

Abstract
The pharmacological suppression of the DNA damage response and DNA repair can increase the therapeutic indices of conventional chemotherapeutics. Replication Protein A (RPA), the major single-stranded DNA binding protein in eukaryotes, is required for DNA replication, DNA repair, DNA recombination, and DNA damage response signaling. Through the use of high-throughput screening of 1500 compounds, we have identified a small molecule inhibitor, 15-carboxy-13-isopropylatis-13-ene-17,18-dioic acid (NSC15520), that inhibited both the binding of Rad9–GST and p53–GST fusion proteins to the RPA N-terminal DNA binding domain (DBD), interactions that are essential for robust DNA damage signaling. NSC15520 competitively inhibited the binding of p53-GST peptide with an IC50 of 10 μM. NSC15520 also inhibited helix destabilization of a duplex DNA (dsDNA) oligonucleotide, an activity dependent on the N-terminal domain of RPA70. NSC15520 did not inhibit RPA from binding single-stranded oligonucleotides, suggesting that the action of this inhibitor is specific for the N-terminal DBD of RPA, and does not bind to DBDs essential for single-strand DNA binding. Computer modeling implicates direct competition between NSC15520 and Rad9 for the same binding surface on RPA. Inhibitors of protein–protein interactions within the N-terminus of RPA are predicted to act synergistically with DNA damaging agents and inhibitors of DNA repair. Novel compounds such as NSC15520 have the potential to serve as chemosensitizing agents.
Keywords: Replication Protein A, Inhibitors, High-throughput screen, EMSA, Docking
1. Introduction
Pharmacological manipulation of the DNA damage response (DDR) and DNA repair pathways are attractive additions to conventional radiation and genotoxic anticancer therapies. The possibility of increasing the efficacy and decreasing the toxic side effects of radiation and chemotherapeutics has stimulated interest in identifying chemical compounds that sensitize cells by targeting DDR and DNA repair pathways. For example, exploiting the homologous recombination repair (HRR) pathway with conventional platinum based therapies has been shown to be successful in BRCA1- and BRCA2-deficient ovarian cancers.1–3 In addition, DNA repair inhibitors such as poly (ADP-ribose) polymerase 1 (PARP1) inhibitors are relatively non-toxic to most cancer cells. However, these drugs display synthetic lethality in tumor cells deficient for BRCA1 or BRCA2 without exogenous DNA damage.4
In support of identifying small molecule inhibitors (SMIs) of DDR pathways, increased radio- and chemo-resistance due to enhanced DDR signaling in leukemia and glioma cancers has been reported.5,6 Additionally, the frequent mutation of p53 in many different human cancers sensitizes cancer cells to the cytotoxic effects of genotoxic chemotherapeutics.7,8 This creates the opportunity to further enhance therapeutic indices by combining chemotherapeutics with compounds that inhibit ATM-Chk2 and ATR-Chk1 checkpoint signaling pathways.9–11 One protein essential to both HRR and DDR signaling is Replication Protein A (RPA). RPA, a heterotrimeric protein that consists of 70 kDa, 32 kDa and 14 kDa subunits, has a high binding affinity for single-strand DNA (ssDNA) and interacts with multiple proteins involved in DNA metabolic processes.12,13 The primary structural feature of RPA is the presence of multiple oligonucleotide/oligosaccharide binding (OB) folds, common DNA binding domains of ssDNA binding proteins.14 These DNA binding domains (DBD) are found on all three subunits.15–19 The DBDs responsible for high affinity ssDNA binding, DBD-A and DBD-B, are centrally located on RPA70.20 DBD-F, connected by an extended flexible linker located at the N-terminus of RPA70, interacts with multiple DDR and DNA repair proteins that include Rad9, ATRIP, Mre11, and p53 (Fig. 1A).21–23 Each of these proteins contains a conserved acidic alpha helical domain, which is, responsible for binding to the basic cleft of DBD-F, (Fig. 1B). The importance of this domain has been demonstrated previously in yeast. A single rfa1-t11 mutation in Saccharomyces cerevisiae, K45E, within the N-terminal basic cleft binding domain of RPA70, equivalent to a mutation at Arg41in human RPA70, leads to chromosomal instability, sensitivity to UV radiation and a loss of recruitment of checkpoint proteins.24–28 Mutations in the Rad9 and Mre11 acidic helical domains cause defects in ATR-Chk1 signaling and the S and G2 checkpoints.22,29 The cells expressing the Rad9 mutant decreased ATR phosphorylation and activation of Chk1 which increased sensitivity to hydroxyurea, an agent that depletes the nucleotide pool.22 The nucleoside analog 5-fluorouracil (5-FU), a drug used in the treatment of colon, breast, skin, stomach and esophageal cancers, is mechanistically comparable to hydroxyurea30 and would potentially synergize with a drug that inhibits Rad9–RPA interactions. Gemcitabine, another nucleoside analog, inhibits DNA polymerase activity as a strand terminator and depletes the nucleotide pool. This drug is commonly used to treat non-small cell lung, pancreatic, bladder and breast cancer.31 In studies where gemcitabine was combined with Chk1 inhibitors, analysis revealed the combination of drugs caused collapse of stalled replication forks with premature entry into mitosis and apoptosis.32 A SMI targeting RPA that inhibits Chk1 activity has the potential to synergize with nucleoside analogs with similar mechanisms of toxicity. Thus, the DBD-F of RPA represents a novel target whose pharmacological inhibition can potentiate tumor cell killing by a wide variety of genotoxic agents.
Figure 1.
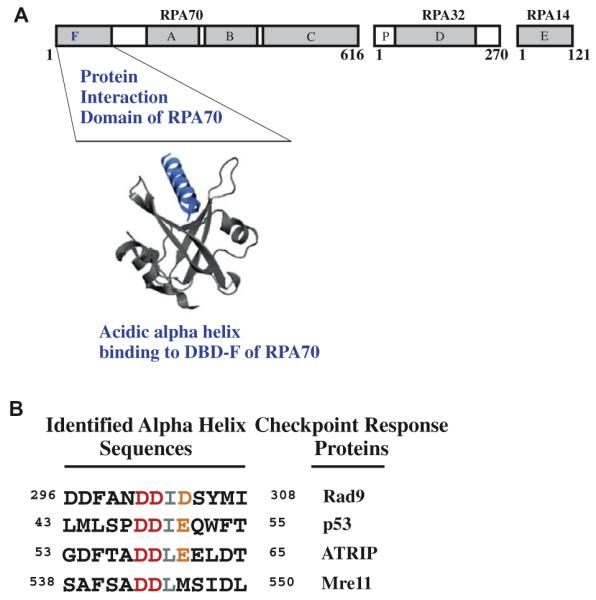
(A) Diagram of RPA heterotrimer and modeled 3D structure of an acidic alpha helix binding to the basic cleft of the N-terminal domain of RPA70. (B) Residues within Rad9, p53, ATRIP and Mre11 that have been identified to interact with the N-terminus of RPA70.
To identify SMIs that target RPA protein interactions, we have developed an efficient and reliable high-throughput screen (HTS). Initial in silico screening of compounds from the NCI Developmental Therapeutics Program repository provided insight into structural requirements and suggested potential binding sites within the basic cleft of DBD-F.
2. Results
2.1. Rad9 does not bind N-terminal truncated RPA70
In designing a HTS for DBD-F, Rad9, a component of the PCNA-like 9-1-1 checkpoint sensor clamp previously shown to bind to DBD-F,22 was used to optimize the system. We initially tested RPA–Rad9 interactions using purified proteins in a pull down assay (Fig. 2A). Full-length RPA bound to biotin-labeled ssDNA binds to GST-Rad9 efficiently when pulled down with streptavidin beads (Fig. 2B, lane 3). In contrast, neither DNA bound to Escherichia coli SSB nor the RPAΔDBD-F mutant protein (deletion of 1–168 of RPA70) interacts with Rad9 (Fig. 2B, lanes 4 and 5). The GST-Rad9 fusion protein satisfied the requirements of the HTS and was further used in testing the 1500 compounds for an RPA interaction inhibitor.
Figure 2.

Purification and interaction of recombinant RPA and GST–Rad9. (A) Proteins were separated by SDS–PAGE and stained with Coomassie blue. (B) Pulldown of Rad9 with ssDNA bound RPA using streptavidin-linked magnetic beads. Lane 1 does not include RPA and ssDNA and Lane 2 does not include RPA. Lanes 3–5 used RPA, mutant RPA (Δ1–168 RPA70), and E. coli SSB protein bound to biotin-labeled ssDNA, respectively.
2.2. Design of a high-throughput screening assay
A plate binding assay, similar to an ELISA, was developed for determining inhibition of RPA–Rad9 interactions. Using streptavidin coated 384-well plates, reagents are added in a stepwise manner, resulting in a positive near-infrared fluorescent signal when all reagents are added to the plate well in the absence of inhibition (Fig. 3A, wells 1 and 7). Lack of any of the components results in a loss of signal (Fig. 3A, wells 2–6). Using this approach, the plate binding assay accentuates the generation of false positives while reducing the possibility of false negatives. A set of positive and negative controls were incorporated into each plate (Fig. 3B, column 4).
Figure 3.

HTS for an RPA inhibitor. (A) Controls used in the 384-well HTS. (B) An example of a 384-well plate used in the assay. Wells in column 4 were used for controls.
2.3. High-throughput and secondary screening results
Compounds screened in the HTS assay were obtained from the NCI Diversity Set, Diversity Set II (http://dtp.nci.nih.gov/branches/dscb/div2_explanation.html). This set contains a diverse collection of compounds with pharmacologically desirable features specifically developed for initial HTS projects. Compounds were initially screened for 50% inhibition of RPA–Rad9 interaction at a concentration of 200 μM. Of the 1500 compounds tested, 44 hits were initially obtained (Supplementary Fig. S1).
One mechanism that decreases detection of RPA–Rad9 interactions in the HTS assay is the inhibition of RPA ssDNA binding activity. A large decrease in RPA ssDNA binding activity can occur when compounds bind to DBD-A and DBD-B, compete for binding to ssDNA or act as a chelating agent on RPA’s zinc finger motif. The zinc binding motif of RPA located in the C-terminal of RPA70 is important in structural stability and ssDNA binding.33 To identify compounds that affect RPA ssDNA binding activity, electrophoretic mobility shift assays (EMSAs) were employed (Fig. 4). Upon an excess of a fluorescent ssDNA probe, RPA binds a fraction of ssDNA, visualized as a ‘shifted’ band of slower mobility. Inhibitors that reduced the ratio of shifted ssDNA to free ssDNA by more than 20% were considered undesirable and were dropped from further testing, which resulted in the elimination of 14 inhibitors (Fig. 4, and Supplementary Fig. S1, shown as red).
Figure 4.

Analyzes of inhibitor activity on ssDNA binding of RPA. RPA was incubated with compounds identified in the HTS (200 μM) and DNA binding of RPA was analyzed by EMSA. The position of free DNA and the RPA–DNA complex is denoted. Compounds labeled with red text inhibited RPA–DNA binding and were excluded from further study.
The remaining 30 inhibitors were retested in two separate HTS assays in triplicate. Of these compounds, four were confirmed as strong inhibitors of RPA–Rad9 interaction (Fig. 5). Compound D, also called NSC15520 (Supplementary Fig. S1), is obtained from wood resins and has been synthesized in both academic and industrial laboratories34 The familiarity and potential abundance of NSC15520 made this compound an ideal candidate for further testing.
Figure 5.
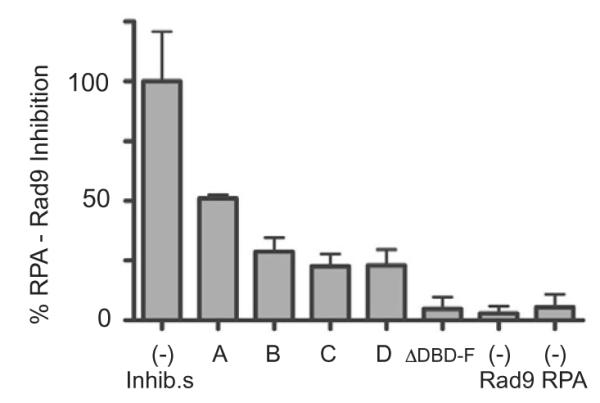
HTS for RPA–Rad9 interactions. RPA bound to biotinylated-ssDNA in a streptavidin coated 384-well plate was mixed with the indicated compounds, A, B, C, and D (D represents NSC15520), and percent inhibition was normalized to reactions that did not contain inhibitors. Results are representative of two independent studies performed in triplicate. Data represent mean ± SEM.
2.4. NSC15520 inhibits the interaction of RPA with GST-p53 peptide
While initial results demonstrated that NSC15520 inhibited RPA–Rad9 interactions without affecting RPA ssDNA binding activity, the analyses did not determine whether NSC15520 was directly binding to Rad9 or RPA. To ensure that the action of NSC15520 was specific for RPA, GST-Rad9 was substituted with a GST-peptide fusion protein containing an 18 amino acid sequence derived from the p53 acidic helical domain, MDDLMLSPDDIEQWFTED, which binds DBD-F.23 Using titrations of the GST-p53, the IC50 of NSC15520 was determined to be 10 μM (Fig. 6).
Figure 6.
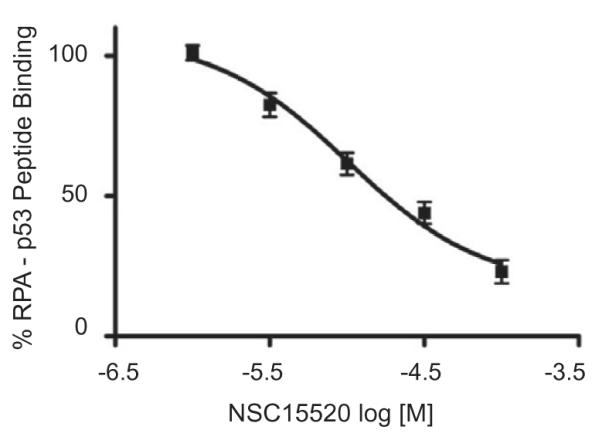
Quantification of RPA–p53 binding as a function of NSC15520 concentration. Data was normalized to a non-inhibited control. Results are representative of two independent studies performed in triplicate. Data represent mean ± SEM.
2.5. NSC15520 inhibits RPA dsDNA binding and helix destabilization activity
In addition to binding ssDNA, RPA can also bind duplex DNA (dsDNA) with lower affinity via a highly efficient helix destabilization process.35,36 This unwinding ability is greatly reduced in the absence of the DBD-F of RPA.37 However, deletion of the DBD-F does not significantly alter ssDNA binding affinity.36 The basic cleft of DBD-F has been shown to contribute to the nucleation of dsDNA, the initial and rate limiting step in DNA unwinding by RPA.37 In the nucleation process, RPA binds to transient ssDNA regions in the dsDNA contributing to the helix destabilization of dsDNA.
Due to the ability of NSC15520 to block protein-protein interactions known to occur at DBD-F, the compound was also tested for its effect on DBD-F activity in dsDNA binding and helix destabilization. NSC15520 inhibits RPA binding of a dsDNA 30-mer at concentrations below 100 μM (Fig. 7A). The concentrations of NSC15520 used did not inhibit the binding activity of RPA to ssDNA (Fig. 7B). These results further implicate NSC15520 as a specific inhibitor of DBD-F, as both protein interactions with DBD-F and DBD-F dsDNA binding and helix destabilization activities are decreased while ssDNA binding activities remain unchanged.
Figure 7.
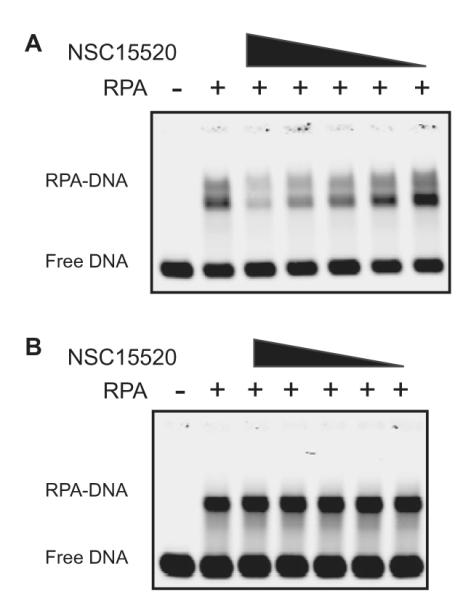
EMSA analyzes of RPA binding activity using dsDNA and ssDNA substrates in the presence of NSC15520. RPA was pre-incubated with inhibitor at 0, 400 μM, 200 μM, 100 μM, 50 μM and 25 μM, respectively. (A) Inhibition of dsDNA binding and helix destabilization by NSC 15520. RPA binds efficiently to the resulting ssDNA after the dsDNA substrate is denatured. (B) NSC15520 does not inhibit binding of RPA to ssDNA substrate.
2.6. Virtual docking implicates NSC15520 as a direct competitor of Rad9 for binding to the DBD-F
To gain further insight into the structural determinants of DBD-F binding by NSC15520, both the 3D structure of the inhibitor and a virtually constructed peptide containing the acidic helical domain of Rad9 were docked to the crystal structure of DBD-F (PDB: 2B29) in silico (Fig. 8A). Using the Autodock Vina software suite,38 a large section of the N-terminal domain of RPA containing the entire basic cleft as well as the adjacent cleft walls were included in the grid for docking of both inhibitor and peptide. Consistent with the HTS analyses that demonstrate NSC15520 inhibiting RPA–Rad9 interactions, the calculated lowest free energy binding state of the peptide overlapped with the two most favorable binding conformations of the inhibitor (Fig. 8A). In this virtual model, a carboxyl group, within the aspartic acid doublet of the Rad9 peptide competes with a carboxyl group of the inhibitor for electrostatic interaction with Arg41 located deep within the cleft (Fig. 8B). The competition of the Rad9 peptide and NSC15520 for Arg41 is consistent with a previous report that demonstrated Arg41 as essential for Rad9 binding to RPA70.22
Figure 8.

Predicted binding models of the N-terminus of RPA70 with NSC15520 and Rad9. (A) RPA is shown in surface representation where carbon, oxygen, nitrogen, and sulfur atoms are colored in gray, red, blue, and yellow, respectively. The carbon atoms in NSC15520 are shown in blue and green and oxygen atoms are red. The Rad9 peptide is shown as a dark blue helix. The two lowest docking energy sites of NSC15520 within the entire crystal structure of the RPA N-terminal domain, PDB: 2B29 are shown. (B) Predicted hydrogen bonding between RPA70 and NSC15520 are depicted as dotted lines in yellow. Hydrogen bond lengths are given in angstroms.
3. Discussion
RPA interactions with other proteins involved in DNA replication, DNA repair and checkpoint signaling create several unique potential mechanisms of synergistic cell killing with radiation and chemotherapeutics. RPA consists of six OB-folds that have similarities and differences in structure.15–19 DBD-A and DBD-B have a short 10-residue linker between the two domains which allows these two domains to work in tandem and promotes ssDNA binding. The extended 70-residue linker between DBD-F and DBD-A enables a large amount of flexibility for DBD-F. In fact, a recent study provides evidence that DBD-F does not bind ssDNA and suggests this OB-fold functions as a protein recruitment module orchestrating protein exchanges analogous to FEN1-PCNA and BRCA2-Rad51.39–41 The flexibility and dynamic nature of DBD-F creates distinctive differences between this domain and other OB-folds of RPA. These differences may partially explain the specificity of NSC15520 for DBD-F.
Targeting distinct OB-folds within RPA has the potential to exploit distinct pathways in which RPA participates. The DBD-F OB-fold is not required for DNA replication yet is important in checkpoint activation and DNA repair.22,42,43 An inhibitor that limits RPA function in checkpoint responses and DNA repair will amplify levels of DNA damage and potentiate cell killing by a wide variety of genotoxic agents. Studies supporting this principle include a Rad9 mutant incapable of binding to RPA showed decreased ATR phosphorylation and activation of Chk1 which increased sensitivity to hydroxyurea.22 Other studies have shown cell cycle regulation defects in cells expressing RPA DBD-F mutants following camptothecin and etoposide exposure that were attributed to deficiencies in DNA repair.42,43
NSC15520 also prevents interaction between p53 and RPA. The p53-RPA interaction has been extensively studied, however, the functional consequences remain to be established. One model is that p53-RPA binding serves to organize a pool of p53 protein which allows for quick release of p53 from RPA to participate either in DNA repair or the transactivation of p53 responsive genes.44 Blocking this interaction with a specific DBD-F inhibitor could disrupt temporal and spatial localization of p53 at critical times in the DDR, causing increased genomic stress and death. Alternatively, sequestering of RPA by p53 has been suggested to regulate HRR;45 however, the precise mechanisms of how p53-RPA interactions affect HRR remain to be elucidated.
Another RPA interacting protein involved in HRR includes BRCA2. RPA binds to the N-terminal acidic alpha helical domain of BRCA2.46 BRCA2 bound to DSS1, a small (70 amino acid) acidic alpha helical protein, stimulates Rad51 binding to RPA covered ssDNA, suggesting a role for BRCA2 and DSS1 interaction with RPA in HRR.47 We are currently investigating the protein interactions of RPA with the N-terminal acidic alpha helical domain of BRCA2 and DSS1.47 Inhibition of RPA interactions with these HRR proteins could potentially sensitize cancer cells to PARP1 inhibitors by blocking BRCA2 function.
Because genetic instability due to oncogene-driven processes is one of the leading causes of tumorigenesis in sporadic non-hereditary cancers, targeting proteins involved in the DDR and DNA repair may improve existing therapies and generate novel treatments.48–50 The result of inhibiting RPA checkpoint and DNA repair activity without affecting DNA replication could be selectively deleterious to cancer cells. Unlike normal cells, activated oncogenes in cancer cells drive replication under inappropriate conditions in a deregulated manner that results in an increased dependency on the DNA damage response.48,51 RPA has already proven to be a valid target for cancer therapy. The identification of SMIs that target the central OB-folds of RPA70, DBD-A and DBD-B, have shown to induce cyto-toxicity and increase the efficacy of genotoxic chemotherapeutics.52 In these previous studies, a series of structurally similar SMIs were analyzed for inhibition of RPA ssDNA binding activity. Those analogs containing an anhydride functional group were highly active in inhibiting RPA binding to ssDNA.53,54 Interestingly, analogs containing dicarboxylic acids were much less active. This is consistent with our data as NSC15520 contains a dicarboxylic acid and does not inhibit RPA ssDNA binding activity. Protein functional groups able to react with anhydrides and form stable products include the e-amine side chain of lysine.55 Acylation with anhydrides of the e-amine group can form stable amide bonds. Two lysines important in RPA DNA binding activity, one located in DBD-A, K263, and the other in DBD-B, K343, are in direct contact with DNA15,16 and may be targets for SMIs containing anhydride functional groups. In contrast, dicarbonyl compounds, such as NSC15520, specifically target the guanidinyl group of arginine.56 This may explain the preference of NSC15520 for DBD-F. The mutation of Arg41 and Arg43 to glutamic acids completely blocked the ability of RPA70 to bind to Rad9.22 The inhibition of Rad9 interaction with RPA by NSC15520 is consistent with NSC15520 targeting arginine residues located within the DBD-F.
In summary, we have identified NSC15520 as a novel inhibitor of functional activities of the RPA N-terminal basic cleft including protein recruitment and helix destabilization. Taken together, our data indicate NSC15520 is an ideal candidate for further optimization and development as a novel therapy for the treatment of cancer.
4. Experimental
4.1. Reagents
DNA oligonucleotides were purchased from Integrated DNA Technologies, Inc. Streptavidin coated plates were purchased from Thermo Scientific. Superflow glutathione-linked agarose was purchased from Qiagen. Antibodies were purchased from Bethyl (Rad9, RPA70 and GST), Labvision Corporation (RPA32), GeneTex, Inc. (RPA14), Invitrogen (Alexa Fluor 680 goat anti-rabbit) and Thermo Scientific (DyLight 800 goat anti-mouse).
4.2. Cloning, expression and purification of RPA, GST–Rad9, GST–p53(40–57)
RPA expressing vectors p11d-tRPA and p11d-tRPAD1-169 were kindly provided by Dr. Marc Wold at the University of Iowa. Recombinant human RPA was purified from E. coli as described previously.57 The GST–Rad9 fusion protein was expressed in the vector pGEX-4T-2, kindly provided by Dr. Sheau-Yann Shieh at the Institute of Biomedical Sciences, Taiwan. The GST-p53 fusion protein was expressed in the vector pRSFDuet-1. After incorporation of GST, the 18 amino acid p53 sequence, 40MDDLMLSPDDIEQWF TED57, was added to the N-terminal end of GST by annealing primers, 5-CATGGATGATCTTATGCTTTCTCCTGATGATATTGAACAAG GTTTACTG AAGATCA-3 and 5-TCGATGATCTTCAGTAAACCATTGTT CAATATCATCAGGAGAAAGCATAAGAT CATC-3 to NcoI/SalI cut pRSFDuet-GST. GST proteins were induced with IPTG (300 μM) in E. coli, and purified through a glutathione-linked agarose column (Bio-Rad).
4.3. Pulldown assay
All incubations and washes were performed in Binding buffer (100 mM NaCl, 10% glycerol, 10 lg/ml BSA, 0.1% NP40). For each sample, 40 μl of streptavidin coated magnetic beads (Invitrogen) were washed 3×, incubated with 500 pmol 3′ biotinylated polyT(30) oligonucleotide for 30 min, then washed 3× and incubated with 300 pmol RPA or SSB (Epicentre) for 30 min. The resulting complex was washed 3×, then incubated with 200 pmol of GST–Rad9 for 1 h on a rotator at 4°C. After washing and removal of buffer, proteins were eluted with SDS sample buffer and each sample was subjected to SDS–PAGE prior to immunoblotting. Membranes were incubated sequentially with Rad9 and RPA primary antibodies, fluorescent secondary antibodies, and visualized on an Odyssey infrared scanner (LiCor).
4.4. High-throughput screening
The majority of compounds used for high-throughput screening were derived from the NCI Developmental Therapeutics Program, Diversity Set II. An additional 200 compounds were selected from the original diversity set based on a criterion of availability and low autofluorescence. All compounds were solubilized in DMSO at a stock concentration of 10 mM.
All incubations were performed at RT with gentle agitation. In the HTS, streptavidin coated plates (384-well) were washed 3× with PBS, and then incubated with 15 pmol of biotinylated oligonucleotides in PBS for 2 h. The plates were then washed 3× with PBS, and then incubated with 20 pmol RPA in PBS for 1.5 h. Each plate was then washed 2× with PBS, 1× Binding buffer, then incubated in Binding buffer in with the presence of 30 pmol GST–Rad9, GST–p53 or GST and 200 μM inhibitor for 30 min. Plates were washed 4× with Binding buffer, then incubated with Rad9 antibody in Binding buffer. Each plate was then washed 5× with Binding buffer, and then incubated with anti-rabbit fluorescent antibody in Binding buffer followed by 5× washing before visualization on an Odyssey infrared scanner.
4.5. Electrophoretic mobility shift assays (EMSAs)
All EMSA incubations were performed in 10 μl Binding buffer (15 mM NaCl was used with dsDNA analyses) with 0.4 pmol RPA at RT. Reactions were loaded on 1% agarose gels in 40 mM Tris acetate, and run for 15 min at 100 V. Gels were scanned and visualized on the Odyssey infrared scanner.
For screening of compounds that disrupt ssDNA bound RPA, RPA was prebound to 2 pmol of polyT(30) oligonucleotide labeled with 5′ IRDYE-700 (Integrated DNA Technologies) for 5 min, then exposed to 2 nmol of inhibitor for 30 min. For measurement of the inhibition of RPA/ssDNA binding, RPA was exposed to varying amounts of inhibitor for 5 min followed by the addition of 2 pmol of ssDNA for 30 min.
For dsDNA binding studies, a fluorescent dsDNA substrate was generated by annealing the fluorescent polyT(30) oligonucleotide to an unlabeled polyA(30) oligonucleotide. The duplex DNA was treated with mung bean nuclease to remove any remaining ssDNA. For detection of dsDNA binding and helix destabilization, RPA was exposed to varying amounts of inhibitor for 5 min before the addition of 2 pmol of dsDNA for 30 min.
4.6. Molecular modeling and docking
Conversion of the available 2D structure of NSC15520 (NSC15520: Pubchem) into a 3D PDB structure was accomplished using the OpenBabel 2.2.3 program. The graphical software tool PyMOL (Schrodinger) was used to generate a 3D structure of the RPA binding sequence of Rad9, 297DFANDDIDS305, and to display the results of the docking simulations.
Molecular docking of both NSC15520 and the Rad9 peptide into the crystal X-ray structure of the RPA N-terminus (PDB code: 2B29) was carried out using the Autodock Vina software package as implemented through the graphical user interface AutoDockTools (ADT 1.4.6). Default parameters were applied for docking simulations.
Supplementary Material
Acknowledgements
Financial support from National Institutes of Health Grant P20-RR01875, American Cancer Society ACS RSG-10-031-01-CCG and Nebraska Department of Health and Human Services 2011–25, is acknowledged.
Abbreviations
- DNA
deoxyribonucleic acid
- DDR
DNA damage response
- BER
base excision repair
- HRR
homologous recombination repair
- SMI
small molecule inhibitor
- RPA
Replication Protein A
- kDa
kilodalton
- ssDNA
single-stranded DNA
- DBD
DNA binding domain
- NCI
National Cancer Institute
- HTS
high-throughput screen
- SSB
single-stranded binding protein
- GST
glutathione s-transferase
- ELISA
enzyme-linked immunosorbent assay
- EMSA
electrophoretic mobility shift assay
- dsDNA
duplex DNA
- PDB
Protein Data Base
- IC50
half maximal inhibitory concentration
- IPTG
Isopropyl b-D-1-thiogalactopyranoside
- FPLC
fast protein liquid chromatography
- BSA
bovine serum albumin
- polyT
polythymidine
- SDS–PAGE
polyacrylamide gel electrophoresis
- SEM
standard error of the mean
- DMSO
dimethyl sulfoxide
Footnotes
The authors declare no competing financial interests.
Supplementary data Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmc.2011.03.012.
References and notes
- 1.Foulkes WD. Fam. Cancer. 2006;5:135. doi: 10.1007/s10689-005-2832-5. [DOI] [PubMed] [Google Scholar]
- 2.Safra T. Womens Health. 2009;5:543. doi: 10.2217/whe.09.40. [DOI] [PubMed] [Google Scholar]
- 3.Tan DS, Rothermundt C, Thomas K, Bancroft E, Eeles R, Shanley S, Ardern-Jones A, Norman A, Kaye SB, Gore ME. J. Clin. Oncol. 2008;26:5530. doi: 10.1200/JCO.2008.16.1703. [DOI] [PubMed] [Google Scholar]
- 4.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Nature. 2005;434:913. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 5.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Nature. 2006;444:756. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 6.Nieborowska-Skorska M, Stoklosa T, Datta M, Czechowska A, Rink L, Slupianek A, Koptyra M, Seferynska I, Krszyna K, Blasiak J, Skorski T. Cell cycle (Georgetown, Tex.) 2006;5:994. doi: 10.4161/cc.5.9.2722. [DOI] [PubMed] [Google Scholar]
- 7.Bertheau P, Turpin E, Rickman DS, Espie M, de Reynies A, Feugeas JP, Plassa LF, Soliman H, Varna M, de Roquancourt A, Lehmann-Che J, Beuzard Y, Marty M, Misset JL, Janin A, de The H. PLoS Med. 2007;4:e90. doi: 10.1371/journal.pmed.0040090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cortinovis DL, Andriani F, Livio A, Fabbri A, Perrone F, Marcomini B, Pilotti S, Mariani L, Bidoli P, Bajetta E, Roz L, Sozzi G. Curr. Cancer Drug Targets. 2008;8:342. doi: 10.2174/156800908785133204. [DOI] [PubMed] [Google Scholar]
- 9.Wilsker D, Bunz F. Mol. Cancer Ther. 2007;6:1406. doi: 10.1158/1535-7163.MCT-06-0679. [DOI] [PubMed] [Google Scholar]
- 10.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. Cancer Res. 2004;64:9152. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 11.Smith J, Tho LM, Xu N, Gillespie DA. Adv. Cancer Res. 2010;108:73. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- 12.Oakley GG, Patrick SM. Front. Biosci. 2010;15:883. doi: 10.2741/3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wold MS. Annu. Rev. Biochem. 1997;66:61. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- 14.Murzin AG. EMBO J. 1993;12:861. doi: 10.1002/j.1460-2075.1993.tb05726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bochkarev A, Pfuetzner RA, Edwards AM, Frappier L. Nature. 1997;385:176. doi: 10.1038/385176a0. [DOI] [PubMed] [Google Scholar]
- 16.Arunkumar AI, Stauffer ME, Bochkareva E, Bochkarev A, Chazin WJ. J. Biol. Chem. 2003;278:41077. doi: 10.1074/jbc.M305871200. [DOI] [PubMed] [Google Scholar]
- 17.Bochkareva E, Frappier L, Edwards AM, Bochkarev A. J. Biol. Chem. 1998;273:3932. doi: 10.1074/jbc.273.7.3932. [DOI] [PubMed] [Google Scholar]
- 18.Bochkareva E, Korolev S, Lees-Miller SP, Bochkarev A. EMBO J. 2002;21:1855. doi: 10.1093/emboj/21.7.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brill SJ, Bastin-Shanower S. Mol. Cell Biol. 1998;18:7225. doi: 10.1128/mcb.18.12.7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wyka IM, Dhar K, Binz SK, Wold MS. Biochemistry. 2003;42:12909. doi: 10.1021/bi034930h. [DOI] [PubMed] [Google Scholar]
- 21.Ball HL, Ehrhardt MR, Mordes DA, Glick GG, Chazin WJ, Cortez D. Mol. Cell. Biol. 2007;27:3367. doi: 10.1128/MCB.02238-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu X, Vaithiyalingam S, Glick GG, Mordes DA, Chazin WJ, Cortez D. Mol. Cell. Biol. 2008;28:7345. doi: 10.1128/MCB.01079-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bochkareva E, Kaustov L, Ayed A, Yi GS, Lu Y, Pineda-Lucena A, Liao JC, Okorokov AL, Milner J, Arrowsmith CH, Bochkarev A. Proc. Natl. Acad. Sci. U.S.A. 2005;102:15412. doi: 10.1073/pnas.0504614102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zou L, Elledge SJ. Science. 2003;300:1542. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 25.Kanoh Y, Tamai K, Shirahige K. Gene. 2006;377:88. doi: 10.1016/j.gene.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 26.Grandin N, Charbonneau M. Nucleic Acids Res. 2007;35:822. doi: 10.1093/nar/gkl1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Umezu K, Sugawara N, Chen C, Haber JE, Kolodner RD. Genetics. 1998;148:989. doi: 10.1093/genetics/148.3.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim HS, Brill SJ. Mol. Cell. Biol. 2001;21:3725. doi: 10.1128/MCB.21.11.3725-3737.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olson E, Nievera CJ, Liu E, Lee AY, Chen L, Wu X. Mol. Cell. Biol. 2007;27:6053. doi: 10.1128/MCB.00532-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Longley DB, Harkin DP, Johnston PG. Nat. Rev. 2003;3:330. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 31.Toschi L, Finocchiaro G, Bartolini S, Gioia V, Cappuzzo F. Future Oncol. 2005;1:7. doi: 10.1517/14796694.1.1.7. [DOI] [PubMed] [Google Scholar]
- 32.McNeely S, Conti C, Sheikh T, Patel H, Zabludoff S, Pommier Y, Schwartz G, Tse A. Cell cycle. 2010;9:995. doi: 10.4161/cc.9.5.10935. [DOI] [PubMed] [Google Scholar]
- 33.Bochkareva E, Korolev S, Bochkarev A. J. Biol. Chem. 2000;275:27332. doi: 10.1074/jbc.M000620200. [DOI] [PubMed] [Google Scholar]
- 34.Kato K, Aburaya K, Matsumoto M, Tohnai N, Miyata M. Chem. Lett. 2003;32:952. [Google Scholar]
- 35.Iftode C, Borowiec JA. Nucleic Acids Res. 1998;26:5636. doi: 10.1093/nar/26.24.5636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lao Y, Lee CG, Wold MS. Biochemistry. 1999;38:3974. doi: 10.1021/bi982371m. [DOI] [PubMed] [Google Scholar]
- 37.Binz SK, Lao Y, Lowry DF, Wold MS. J. Biol. Chem. 2003;278:35584. doi: 10.1074/jbc.M305388200. [DOI] [PubMed] [Google Scholar]
- 38.Trott O, Olson AJ. J. Comput. Chem. 2010;31:455. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pretto DI, Tsutakawa S, Brosey CA, Castillo A, Chagot ME, Smith JA, Tainer JA, Chazin WJ. Biochemistry. 2010;49:2880. doi: 10.1021/bi9019934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chapados BR, Hosfield DJ, Han S, Qiu J, Yelent B, Shen B, Tainer JA. Cell. 2004;116:39. doi: 10.1016/s0092-8674(03)01036-5. [DOI] [PubMed] [Google Scholar]
- 41.Shin DS, Pellegrini L, Daniels DS, Yelent B, Craig L, Bates D, Yu DS, Shivji MK, Hitomi C, Arvai AS, Volkmann N, Tsuruta H, Blundell TL, Venkitaraman AR, Tainer JA. EMBO J. 2003;22:4566. doi: 10.1093/emboj/cdg429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oakley GG, Tillison K, Opiyo SA, Glanzer JG, Horn JM, Patrick SM. Biochemistry. 2009;48:7473. doi: 10.1021/bi900694p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haring SJ, Mason AC, Binz SK, Wold MS. J. Biol. Chem. 2008;283:19095. doi: 10.1074/jbc.M800881200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abramova NA, Russell J, Botchan M, Li R. Proc. Natl. Acad. Sci. U.S.A. 1997;94:7186. doi: 10.1073/pnas.94.14.7186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Romanova LY, Willers H, Blagosklonny MV, Powell SN. Oncogene. 2004;23:9025. doi: 10.1038/sj.onc.1207982. [DOI] [PubMed] [Google Scholar]
- 46.Wong JM, Ionescu D, Ingles CJ. Oncogene. 2003;22:28. doi: 10.1038/sj.onc.1206071. [DOI] [PubMed] [Google Scholar]
- 47.Liu J, Doty T, Gibson B, Heyer WD. Nat. Struct. Mol. Biol. 2010;17:1260. doi: 10.1038/nsmb.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Halazonetis TD, Gorgoulis VG, Bartek J. Science. 2008;319:1352. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 49.Negrini S, Gorgoulis VG, Halazonetis TD. Nat. Rev. Mol. Cell Biol. 2010;11:220. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- 50.Durkin SG, Glover TW. Annu. Rev. Genet. 2007;41:169. doi: 10.1146/annurev.genet.41.042007.165900. [DOI] [PubMed] [Google Scholar]
- 51.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J. Nature. 2005;434:864. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 52.Shuck SC, Turchi JJ. Cancer Res. 2010;70:3189. doi: 10.1158/0008-5472.CAN-09-3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Granadillo V. J. Anciano, Earley JN, Shuck SC, Georgiadis MM, Fitch RW, Turchi JJ. J. Nucleic Acids. 2010;2010:304035. doi: 10.4061/2010/304035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bonetti A. Platinum and Other Heavy Metal Compounds in Cancer Chemotherapy: Molecular Mechanisms and Clinical Applications. Humana Press; New York: 2009. [Google Scholar]
- 55.Hermanson GT. Bioconjugate Techniques. Academic Press; San Diego: 1996. [Google Scholar]
- 56.Patthy L, Smith EL. J. Biol. Chem. 1975;250:557. [PubMed] [Google Scholar]
- 57.Henricksen LA, Wold MS. J. Biol. Chem. 1994;269:24203. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.