitted nicely with a single exponential function, as shown in Fig. 1C. The summarized inactivation time constant was 509.8± 51.2 ms from 6 individual Daudi cells and 347.3±35.4 ms from 6 individual normal lymphocytes (Fig. 1D). Western blot experiments showed that in contrast to Daudi B cells, normal lymphocytes expressed less Kv1.3 channels (Fig. 1E). These data suggest that the gating and expression of Kv1.3 channels are upregulated in malignant Daudi B cells. However, it remains to be determined whether the upregulation of Kv1.3 channels is related to the malignancy of Daudi B cells.
Fig. 1.

Kv1.3 inactivation and expression are different between Daudi cells and normal lymphocytes. (A) Representative whole-cell recordings from a Daudi cell before (left) and after application of 10 nM MgTX to the bath (right). (B) Representative whole-cell recordings from a normal lymphocyte before (left) and after application of 10 nM MgTX to the bath (right). (C) Representative Kv1.3 currents (black lines behind red and green line) induced by a voltage-step pulse from the holding potential of −60 mV to +60 mV were fitted with a single exponential function, yielding a time constant (τ) of either 511.0 ms in a Daudi cell (red line) or 314.7 ms in a normal lymphocyte (green line). (D) Summary plot of τ in either Daudi cells (open bar) or normal lymphocyte (solid bar). (E) Western blot of Daudi cells (lane 1) and normal lymphocytes (lane 2), showing that Daudi cells express higher levels of Kv1.3 channels than normal lymphocytes. Detection of β-actin was used to show equal loading of protein in each lane. The data represent three individual experiments showing consistent results. In (A) and (B), a voltage-step protocol from −100 mV to +60 mV with an increment of +20 mV at the holding potential of −60 mV was used to induce Kv1.3 current. Patch pipettes were filled with KCl pipette solution while NaCl bath solution was used for the bath. No leaky subtraction was applied to this figure and any other patch-clamp recordings.
3.2. Rituximab promotes inactivation of Kv1.3 channels via FcγRIIB receptors
To examine the effect of rituximab on Kv1.3 channel gating, Kv1.3 currents induced by a voltage-step protocol before and after application of rituximab were recorded as shown in Fig. 2A. Rituximab seemed to reduce the current amplitude by promoting the channel inactivation. Therefore, the decay rate of the current induced by a voltage-step pulse from the holding potential of −60 mV to +60 mV before rituximab was compared with that after rituximab. Fig. 2C showed that the representative currents before and 3 min after 100 μg/ml rituximab were fitted nicely with a single exponential function. After application of rituximab, the mean inactivation time constant (τ) was significantly reduced, from 517.3±54.6 ms to 169.0±26.1 ms (p<0.001; n=6) (Fig. 2E). However, in the cells pre-treated with 2 μg/ml anti-FcγRIIB antibody to block the FcγRIIB receptor, rituximab no longer affected Kv1.3 currents (Fig. 2B). Fig. 2D showed that the representative currents induced by a voltage-step pulse from the holding potential of −60 mV to +60 mV before and 3 min after 100 μg/ml rituximab in these pretreated cells were fitted well with a single exponential function. The mean inactivation time constant (τ) remained unchanged, 503.5±57.1 ms (before) versus 485.8±47.8 ms (after rituximab) (p=0.129; n=6) (Fig. 2E). To confirm the involvement of the FcγRIIB receptor, we performed Western blotting experiments. The data showed that the FcγRIIB receptor is expressed in Daudi B cells (Fig. 2F). To examine the effect of rituximab on Kv1.3 channel at the single-channel level, we also performed cell-attached patch-clamp recordings. Consistent with our previous report [44], a 19-pS Kv channel was observed in Daudi cells and did not completely inactivate in response to prolonged depolarization (Fig. 3A). Like Kv1.3 whole-cell currents, the single-channel activity was completely blocked with 10 nM MgTX in the patch pipette (Fig. 3B). Fig. 3C showed that at 5 min after application of 100 μg/ml rituximab to the bath, the single-channel events were almost only observed during the initial second, indicating that rituximab promotes Kv1.3 channel inactivation. However, the unit conductance was unchanged (19.5±0.6 pS) (p=0.694, n=4), compared to that before rituximab (19.4±0.9 pS). The effect of rituximab on Kv1.3 channel inactivation was abolished in the cells pretreated with 2 μg/ml anti-FcγRIIB antibody to selectively block the FcγRIIB receptor (Fig. 3D). These data suggest that rituximab promotes Kv1.3 channel inactivation via the FcγRIIB receptor.
Fig. 2.

Rituximab promotes Kv1.3 channel inactivation in Daudi cells via FcγRIIB receptors. (A) Representative whole-cell recordings from a Daudi cell before (left) and after application of 100 μg/ml rituximab to the bath (right). (B) Representative whole-cell recordings from a Daudi cell pretreated with 2 μg/ml goat anti-human FcγRIIB antibody for 5 min before (left) and after application of 100 μg/ml rituximab to the bath (right). (C) Representative Kv1.3 currents (black lines behind red and green line) induced by a voltage-step pulse from the holding potential of −60 mV to +60 mV in a control Daudi cells were fitted with a single exponential function, yielding a time constant (τ) of either 529.2 ms under control conditions before rituximab (red line) or 135.9 ms after application of 100 μg/ml rituximab to the bath (green line). (D) Representative Kv1.3 currents induced by a voltage-step pulse from the holding potential of −60 mV to +60 mV in a Daudi cells pretreated with 2 μg/ml goat anti-human FcγRIIB antibody for 5 min were fitted with a single exponential function, yielding a time constant (τ) of either 497.1 ms under control conditions before rituximab (red line) or 486.3 ms after application of 100 μg/ml rituximab to the bath (green line). (E) Summary plot of τ either in control Daudi cells (left bars) or Daudi cells pretreated with 2 μg/ml goat anti-human FcγRIIB antibody for 5 min (right bars). (F) Western blot shows that FcγRIIB is expressed in Daudi B cells. The voltage-step protocol and solutions were the same as used in Fig. 1.
Fig. 3.

Rituximab promotes Kv1.3 channel inactivation in Daudi cells via FcγRIIB receptors in cell-attached patches. (A) 5 superimposed single-channel currents from 3 cell-attached recordings show that a voltage-step pulse from 0 mV to +80 mV (−Vpipette) induced Kv1.3 channel activity which does not inactivate completely. (B) 5 superimposed single-channel currents from 5 cell-attached recordings show that Kv1.3 channel activity was completely blocked by 10 nM MgTX in the patch pipette. (C) 5 superimposed single-channel currents from 4 cell-attached recordings either before (left) or after addition of 100 μg/ml rituximab to the bath (right) show that rituximab promoted the channel inactivation. (D) 5 superimposed single-channel currents from 5 cell-attached recordings in Daudi cells pretreated with 2 μg/ml goat anti-human FcγRIIB antibody for 5 min show that blockade of FcγRIIB receptor abolished the effect of rituximab. Inset panels A1, B1, C1, C2, or D1 shows a representative trace in each superimposed traces. A voltage-step pulse from a holding potential of 0 mV to +80 mV (−Vpipette =+80 mV). NaCl bath solution was used both for filling patch pipettes and for the bath.
3.3. Kv1.3 channel in Daudi cells can be activated by a slow depolarization
Since lymphocytes are not excitable cells, the membrane potential may alter gradually. To determine whether a slow depolarization activates Kv1.3 channel, a voltage-ramp protocol was used to quickly get the I–V relationship. The voltage-ramp protocol from −120 to +60 mV with the holding potential of −60 mV was given at a rate of 1 min. A voltage-dependent outward current was induced by the voltage-ramp protocol. The threshold potential for activation of this current was approximately −40 mV. This current was completely blocked by addition of 100 μM quinine, a generic K+ channel blocker, to the bath in all of 11 recordings. The blockade was almost completely reversed every time when quinine was washed out of the bath (Fig. 4A). The current was also blocked by addition of either 100 nM charybdotoxin (ChTX), a voltage-dependent K+ channel blocker, to the bath in all of 8 recordings, or 10 nM margatoxin (MgTX), a Kv1.3 channel blocker, to the bath in all of 6 recordings. The blockade by quinine was completely reversible while the blockade by ChTX or MgTX was either partially reversible or irreversible. This is consistent with the fact that MgTX has a slow dissociation rate [15]. Replacement of 145 mM Na+ in the bath with 145 mM K+ shifted the reversal potential to 0 mV (Fig. 4B), indicating that this current is carried by K+. Furthermore, the replacement of cytoplasmic K+ with 145 mM Na+ by whole-cell dialysis of the cell eliminated the K+ current. These data together with our previous report [44] suggest that a slow depolarization can also activate Kv1.3 channels in Daudi B cells.
Fig. 4.

Kv1.3 channel is induced by a slow voltage-ramp pulse. (A) Addition of 100 μM quinine (upper left), 100 nM ChTX, a voltage-dependent K+ channel blocker (upper right), or 10 nM MgTX, a Kv1.3 channel blocker (supper left), to the bath completely blocked whole-cell currents induced by a voltage-ramp protocol. Immediately after the blockade was observed, ChTX and MgTX were washed out of the bath. The blockade by ChTX was almost completely reversed in 1 out of total 8 recordings (trace c, upper right), but was partially reversed in the other 7 recordings. The blockade by MgTX was partially reversed in 2 out of total 6 recordings (trace c, supper left) or irreversible in the other 4 recordings. (B) The ChTX-sensitive, voltage-dependent current was reversed by replacement of 145 mM Na+ in the bath with 145 mM K+; the reversal potential was shifted to 0 mV (left), and disappeared after whole-cell dialysis of the cells with 145 mM Na+ (right). A voltage-ramp protocol was used to induce the voltage-dependent Kv1.3 currents, in which the voltage immediately went down to −120 mV from a holding potential of −60 mV and then gradually went up to +60 mV. As indicated, patch pipettes were filled with either KCl pipette solution containing 145 mM K+ or NaCl pipette solution containing 145 mM Na+; the bath was washed with either KCl bath solution or NaCl bath solution. Upward tracings show outward currents, representing K+ efflux.
3.4. Rituximab inhibits Kv1.3 currents induced by a slow depolarization via activation of FcγRIIB receptors
To determine how rituximab affects Kv1.3 currents in response to a slow membrane depolarization, the voltage-ramp protocol was also used in the following experiments. Since rituximab cross-links CD20 molecules with high binding affinities (Kd is 0.3–6 nM) [12,14], we first exposed Daudi B cells to rituximab at 10 μg/ml (=69 nM), a concentration which was over 10 times greater than the Kd for rituximab to bind to CD20. However, the data from our whole-cell patch-clamp experiments demonstrated that at this concentration rituximab failed to regulate Kv1.3 channels in Daudi B cells (Fig. 5A). But at a high concentration (200 μg/ml=1.4 μM) which can stimulate the low affinity Fc receptors (Kd is 0.6–2.5 μM, [24]), rituximab significantly reduced Kv1.3 whole-cell currents within 2 min after it was added to the bath (Fig. 5B). We hypothesized that rituximab may stimulate the low affinity FcγRIIB receptor which is highly expressed on the surface of B cells [34]. Since anti-FcγRIIB antibody can be used for selectively blocking FcγRIIB receptors [11], Daudi B cells were pretreated with 2 μg/ml goat anti-human FcγRIIB antibody to selectively block the FcγRIIB receptor on Daudi cells. After the pretreatment, application of 200 μg/ml rituximab to the bath no longer reduced Kv1.3 currents (Fig. 5C). Compared to rituximab which has a human IgG1 Fc region [19], rabbit IgG should hardly bind to human Fc receptors on Daudi cells. Therefore, rabbit IgG was used as a control. Kv1.3 currents in Daudi B cells were only slightly reduced at 3 min after addition of 200 μg/ml rabbit IgG to the bath (Fig. 5D). Percent inhibition of the peak Kv1.3 currents under each condition was plotted in Fig. 5E. 10 μg/ml rituximab did not alter the peak Kv1.3 current during the 3-min period in 8 whole-cell recordings (p>0.05, n=8). However, 200 μg/ml rituximab reduced the peak Kv1.3 current by more than 70% within 2 min (p<0.001, n=9). In contrast, in Daudi B cells pre-treated with 2 μg/ml anti-FcγRIIB antibody, 200 μg/ml rituximab failed to reduce Kv1.3 currents (p>0.05, n=8). As a control, 200 μg/ml rabbit IgG tended to reduce Kv1.3 currents in Daudi B cells at 3 min after the addition, but the effect is only marginally significant (p=0.05, n=7).
Fig. 5.

Rituximab reduces Kv1.3 currents induced by a slow voltage-ramp pulse in an FcγRIIB receptor-dependent manner. (A) Representative whole-cell Kv1.3 currents induced by a voltage-ramp protocol; the protocol was given at a rate of 1 min. After the first recording (before) was made, 10 μg/ml rituximab was added to the bath. Then, the voltage-ramp protocol was given to the cell for additional three times, respectively at 1, 2, or 3 min after rituximab. The four current traces in response to the voltage-ramp protocol were superimposed and showed no changes. (B) A remarkable decrease in Kv1.3 currents occurred at 2 and 3 min after addition of 200 μg/ml rituximab to the bath. (C) 200 μg/ml rituximab failed to reduce Kv1.3 currents in a Daudi B cell pretreated with 2 μg/ml goat anti human FcγRIIB antibody to selectively block FcγRIIB receptors. (D) Addition of 200 μg/ml rabbit IgG to the bath slightly reduced Kv1.3 currents only at 3 min after the addition. (E) Summary plots of relative inhibition of the peak Kv1.3 currents under the conditions used in A through D. Patch pipettes were filled with KCl pipette solution while NaCl bath solution was used for the bath.
To examine the concentration-dependent effects of rituximab, five concentrations of rituximab (12.5, 25, 50, 100, and 200 μg/ml) were used in 6 whole-cell recordings. The representative recordings of Kv1.3 currents before and at 1, 2, and 3 min after addition of 12.5, 50, or 200 μg/ml rituximab were plotted in Fig. 6A. As shown in Fig. 6B, the percentages of Kv1.3 current inhibition versus rituximab concentrations were fitted with the Hill function; the IC50 for rituximab to inhibit Kv1.3 current was 80 μg/ml. These results suggest that the effect of rituximab on Kv1.3 channels is dependent on the activity of the low affinity Fc receptor FcγRIIB rather than the high affinity B cell surface marker CD20. The present study for the first time suggests that the Kv1.3 channel in B lymphocytes is regulated by the FcγRIIB receptor. However, the underlying signal transduction pathway remains to be determined.
Fig. 6.

Rituximab inhibits Kv1.3 currents in a dose-dependent manner. (A) Representative recordings of Kv1.3 currents. Four traces were superimposed showing Kv1.3 currents before and 1, 2, or 3 min after addition of rituximab to the bath at 12.5 (left), 50 (middle) or 200 μg/ml (right). (B) Relative inhibition of the peak Kv1.3 currents at 3 min after 12.5, 25, 50, 100, or 200 μg/ml rituximab was added to the bath (solid circles). Data are shown as mean±SD. Patch pipettes were filled with KCl pipette solution while NaCl bath solution was used for the bath.
3.5. Rituximab reduces the membrane potential of Daudi B cells via FcγRIIB receptor inhibition of Kv1.3 channels
We have shown that Kv1.3 channels play an important role in regulating the membrane potential of Daudi B cells [44]. Since rituximab could inhibit Kv1.3 channels, theoretically, the membrane potential of Daudi B cells should be reduced after application of rituximab. Therefore, after forming the whole-cell configuration, the membrane potential of Daudi B cells was measured with the current-clamp mode when the current was clamped at zero. The data showed that application of 100 μg/ml rituximab significantly reduced the membrane potential from −48.6±4.4 mV to −37.7±5.4 mV (p<0.001, n=6) when Daudi B cells were under control conditions (Fig. 7). In contrast, when Daudi B cells were pretreated with 2 μg/ml anti-FcγRIIB anti-body to selectively block the FcγRIIB receptor, application of 100 μg/ml rituximab no longer affected the membrane potential, −48.5± 5.0 mV versus −48.2±4.8 mV (p=0.679, n=6). Application of 10 nM MgTX to the bath strongly reduced the membrane potential from −49.2±4.5 mV to −9.2±3.4 mV (p<0.001, n=6), but in the presence of MgTX, 100 μg/ml rituximab did not further reduce the membrane potential (−9.2±3.4 mV; p=0.695, n=6). These data suggest that rituximab reduces the membrane potential of Daudi B cells by inhibiting Kv1.3 channels through the FcγRIIB receptor.
Fig. 7.
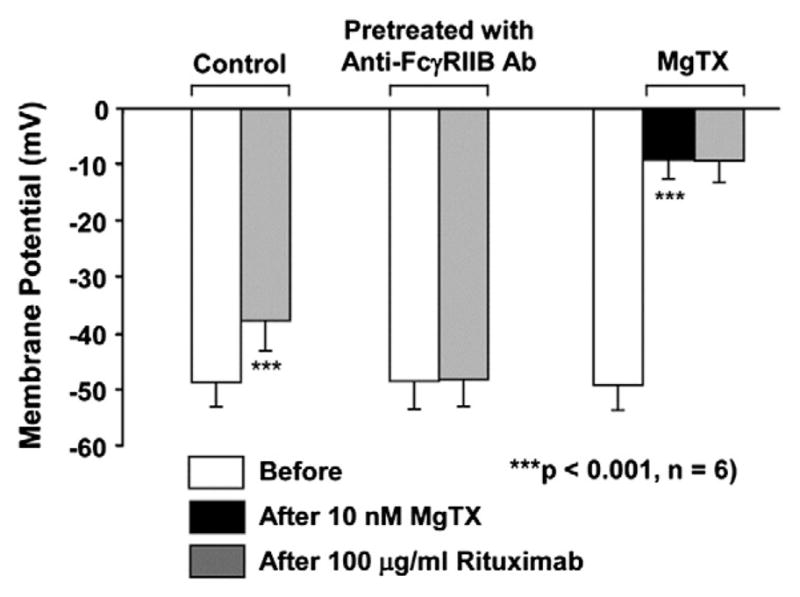
Rituximab reduces the membrane potential of Daudi B cells via FcγRIIB receptor inhibition of Kv1.3 channels. The experiments were performed either under control conditions or after pretreatment with 2 μg/ml goat anti-human FcγRIIB antibody for 5 min. Left bars show that rituximab reduced the membrane potential in control Daudi B cells. Middle bars show that rituximab did not alter the membrane potential in Daudi B cells pretreated with 2 μg/ml goat anti-human FcγRIIB antibody for 5 min. Right bars show that MgTX reduced the membrane potentials in control Daudi B cells and that after MgTX, rituximab did not further reduce the membrane potential. The membrane potential from the same whole-cell recording before MgTX or rituximab (open bars) was used as its own control for comparison with those 5 min after application of either 10 nM MgTX (black bars) or rituximab (gray bars). Membrane potentials of Daudi cells were measured using the current-clamp mode (I=0).
3.6. Rituximab induces apoptosis of Daudi B cells partially via FcγRIIB receptors
Recent studies have shown that the FcγRIIB receptor plays a role in mediating antibody-induced apoptosis of malignant B cells [27]. To determine whether the FcγRIIB receptor acts as one of the pathways for rituximab to induce apoptosis, Daudi B cells were stained with FITC-conjugated annexin-V (AV) and propidium iodide (PI). First, confocal microscopy experiments were performed, as shown in Fig. 8A. In untreated Daudi cells, only occasionally we could see AV- and PI-positive cells (apoptotic cells). In contrast, in the cells treated with 200 μg/ml rituximab for 30 min, a significant portion of cells underwent apoptosis. In a parallel with the inhibition of Kv1.3 channels observed in patch-clamp experiments, pretreatment of the cells with 2 μg/ml anti-FcγRIIB antibody attenuated the rituximab-induced apoptosis. To determine whether inhibition of Kv1.3 channels by Kv1.3 channel blockers could result in apoptosis, Daudi B cells were treated with 10 nM MgTX for 30 min. MgTX seemed to induce a portion of cells to undergo apoptosis. To quantify these effects, we performed six separate flow cytometry experiments for each group. The representative data were shown in Fig. 8B. The summarized results were plotted in Fig. 8C. Under control conditions, only 3±1% (mean±SD, n=6) cells were automatically apoptotic. In contrast, after the cells were treated with 200 μg/ml rituximab for 30 min, the number of apoptotic cells was increased to 24%±6% (p<0.001, n=6). This effect was attenuated in the cells pretreated with 2 μg/ml anti-FcγRIIB antibody to selectively block the FcγRIIB receptor; the number of apoptotic cells was 14±5%, which is significantly less than that in the cells treated with rituximab alone (p<0.001, n=6), but still greater than that in the cells under control conditions (p<0.001). This indicates that rituximab induces apoptosis through the FcγRIIB receptor, but the FcγRIIB receptor should not be the only pathway for rituximab-induced apoptosis. After the cells were treated with 10 nM MgTX for 30 min, the number of apoptotic cells was increased to 8%±3% (p<0.001, n=6), but less than that in the cells treated with rituximab (p<0.001). These data suggest that a high concentration of rituximab could induce apoptosis of Daudi B cells partially by stimulating the FcγRIIB receptor and that inhibition of Kv1.3 channels may contribute to the rituximab-induced apoptosis of B cells.
Fig. 8.

Blockade of FcγRIIB receptors attenuates rituximab-induced apoptosis of Daudi B cells. (A) Representative confocal microscopy data showed that 200 μg/ml rituximab induced apoptosis of Daudi B cells in both FcγRIIB receptor- and Kv1.3 channel-dependent manner. Apoptotic cells were stained with FITC-conjugated Annexin V (shown in green, upper left panels in each image) due to phosphatidylserine externalization and propidium iodide (shown in red, upper rights in each image) due to permeable nuclear membrane. DIC images were taken to show the total Daudi cells in the field (lower left panels in each image). Lower right panels in each image show the overlay of AV, PI and DIC images. (B) Representative flow cytometry data showed that 200 μg/ml rituximab increased the number of apoptotic cells in an FcγRIIB receptor-dependent manner. In lower left gate, cells were both AV- and PI-negative; in lower right gate, cells were AV-positive; in the upper left gate, cells were PI-positive; in the upper right gate, cells were both AV- and PI-positive. The experiments were performed in the cells under the conditions used for the confocal microscopy experiments. (C) Summary plots of percent apoptotic cells under each condition from six separate flow cytometry experiments, showing that 200 μg/ml rituximab significantly increased the number of apoptotic cells partially through FcγRIIB receptors and Kv1.3 channels. As indicated, Daudi B cells were under control conditions, treated with 200 μg/ml rituximab for 30 min either without or with pretreatment with 2 μg/ml goat anti-human FcγRIIB antibody for 5 min, or treated with 10 nM MgTX for 30 min.
4. Discussion
We have previously shown that Kv1.3 channel is expressed in Daudi B cells and does not inactivate completely in response to prolonged depolarization [44]. The present study shows that the expression levels of Kv1.3 channel in Daudi B cells are higher than those in normal lymphocytes and that unlike the Kv1.3 channel in Daudi B cells, the Kv1.3 channel in normal lymphocytes does inactivate completely. However, it is unlikely that defective inactivation and enhanced expression of Kv1.3 channels account for the malignant proliferation of Daudi B cells, because our recent studies suggest that an intermediate Ca2+-activated K+ channel rather than Kv1.3 channel promotes Daudi cell proliferation [41]. It has been suggested that Kv1.3 channel plays an important role in regulating apoptosis in T cells [39]. The present study shows that Kv1.3 channel is downregulated by the anti-lymphoma drug, rituximab and that the downregulation appears to contribute to rituximab-induced apoptosis. These studies together suggest that the incomplete inactivation and the enhanced expression of Kv1.3 channel observed in this study may be more important for Daudi B cells to escape from apoptosis rather than to become highly proliferative. Crosslinking of the CD20 receptor with its antibodies including rituximab could induce apoptosis of malignant B cells [16,36]. However, our results shows that rituximab inhibits Kv1.3 channel with an IC50 of 80 μg/ml (552 nM), which is almost 100 times higher than the Kd (6 nM) for rituximab to bind to CD20 [12], indicating that the effect is not mediated by the CD20 receptor. Since the Kd for IgG to stimulate the low affinity Fc receptors is in the range from 0.6 to 2.5 μM [24] and the FcγRIIB receptor is expressed in Daudi B cells, rituximab should activate the FcγRIIB receptor. Indeed, we show that selective blockade of the FcγRIIB receptor with its antibody is able to abolish the inhibition of Kv1.3 channels by rituximab. These results for the first time suggest that the Kv1.3 channel in B lymphocytes is regulated by the FcγRIIB receptor.
Recent studies suggest that the Kv1.3 channel in T lymphocytes acts as a therapeutic target for treatments of autoimmune diseases [2,35] such as type-1 diabetes mellitus or rheumatoid arthritis [4] and multiple sclerosis [3,31,42]. Since the FcγRIIB receptor plays an important role in down-regulating immune responses in B lymphocytes [17,20,29,37], inhibition of Kv1.3 channels by activation of the FcγRIIB receptor may complement the possible clinical use of Kv1.3 channel blockers to treat autoimmune diseases. Our results show that rituximab strongly inhibits Kv1.3 channels in B cells and induces apoptosis by stimulating the FcγRIIB receptor with its human Fc region. Therefore, in addition to the previously described complement-dependent cytotoxicity [26,33,45], antibody-dependent cytotoxicity [6,21,30], or phagocytosis [7,40], rituximab may directly target B cells by stimulating an FcγRIIB receptor-dependent pathway associated with inhibition of Kv1.3 channels. Although crosslinking of CD20 does serve a pathway for rituximab and other anti-CD20 antibodies to induce B cell apoptosis, CD20 antibodies alone at a concentration (10 μg/ml) which could cross-link CD20 molecules are unable to produce the effect unless a secondary antibody is present to further cross-link the CD20 antibodies [16,36]. Since the serum concentration of rituximab in the patients can reach 96.8 μg/ml (0.7 μM) [5], rituximab should not only stimulate CD20, but also activate the low affinity FcγRIIB receptor. The present in vitro study shows that rituximab directly induces apoptosis of cultured Daudi cells by stimulating the FcγRIIB receptor. However, the FcγRIIB receptor is also expressed in monocytes, macrophages, and some natural killer cells [13,18,38]. The FcγRIIB receptor in these effector cells negatively modulates antibody-dependent cytotoxicity against melanoma and breast tumor [8]. The FcγRIIB receptor may reduce clinical efficacy of rituximab by promoting its internalization [23]. Nevertheless, controversial results also exist, showing that the FcγRIIB receptor may be a target of monoclonal antibody therapy of B-cell lymphoma [32]. In addition, the present study indicates that the FcγRIIB receptor may contribute to the multiple pathways initiated by rituximab for depleting B cells. Therefore, the role of the FcγRIIB receptor in anti-lymphoma effect of rituximab remains to be determined.
Acknowledgments
We thank Dr. David G. Warnock (University of Alabama at Birmingham) for his idea to initiate this study. This work was supported by Department of Health and Human Services, National Institutes of Health grants (5R01-DK067110 to He-Ping Ma and 5R37-DK037963 to Douglas C. Eaton).
Abbreviations
- AV
annexin
- V PI
propidium iodide
- CTX
charybdotoxin
- MgTX
margatoxin
- SHIP
Src homology region 2-containing inositol 5-phosphatase
References
- 1.Amoroso A, Hafsi S, Militello L, Russo AE, Soua Z, Mazzarino MC, Stivala F, Libra M. Understanding rituximab function and resistance: implications for tailored therapy. Front Biosci. 2011;16:770–782. doi: 10.2741/3719. [DOI] [PubMed] [Google Scholar]
- 2.Beeton C, Pennington MW, Wulff H, Singh S, Nugent D, Crossley G, Khaytin I, Calabresi PA, Chen CY, Gutman GA, Chandy KG. Targeting effector memory T cells with a selective peptide inhibitor of Kv1.3 channels for therapy of autoimmune diseases. Mol Pharmacol. 2005;67:1369–1381. doi: 10.1124/mol.104.008193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beeton C, Wulff H, Barbaria J, Clot-Faybesse O, Pennington M, Bernard D, Cahalan MD, Chandy KG, Beraud E. Selective blockade of T lymphocyte K+ channels ameliorates experimental autoimmune encephalomyelitis, a model for multiple sclerosis. Proc Natl Acad Sci U S A. 2001;98:13942–13947. doi: 10.1073/pnas.241497298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beeton C, Wulff H, Standifer NE, Azam P, Mullen KM, Pennington MW, Kolski-Andreaco A, Wei E, Grino A, Counts DR, Wang PH, LeeHealey CJ, Andrews S, San-karanarayanan A, Homerick D, Roeck WW, Tehranzadeh J, Stanhope KL, Zimin P, Havel PJ, Griffey S, Knaus HG, Nepom GT, Gutman GA, Calabresi PA, Chandy KG. Kv1.3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc Natl Acad Sci U S A. 2006;103:17414–17419. doi: 10.1073/pnas.0605136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berinstein NL, Grillo-Lopez AJ, White CA, Bence-Bruckler I, Maloney D, Czuczman M, Green D, Rosenberg J, McLaughlin P, Shen D. Association of serum rituximab (IDEC-C2B8) concentration and anti-tumor response in the treatment of recurrent low-grade or follicular non-Hodgkin’s lymphoma. Ann Oncol. 1998;9:995–1001. doi: 10.1023/A:1008416911099. [DOI] [PubMed] [Google Scholar]
- 6.Beum PV, Lindorfer MA, Taylor RP. Within peripheral blood mononuclear cells, antibody-dependent cellular cytotoxicity of rituximab-opsonized Daudi cells is promoted by NK cells and inhibited by monocytes due to shaving. J Immunol. 2008;181:2916–2924. doi: 10.4049/jimmunol.181.4.2916. [DOI] [PubMed] [Google Scholar]
- 7.Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S, Jan M, Cha AC, Chan CK, Tan BT, Park CY, Zhao F, Kohrt HE, Malumbres R, Briones J, Gascoyne RD, Lossos IS, Levy R, Weissman IL, Majeti R. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell. 2010;142:699–713. doi: 10.1016/j.cell.2010.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6:443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 9.Czuczman MS, Gregory SA. The future of CD20 monoclonal antibody therapy in B-cell malignancies. Leuk Lymphoma. 2010;51:983–994. doi: 10.3109/10428191003717746. [DOI] [PubMed] [Google Scholar]
- 10.Daniels I, Abulayha AM, Thomson BJ, Haynes AP. Caspase-independent killing of Burkitt lymphoma cell lines by rituximab. Apoptosis. 2006;11:1013–1023. doi: 10.1007/s10495-006-6314-5. [DOI] [PubMed] [Google Scholar]
- 11.Dhodapkar KM, Banerjee D, Connolly J, Kukreja A, Matayeva E, Veri MC, Ravetch JV, Steinman RM, Dhodapkar MV. Selective blockade of the inhibitory Fcγ receptor (FcγRIIB) in human dendritic cells and monocytes induces a type I interferon response program. J Exp Med. 2007;204:1359–1369. doi: 10.1084/jem.20062545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dias CR, Jeger S, Osso JA, Jr, Muller C, De PC, Hohn A, Waibel R, Schibli R. Radiolabeling of rituximab with (188)Re and (99m)Tc using the tricarbonyl technology. Nucl Med Biol. 2011;38:19–28. doi: 10.1016/j.nucmedbio.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 13.Dutertre CA, Bonnin-Gelize E, Pulford K, Bourel D, Fridman WH, Teillaud JL. A novel subset of NK cells expressing high levels of inhibitory FcγRIIB modulating antibody-dependent function. J Leukoc Biol. 2008;84:1511–1520. doi: 10.1189/jlb.0608343. [DOI] [PubMed] [Google Scholar]
- 14.Ernst JA, Li H, Kim HS, Nakamura GR, Yansura DG, Vandlen RL. Isolation and characterization of the B-cell marker CD20. Biochemistry. 2005;44:15150–15158. doi: 10.1021/bi0511078. [DOI] [PubMed] [Google Scholar]
- 15.Garcia-Calvo M, Leonard RJ, Novick J, Stevens SP, Schmalhofer W, Kaczorowski GJ, Garcia ML. Purification, characterization, and biosynthesis of marga-toxin, a component of Centruroides margaritatus venom that selectively inhibits voltage-dependent potassium channels. J Biol Chem. 1993;268:18866–18874. [PubMed] [Google Scholar]
- 16.Hofmeister JK, Cooney D, Coggeshall KM. Clustered CD20 induced apoptosis: src-family kinase, the proximal regulator of tyrosine phosphorylation, calcium influx, and caspase 3-dependent apoptosis. Blood Cells Mol Dis. 2000;26:133–143. doi: 10.1006/bcmd.2000.0287. [DOI] [PubMed] [Google Scholar]
- 17.Hunter S, Indik ZK, Kim MK, Cauley MD, Park JG, Schreiber AD. Inhibition of Fcγ receptor-mediated phagocytosis by a nonphagocytic Fcγ receptor. Blood. 1998;91:1762–1768. [PubMed] [Google Scholar]
- 18.Ichiyama T, Ueno Y, Hasegawa M, Ishikawa Y, Matsubara T, Furukawa S. Intravenous immunoglobulin does not increase FcγRIIB expression on monocytes/macrophages during acute Kawasaki disease. Rheumatology (Oxford) 2005;44:314–317. doi: 10.1093/rheumatology/keh488. [DOI] [PubMed] [Google Scholar]
- 19.Idusogie EE, Presta LG, Gazzano-Santoro H, Totpal K, Wong PY, Ultsch M, Meng YG, Mulkerrin MG. Mapping of the C1q binding site on rituxan, a chimeric antibody with a human IgG1 Fc. J Immunol. 2000;164:4178–4184. doi: 10.4049/jimmunol.164.8.4178. [DOI] [PubMed] [Google Scholar]
- 20.Jacob A, Cooney D, Tridandapani S, Kelley T, Coggeshall KM. FcγRIIb modulation of surface immunoglobulin-induced Akt activation in murine B cells. J Biol Chem. 1999;274:13704–13710. doi: 10.1074/jbc.274.19.13704. [DOI] [PubMed] [Google Scholar]
- 21.Kohrt HE, Houot R, Goldstein MJ, Weiskopf K, Alizadeh AA, Brody J, Muller A, Pachynski R, Czerwinski D, Coutre S, Chao MP, Chen L, Tedder TF, Levy R. CD137 stimulation enhances the antilymphoma activity of anti-CD20 antibodies. Blood. 2011;117:2423–2432. doi: 10.1182/blood-2010-08-301945. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Lefebvre ML, Krause SW, Salcedo M, Nardin A. Ex vivo-activated human macrophages kill chronic lymphocytic leukemia cells in the presence of rituximab: mechanism of antibody-dependent cellular cytotoxicity and impact of human serum. J Immunother. 2006;29:388–397. doi: 10.1097/01.cji.0000203081.43235.d7. [DOI] [PubMed] [Google Scholar]
- 23.Lim SH, Vaughan AT, Ashton-Key M, Williams EL, Dixon SV, Chan CH, Beers SA, French RR, Cox KL, Davies AJ, Potter KN, Mockridge CI, Oscier DG, Johnson PW, Cragg MS, Glennie MJ. Fc gamma receptor IIb on target B cells promotes rituximab internalization and reduces clinical efficacy. Blood. 2011;118:2530–2540. doi: 10.1182/blood-2011-01-330357. [DOI] [PubMed] [Google Scholar]
- 24.Maenaka K, van der Merwe PA, Stuart DI, Jones EY, Sondermann P. The human low affinity Fcgamma receptors IIa, IIb, and III bind IgG with fast kinetics and distinct thermodynamic properties. J Biol Chem. 2001;276:44898–44904. doi: 10.1074/jbc.M106819200. [DOI] [PubMed] [Google Scholar]
- 25.Maloney DG, Grillo-Lopez AJ, White CA, Bodkin D, Schilder RJ, Neidhart JA, Janakiraman N, Foon KA, Liles TM, Dallaire BK, Wey K, Royston I, Davis T, Levy R. IDEC-C2B8 (rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood. 1997;90:2188–2195. [PubMed] [Google Scholar]
- 26.Natsume A, Shimizu-Yokoyama Y, Satoh M, Shitara K, Niwa R. Engineered anti-CD20 antibodies with enhanced complement-activating capacity mediate potent anti-lymphoma activity. Cancer Sci. 2009;100:2411–2418. doi: 10.1111/j.1349-7006.2009.01327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen TH, Havari E, McLaren R, Zhang M, Jiang Y, Madden SL, Roberts B, Kaplan J, Shankara S. Alemtuzumab induction of intracellular signaling and apoptosis in malignant B lymphocytes. Leuk Lymphoma. 2011 doi: 10.3109/10428194.2011.623253. [DOI] [PubMed] [Google Scholar]
- 28.Partiseti M, Korn H, Choquet D. Pattern of potassium channel expression in proliferating B lymphocytes depends upon the mode of activation. J Immunol. 1993;151:2462–2470. [PubMed] [Google Scholar]
- 29.Piccioni M, Monari C, Bevilacqua S, Perito S, Bistoni F, Kozel TR, Vecchiarelli A. A critical role for FcgammaRIIB in up-regulation of Fas ligand induced by a microbial polysaccharide. Clin Exp Immunol. 2011;165:190–201. doi: 10.1111/j.1365-2249.2011.04415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pievani A, Belussi C, Klein C, Rambaldi A, Golay J, Introna M. Enhanced killing of human B-cell lymphoma targets by combined use of cytokine-induced killer cell (CIK) cultures and anti-CD20 antibodies. Blood. 2011;117:510–518. doi: 10.1182/blood-2010-06-290858. [DOI] [PubMed] [Google Scholar]
- 31.Rangaraju S, Chi V, Pennington MW, Chandy KG. Kv1.3 potassium channels as a therapeutic target in multiple sclerosis. Expert Opin Ther Targets. 2009;13:909–924. doi: 10.1517/14728220903018957. [DOI] [PubMed] [Google Scholar]
- 32.Rankin CT, Veri MC, Gorlatov S, Tuaillon N, Burke S, Huang L, Inzunza HD, Li H, Thomas S, Johnson S, Stavenhagen J, Koenig S, Bonvini E. CD32B, the human inhibitory Fc-gamma receptor IIB, as a target for monoclonal antibody therapy of B-cell lymphoma. Blood. 2006;108:2384–2391. doi: 10.1182/blood-2006-05-020602. [DOI] [PubMed] [Google Scholar]
- 33.Reff ME, Carner K, Chambers KS, Chinn PC, Leonard JE, Raab R, Newman RA, Hanna N, Anderson DR. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood. 1994;83:435–445. [PubMed] [Google Scholar]
- 34.Sarmay G, Koncz G, Gergely J. Human type II Fcgamma receptors inhibit B cell activation by interacting with the p21(ras)-dependent pathway. J Biol Chem. 1996;271:30499–30504. doi: 10.1074/jbc.271.48.30499. [DOI] [PubMed] [Google Scholar]
- 35.Schmitz A, Sankaranarayanan A, Azam P, Schmidt-Lassen K, Homerick D, Hansel W, Wulff H. Design of PAP-1, a selective small molecule Kv1.3 blocker, for the suppression of effector memory T cells in autoimmune diseases. Mol Pharma-col. 2005;68:1254–1270. doi: 10.1124/mol.105.015669. [DOI] [PubMed] [Google Scholar]
- 36.Shan D, Ledbetter JA, Press OW. Apoptosis of malignant human B cells by ligation of CD20 with monoclonal antibodies. Blood. 1998;91:1644–1652. [PubMed] [Google Scholar]
- 37.Smith KG, Clatworthy MR. FcgammaRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat Rev Immunol. 2010;10:328–343. doi: 10.1038/nri2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Su K, Yang H, Li X, Li X, Gibson AW, Cafardi JM, Zhou T, Edberg JC, Kimberly RP. Expression profile of FcgammaRIIb on leukocytes and its dysregulation in systemic lupus erythematosus. J Immunol. 2007;178:3272–3280. doi: 10.4049/jimmunol.178.5.3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Szabo I, Gulbins E, Apfel H, Zhang X, Barth P, Busch AE, Schlottmann K, Pongs O, Lang F. Tyrosine phosphorylation-dependent suppression of a voltage-gated K+ channel in T lymphocytes upon Fas stimulation. J Biol Chem. 1996;271:20465–20469. doi: 10.1074/jbc.271.34.20465. [DOI] [PubMed] [Google Scholar]
- 40.Turzanski J, Daniels I, Haynes AP. Involvement of macroautophagy in the caspase-independent killing of Burkitt lymphoma cell lines by rituximab. Br J Haematol. 2009;145:137–140. doi: 10.1111/j.1365-2141.2008.07555.x. [DOI] [PubMed] [Google Scholar]
- 41.Wang J, Xu YQ, Liang YY, Gongora R, Warnock DG, Ma HP. An intermediate-conductance Ca2+-activated K+ channel mediates B lymphoma cell cycle progression induced by serum. Pflugers Arch. 2007;454:945–956. doi: 10.1007/s00424-007-0258-7. [DOI] [PubMed] [Google Scholar]
- 42.Wulff H, Calabresi PA, Allie R, Yun S, Pennington M, Beeton C, Chandy KG. The voltage-gated Kv1.3 channel in effector memory T cells as new target for MS. J Clin Invest. 2003;111:1703–1713. doi: 10.1172/JCI16921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wulff H, Knaus HG, Pennington M, Chandy KG. K+ channel expression during B cell differentiation: implications for immunomodulation and autoimmunity. J Immunol. 2004;173:776–786. doi: 10.4049/jimmunol.173.2.776. [DOI] [PubMed] [Google Scholar]
- 44.Zhou ZH, Unlap T, Li L, Ma HP. Incomplete inactivation of voltage-dependent K+ channels in human B lymphoma cells. J Membr Biol. 2002;188:97–105. doi: 10.1007/s00232-001-0176-0. [DOI] [PubMed] [Google Scholar]
- 45.Zhuang Y, Xu W, Shen Y, Li J. Fcgamma receptor polymorphisms and clinical efficacy of rituximab in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Clin Lymphoma Myeloma Leuk. 2010;10:347–352. doi: 10.3816/CLML.2010.n.067. [DOI] [PubMed] [Google Scholar]