

Abstract
Cyclotides are an abundant and diverse group of ribosomally synthesized plant peptides containing a cyclic cystine-knotted structure that confers them with remarkable stability. They are explored for their distribution in plants, although little is known about the individual peptide content of a single species. Therefore, we chemically analyzed the crude extract of the coffee-family plant Oldenlandia affinis using a rapid peptidomics workflow utilizing nano-LC-MS, peptide reconstruct with database identification, and MS/MS automated sequence analysis to determine its cyclotide content. Biologically, cyclotides are mainly explored for applications in agriculture and drug design; here we report their growth-inhibiting effects on primary cells of the human immune system using biological and immunological end points in cell-based test systems. LC-MS quantification of the active O. affinis plant extract triggered the characterization of the antiproliferative activity of kalata B1, one of the most abundant cyclotides in this extract, on primary activated human lymphocytes. The effect has a defined concentration range and was not due to cytotoxicity, thus opening a new avenue to utilize native and synthetically optimized plant cyclotides for applications in immune-related disorders and as immunosuppressant peptides.
The body’s immune system is a powerful weapon against pathogens, but malfunctioning can cause an over-reactivity of this defense machinery and in some instances lead to autoimmune diseases such as rheumatoid arthritis (RA) or Crohn’s disease. Immunosuppression, the targeted reduction of the activation or efficacy of the immune system, is an option for the treatment of these conditions. Because T-lymphocytes have the greatest impact during this defense response, most immunosuppressive medications aim to act on these cells. A representative is the drug cyclosporine A, a cyclic nonribosomal depsipeptide of fungal origin, which is the method of choice for severe cases of RA,1,2 although it has many and sometimes severe side effects.3
Other naturally occurring circular peptides with potential pharmaceutical applications have been found in various organisms (summarized in Craik4), such as bacteria (e.g., bacteriocin AS-485 and microcin J256), plants (e.g., sunflower trypsin inhibitors7,8), and animals (e.g., rhesus monkey θ-defensins9). One of the largest, but mostly unexplored groups of natural circular peptides are the so-called plant cyclotides.10
Cyclotides are an exceptional family of head-to-tail cyclized peptides. The circular cyclotide chain consists of ~30 amino acids, including six conserved cysteines that form three disulfide bonds arranged in a cyclic cystine-knot (CCK) motif,11 whereas the intercysteine sequences can tolerate a wide range of residue substitutions, and hence the cyclotide scaffold serves as a natural combinatorial peptide template12 (Figure 1). These remarkable structural features make cyclotides extremely resistant to enzymatic, chemical, and thermal degradation.13 In contrast to non-ribosomal-synthesized plant metabolites, cyclotides are true gene products, and their biosynthesis involves ribosomal precursor synthesis, enzymatic processing,14,15 and protein-folding events.16 Since their discovery in the coffee family (Rubiaceae), cyclotides have been extensively studied in the violets (Violaceae) and have recently been found in legumes (Fabaceae).17-19 Although there is an increasing effort to screen plants of different families for the occurrence and distribution of cyclotides,20 we still lack information on the individual cyclotide content of single plant species, mainly due to difficulties associated with the analytical analysis and the identification of cyclotides. Cyclotides possess a wide range of biological activities, e.g., insecticidal,21,22 nematocidal,23,24 antifouling,25 and anti-HIV26,27 activities, as well as cytotoxicity to lymphoma cell lines.28,29 The discovery of the first cyclotide, kalata B1, was based on its presence in Oldenlandia affinis (R&S) DC., which has been used in African indigenous medicine to accelerate childbirth by inducing uterine contractions following its ingestion as a tea.20,30,31 Today it is evident that many other cyclotides exist. Recently it has been estimated that there are at least 50 000 novel cyclotides to be discovered in Rubiaceae32 and another ~9000 in Violaceae,33,34 but we are only beginning to understand their variety and distribution in plants.35 Increasing knowledge about their occurrence will ultimately lead to the discovery and isolation of novel cyclotides with activity on a variety of molecular targets that are potentially important for diverse applications as agrochemicals22,36 as well as drug design due to their enormous stability.37-40
Figure 1.
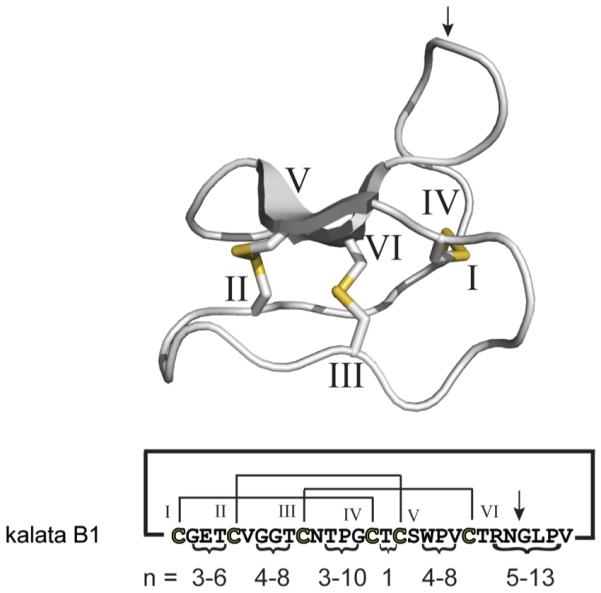
Structure and sequence diversity of cyclotides. The structure of the typical cyclotide kalata B1 is shown in gray. The six conserved cysteines are labeled with roman numerals, and the resulting cystine knot disulfide connectivity (CI–CIV, CII–CV, and CIII–CVI) is shown in yellow. The amino acid sequence and disulfide connectivity of kalata B1 are shown below the structure. The numbers (n) indicate the possible length (in amino acids) of the intercysteine loops comprising all currently known cyclotides (according to Ireland et al.61). The intercysteine loops can tolerate a wide variety of amino acid substitutions and are an indicator of the combinatorial diversity of the cyclotide scaffold.
Here we report an optimized protocol for the analysis of cyclotide plant extracts by combining nanoflow LC-MS/MS and automated database analysis to determine the content of distinct peptides (by molecular weight and peptide sequence) in the cyclotide-containing plant O. affinis. In addition we have tested this crude cyclotide extract of O. affinis as well as one of the most abundant cyclotides from this plant, namely, kalata B1, for its antiproliferative capacity toward activated primary human lymphocytes using relevant biological and immunological end points in cell-based test systems. We characterized for the first time the immunosuppressive properties of plant cyclotides in vitro. Combined with their unique structural features and enormous stability, these results may open new avenues for the application of cyclotides in immune-related disorders.
RESULTS AND DISCUSSION
The purpose of this study was to characterize the antiproliferative and cytotoxic effects of an O. affinis cyclotide-containing plant extract toward activated primary human lymphocytes. To identify the individual molecular peptide components of this immunosuppressive cyclotide mixture, we combined biological in vitro analysis with chemical characterization of the content of individual peptides (cyclotides) in the crude extract of this plant using an optimized rapid peptidomics workflow. This triggered the immunosuppressant characterization of kalata B1 using biological and immunological cell-based test systems.
Chemical Analysis of Oldenlandia affinis Plant Extract
We have grown O. affinis plants and isolated the aerial parts according to well-known laboratory protocols using overnight extraction with CH2Cl2 and MeOH followed by C18 solid-phase extraction of the aqueous part. This standard procedure commonly yields many grams of crude cyclotide-extract per kilogram of fresh plant leaf weight,22,30 while the content of various cyclotides depends on the growth conditions, e.g., habitats of the plants and other environmental factors.34,41 Generally, amino acid sequencing is only feasible from pure or semipurified cyclotide fractions. Therefore, we have used an alternative peptidomics approach to dissect the cyclotide content from a crude plant extract by combining nanoflow LC-MS and peptide reconstruction, identification by molecular weight, as well as proteolytic digestion, LC-MS/MS, and automated sequencing by database analysis using the recently reported ERA cyclotide database tool.42 The crude cyclotide extract was analyzed with various linear gradients on reversed-phase C18 nano LC coupled online to an electrospray ionization hybrid triple-quadrupole/linear ion-trap (ESI-QqLIT) mass spectrometer, which was operated in enhanced MS mode with scan speeds of 1000 and 4000 amu/s, respectively. Application of an automated LC-MS reconstruct tool yielded initially a few hundred peptide masses in the range from 2700 to 3500 Da, a typical MW range for cyclotides. The high number likely accounts for some false-positive hits due to the inclusion of low-abundant data in the calculation. Hence, we adjusted the S/N factor in the algorithm and obtained usually between 50 and 100 reconstructed peptide masses with significant scores above 0.99. Representative LC-MS reconstructed data of at least three independent experiments are listed in file S1. We identified a total of 72 peptide masses in the range from 2700 to 3500 Da. By comparing those peptide masses to the database of cyclotides (CyBase43), we identified 23 known O. affinis cyclotides, 24 peptide masses that correspond to peptides from other cyclotide plant species, and 25 new cyclotide masses. LC-MS experiments were further analyzed with manual peptide reconstruction by extracting the doubly and triply charged ions of respective cyclotide peaks and by calculation of the average molecular weight (unpublished data). The manual analysis was useful as an internal control to ensure the integrity of the generated automated data.
In addition to the analysis of O. affinis cyclotides by molecular weight and database comparison, a number of chemical modifications of the crude extract, i.e., reduction and alkylation followed by trypsin and endo-GluC proteolysis, were performed. Owing to the structural nature and high stability of cyclotides, these chemical modifications are necessary to yield amenable precursor ions for MS/MS sequencing. The modified and digested mixtures were analyzed with a peptidomics workflow utilizing nano LC and peptide sequencing by information-dependent acquisition (for further details see the Experimental Section). The resulting MS and MS/MS data were used for automated cyclotide identification using the Paragon algorithm with a custom-made ERA cyclotide database, a tool that is freely available on the Web. Using this cyclotide peptidomics analysis, 14 known cyclotides could be identified by amino acid sequence (file S2). In summary, using the above-described peptidomics workflow nearly all currently known cyclotides and an even greater number of novel peptide masses corresponding to other known or novel cyclotides could be identified in a crude cyclotide extract from the plant O. affinis (Table 1).
Table 1.
Cyclotides from O. affinis Extract Identified by Nano LC-MS and MS/MS
| cyclotidea | MW (av), Dab | MW (mono.), Dab | scorec | evidenced | theoretical MW, Dae | ΔMW, Daf |
|---|---|---|---|---|---|---|
| kalata B1 | 2892.85 | 2890.39 | 1 | ICP | 2892.33 | 0.52 |
| kalata B2 | 2956.14 | 2953.74 | 1 | ICS | 2955.38 | 0.76 |
| kalata B3 | 3083.31 | 3080.64 | 1 | ICS | 3082.48 | 0.83 |
| kalata B4 | 2893.24 | 2890.56 | 1 | IS | 2893.31 | 0.07 |
| kalata B5 | P | 3061.59 | ||||
| kalata B6 | 3029.96 | 3027.66 | 0.9999 | IS | 3029.42 | 0.54 |
| kalata B7 | 3072.26 | 3069.74 | 0.9998 | IS | 3071.59 | 0.67 |
| kalata B8 | 3284.34 | 3281.75 | 1 | ICS | 3283.79 | 0.55 |
| kalata B9 | P | 3272.72 | ||||
| kalata B9 lin | P | 3290.74 | ||||
| kalata B10 | 3030.21 | 3027.53 | 1 | ICS | 3030.41 | 0.20 |
| kalata B10 lin | 3048.54 | 3046.50 | 1 | ICS | 3048.43 | 0.11 |
| kalata B11 | 2884.48 | 2881.44 | 0.9999 | I | 2884.26 | 0.22 |
| kalata B12 | P | 2880.27 | ||||
| kalata B13 | 3036.06 | 3033.58 | 1 | IC | 3036.46 | 0.40 |
| kalata B14 | 3023.74 | 3021.17 | 0.9987 | I | 3022.43 | 1.31 |
| kalata B15 | 2977.00 | 2974.56 | 1 | ICS | 2976.40 | 0.60 |
| kalata B18 | 3147.33 | 3145.02 | 0.9977 | I | 3145.67 | 1.66 |
| kalata S | 2878.81 | 2875.93 | 0.9993 | I | 2878.30 | 0.51 |
| Oak6 cyclotide 1 | 3035.87 | 3033.49 | 1 | IC | 3035.47 | 0.40 |
| [G-A] kalata B1g | 2906.47 | 2904.75 | 0.9995 | I | 2906.35 | 0.12 |
| kalata b1–1 | 2724.12 | 2722.28 | 1 | IC | 2724.18 | 0.06 |
| [L2A] kalata B1 | 2851.88 | 2849.54 | 1 | IC | 2850.25 | 1.63 |
| Ac-[desGly]-KB1-Am | 2854.31 | 2851.68 | 0.9996 | I | 2853.30 | 1.01 |
| acyclic kalata B1 | 2911.32 | 2908.36 | 1 | IC | 2910.35 | 0.97 |
| Oak6 cyclotide 2 | 3093.29 | 3090.61 | 1 | IC | 3092.56 | 0.73 |
Identification by LC-MS reconstruct of at least three representative LC-MS experiments (±1 Da, 20–70 min, EMS 1000 2 scans) or identification by digest (trypsin or endo-GluC), nano LC-MS/MS, and database search (ERA).
Observed molecular weight.
Score indicating the quality of LC-MS reconstructed peptide MW (≤1.0).
Evidence for identified cyclotides, I = isotope pattern, C = charge pattern, S = full sequence, P = partial sequence or sequence tag.
Data taken from CyBase.43
Δ determined from average molecular weight.
Amino acid position (G-A replacement) not specified.
The combination of nano LC-MS/MS and LC-MS reconstruction, as well as automated database searching, is a rapid and useful technique for the identification of cyclotides in crude extracts. Compared to an earlier study by Plan et al.,44 describing the first cyclotide fingerprint of O. affinis using classical peptide purification via analytical HPLC and offline MS/MS sequencing, we have identified eight additional known cyclotides and are able to provide a list of ~50 peptide masses corresponding to cyclotides of which some can be identified by peptide fingerprint analysis in CyBase.43 This suggests that the number of cyclotides to be found in a single species may be >70 and is, therefore, at least twice the number as earlier anticipated, i.e., on average 34 cyclotides per species.32 This, of course, has a huge impact on the determination of the overall number of cyclotides in the plant kingdom and consequently would lead to a necessary revision of the number of novel cyclotides to be discovered in plants.
Antiproliferative Effects of O. affinis Cyclotide Extract
By using flow cytometric-based forward-side-scatter analysis, we demonstrated that the O. affinis cyclotide extract exhibits a dose-dependent (50–100 μg/mL) decrease of activated proliferating human peripheral blood mononuclear cells (PBMC) compared to untreated stimulated control (Figure 2A and B). Simultaneously, we observed a constant content of viable, resting PBMC, without accumulation of dead cells, showing that the applied concentrations of the cyclotide extract are not harmful to the cells. Above 100 μg/mL the extract showed an increasing cytotoxic effect. Camptothecin (CPT, 30 μg/mL), used as positive inhibitory proliferation control, induced a high proportion of dead cells, indicating that the observed antiproliferative effect, in contrast to the O. affinis cyclotide extract, was mainly due to cytotoxicity.
Figure 2.

Effects of the O. affinis cyclotide extract on cell proliferation of activated human peripheral blood mononuclear cells. CFSE-labeled primary human PBMC were antibody-activated (anti-CD3/CD28 mAbs) and cultured in the presence of medium (ctrl), camptothecin (CPT, 30 μg/mL), or different concentrations (50–100 μg/mL) of O. affinis cyclotide extract. The cells were further analyzed for cell viability and proliferation capacity using flow forward-side-scatter-based flow cytometric analysis (A and B). Cell division analyses were assessed by FACS and illustrated as representative dot plots (C). Results are summarized from three independent experiments in (D), and data are presented as mean ± SEM.
We further evaluated the impact of the crude O. affinis cyclotide preparation on the cell division level of activated PBMC. Therefore, we labeled the cells with the dye carboxyfluorescein diacetate succinimidyl ester (CFSE), which does not influence the viability of the stained cells and is inherited by daughter cells after cell division, and each dividing cell consequently loses fluorescent intensity. These data, shown in Figure 2C and D, indicate that the extract caused a dose-dependent inhibition of cell division of activated PBMC, which confirms that the crude O. affinis cyclotide preparation has the ability to inhibit PBMC proliferation without cell damage.
Relative Quantification of Cyclotides and Isolation of Kalata B1
Since we obtained promising antiproliferative activity of the total cyclotide extract from O. affinis, we determined the relative amount of the major cyclotides and further purified the main components for biological characterization. For this purpose we used the crude cyclotide extract for quantitative nano LC-MS analysis, as described above. Diluted aliquots of the extract were separated by nano C18 RP-HPLC coupled online to the mass spectrometer. Eluted peptides were monitored both with absorbance at 214 nm and by molecular weight. The area under the curve of the major cyclotide peaks in O. affinis was determined by automated integration and, if necessary, manual postprocessing. The relative quantification analysis of the cyclotide content has been carried out from five independent LC-MS experiments (file S3), and a representative O. affinis elution profile, indicating the major cyclotide peaks and their relative abundance (mean ± SEM), is shown in Figure 3.
Figure 3.
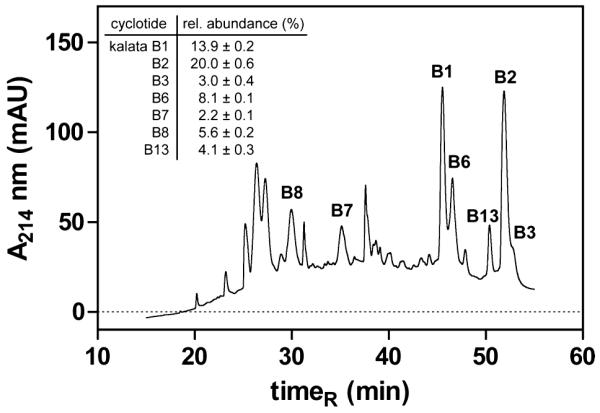
Nano LC-MS chromatogram of O. affinis cyclotides. The nanoflow elution profile of cyclotides from O. affinis was monitored with UV absorbance at 214 nm and mass spectrometry. The HPLC graph of a representative crude cyclotide extract is shown, and its major cyclotides are indicated by name and relative abundance. The relative cyclotide content (mean ± SEM) was determined by peak integration of five independent experiments (file S3). HPLC and MS conditions for cyclotide analysis are shown in the Experimental Section.
In agreement with earlier studies,44 the cyclotides kalata B1 and kalata B2 are the main peptide components, accounting for approximately 34% of the overall cyclotide content in O. affinis. Kalata B1 and B2 differ by only five amino acid positions (Figure S1), namely, Val to Phe (loop 2) and conservative replacements of Thr with Ser (loop 4), Ser with Thr (loop 5), Val with Ile (in loop 5), and Asn with Asp (in loop 6) in kalata B2. Since these substitutions have no significant structural consequences based on the root-mean-square deviation of the backbone atoms (rmsdbackbone kB1/kB2 = 0.599 Å, Figure S1) and since the two peptides have a similar bioactivity profile,22 we decided to use kalata B1 (comprising ~14% of total extract) for further biological analysis and its antiproliferative potential on activated human primary PBMC.
Antiproliferative and Cytotoxic Effects of Kalata B1
After exposure of PBMC to kalata B1 concentrations in the range from 1.8 to 14 μM, a dose-dependent decrease of the cell division capacity was observed with the CFSE assay, as compared to untreated stimulated PBMC controls, as shown in Figure 4. The inhibitory concentration IC50 for the antiproliferative effect of kalata B1 was 3.9 ± 0.5 μM (Figure S2), which compares to other effects of kalata B1, such as nematocidal45 and cytotoxic activities,28,29,46 as has been summarized in Table S1.
Figure 4.
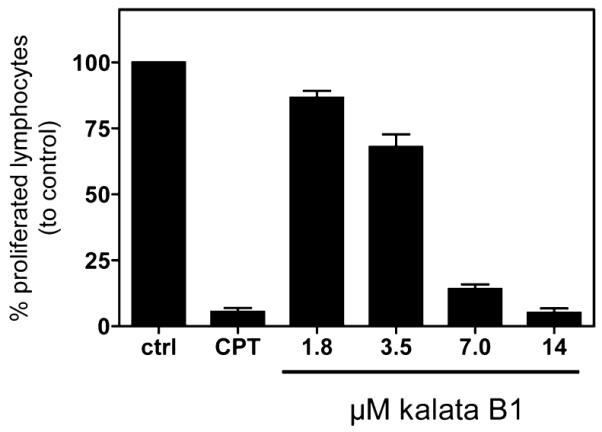
Effects of kalata B1 on cell proliferation of activated human peripheral blood mononuclear cells. The influence of medium (ctrl), camptothecin (CPT, 30 μg/mL), or different concentrations of kalata B1 (1.8–14 μM) on proliferation of CFSE+ anti-CD3/CD28 mAbs-activated human primary PBMC was measured by cell division analysis using flow cytometry. Data are presented as mean ± SEM of four independent experiments.
To analyze whether the antiproliferative effect was due to cell damage, we examined the influence of kalata B1 on the induction of PBMC apoptosis or necrosis (Figure 5) by using an internucleosomal DNA fragmentation (subG1+ cells) assay and phosphatidylserine surface analysis through annexin V and propidium iodide staining. This double staining process allowed the discrimination between viable (annexin−/PI−), apoptotic (annexin+/PI− and annexin+/PI+), and necrotic (annexin−/PI+) cells. The data shown in Figure 5A–C demonstrate that kalata B1 had no significant influence on the induction of apoptosis. Necrosis was slightly increased at higher concentrations (14 μM) of kalata B1, compared to the untreated control (Figure 5D). The positive controls for apoptosis and necrosis, CPT (30 μg/mL) and detergent (Triton-X 100), respectively, significantly increased the fractions of these cells.
Figure 5.

Effects of kalata B1 on cytotoxicity of activated human peripheral blood mononuclear cells. Human primary PBMC were activated with anti-CD3/CD28 mAbs in the presence of medium (ctrl), camptothecin (CPT, 30 μg/mL), Triton-X 100 (T-x), or different concentrations of kalata B1 (1.8–14 μM) and analyzed for “subG1” DNA content (A) by flow cytometry. Cells were stained with annexin V and propidium iodide (PI) to assess the percentages of viable (annexin V−/PI−), apoptotic (annexin V+/PI− or annexin V+/PI+), and necrotic (annexin V−/PI+) cells. Dot plots were analyzed, and representative graphs are shown (B). Results from three independent experiments are summarized, and data are presented as mean ± SEM (C and D). n.d. = not detectable.
The antiproliferative activity of kalata B1 triggered validation and control experiments to determine the nature of the observed effect. Cytometric-based forward-side-scatter analysis (data not shown) provided solid evidence that the antiproliferative effect induced by the cyclotide does not cause cell death by either apoptosis or necrosis, but inhibits the growth of the lymphocytes in a cytostatic fashion. Concentrations higher than 14 μM of the peptide are cytotoxic to the cells (data not shown). This was expected since kalata B1 has earlier been reported to cause hemolysis and membrane disruption at concentrations above ~50 μM.47,48 Therefore, we performed control experiments with the honeybee venom component melittin, a commonly used strong membrane-disrupting peptide agent.
We tested concentrations at which cytotoxic effects on human lymphocytes were described to ensure that our experimental setup was sensitive enough to detect possible cytotoxic effects of kalata B149 (Figure S3). The data revealed that, in contrast to kalata B1, melittin induced a decrease of proliferating PBMC at 1.6 μM (Figure S3A), but this effect was mainly due to the induction of apoptosis, as indicated by the results of the internucleosomal DNA fragmentation analysis (Figure S3B) and by induction of specific apoptotic cells at these concentrations (Figure S3C). In addition, there was a slight necrosis induction at high concentrations of melittin (Figure S3D).
From these control data we conclude that kalata B1, in contrast to melittin, has an antiproliferative capacity, which is not due to cytotoxic effects and the membrane lysing capacity of kalata B1, as otherwise one would have expected similar observations from the much more potent cytotoxic peptide melittin. The proof of antiproliferative effects by holding the cells in an “inactive” state at which they are still viable but are not able to grow and proliferate without causing cell death in a certain dose range is a crucial precondition to classify a substance as immunosuppressant, because cytotoxicity would cause side effects.
Potential of Cyclotides as Immunosuppressive Peptide Templates
Several additional characteristics regarding drug delivery contribute to the above-described immunosuppressant potential of cyclotides: (i) retained activity upon oral administration as tea in humans,30 (ii) great stability in plasma and against gastrointestinal proteases,13,50 and (iii) the presence of surface-exposed hydrophobic patches.12 Generally, therapeutic peptides lack oral bioavailability due to fast degradation upon ingestion and have poor drug permeation due to their hydrophilic nature.51,52 Hence, cyclotides and related cystine-knot peptides are likely to overcome these problems.53 As a proof of concept, a synthetically engineered cyclic conotoxin has recently been confirmed as an oral active circular peptide drug for the treatment of neuropathic pain in vivo.54 Another feature of cyclotides with respect to applications as peptide drugs is that they are ribosomally synthesized gene products55 and can therefore be produced in large quantity by recombinant techniques56 or in plant suspension cultures.41 These cyclotide production techniques and the availability of solid-phase peptide synthesis strategies57 offer opportunities for the optimization of the cyclotide framework, which is amenable to a wide range of amino acid substitutions (Figure 1).
One of the clinically used immunosuppressive drugs of choice to treat or suppress an “overactivity” of lymphocytes, for example, after transplantation surgery or in cases of severe RA, is the fungal cyclodepsipeptide cyclosporine A.3 It is yet too early to compare those two compounds toward clinical efficacy due to a lack of clinical in vivo data for kalata B1, although they are related in the sense that cyclotides and cyclosporine A are of natural origin and contain a circular peptide structure. In conclusion, we described the antiproliferative effects of an extract from the coffee-family plant O. affinis on cells of the human immune system (lymphocytes) and specifically identified kalata B1 as an active component that is responsible for the observed cytostatic effects in a defined concentration range, which does not cause cell death by apoptosis or necrosis. However, the therapeutic window is limited due to cytotoxic effects of kalata B1 at higher doses. As a consequence, these results open new avenues to utilize native and synthetically optimized plant cyclotides for applications in immune-related disorders and as immunosuppressants. The presented results have further impact on the general field of cystine-knot peptides and greatly enhance the possibilities for their potential therapeutic applications. The presumable oral bioavailability of cyclotides and cystine-knot peptides, the availability of recombinant and synthetic production techniques, and the plasticity of the cystine-knot framework, which is amenable to a wide range of amino acid substitutions, provide a promising basis for future mechanistic studies of their activity on lymphocytes and in vivo applications in model systems related to malfunctioning of immune cells in general and in particular the over-reactivity of lymphocytes.
EXPERIMENTAL SECTION
Extraction, Preparation, and Purification of Plant Cyclotides
Oldenlandia affinis (R&S) DC. plants were grown in the glass house at the Department of Pharmacognosy, University of Vienna, from seeds that were obtained as a gift from David Craik, Institute for Molecular Biosciences, University of Queensland. Aerial parts of the plants have been harvested and dried. Plant material was pulverized using a rotor grinder and extracted twice overnight in CH2Cl2/MeOH (1:1 v/v). The extracts were concentrated on a rotoevaporator and were lyophilized. The dried extracts were dissolved in solvent A (ddH2O with 0.1% TFA) and in-batch prepurified with C18 solid-phase extraction (ZEOprep 60 Å, C18 irregular 40-63 μm; ZEOCHEM, Uetikon, Switzerland). To separate the hydrophilic noncyclotide compounds from the hydrophobic cyclotide compounds, the C18 beads were washed with 10% solvent B (90% MeCN in ddH2O with 0.08% TFA) and eluted with 80% solvent B. The eluate containing cyclotides was analyzed by MALDI-TOF MS and reconstituted in ddH2O at 10 mg/mL for biological assays or used for nano LC-MS/MS analysis and further purification. Kalata B1 was purified from crude O. affinis extract by HPLC using a Perkin-Elmer Series 200 system with preparative (Phenomenex Jupiter, 10 μm, 300 Å, 250 × 21.2 mm; 8 mL/min) and semipreparative (Kromasil C18, 5 μm, 100 Å, 250 × 10 mm; 3 mL/min) RP-C18 HPLC columns and linear gradients from 0 to 80% solvent B in 80 min. Eluting peptides were monitored with UV absorbance (A280), collected manually, and lyophilized. Purity and quality of kalata B1 were assessed by analytical HPLC and MALDI-TOF MS.
Nano LC-MS and LC-MS/MS Analysis
Crude, ZipTip prepared, or digested plant extracts (C18 prepurified O. affinis extract, see above) were analyzed by nano LC-MS or LC-MS/MS on an Ultimate 3000 nano HPLC system controlled by Chromeleon 6.8 software (Dionex, Amsterdam, The Netherlands). For LC analysis, 1–5 μL samples of O. affinis extract were injected, preconcentrated using Dionex PepMap C18 cartridges (300 μm × 5 mm, 5 μm, 100 Å), and separated by nano-RP-HPLC prior to online MS analysis using a Dionex Acclaim PepMap C18 column (150 mm × 75 μm, 3 μm, 100 Å; 300 nL/min). The mobile phase consisted of solvent C (0.1% HCO2H) and solvent D (90/10 MeCN/0.08% HCO2H). Peptides were eluted using a linear gradient of 4–90% D in 35 min, 5 min hold at 90% D, followed by a return to 4% D for a 20 min equilibration. For LC-MS/MS analysis aliquots (1–10 μL) of tryptic or endo-GluC digested plant extracts were preconcentrated and separated by C18 nano LC as described above, using several LC gradients of up to 120 min duration (e.g., 4–60% B in 100 min, 60–90% B in 1 min, and finally a 5 min hold at 90% B, followed by a return to 4% B for a 10 min equilibration). Eluated peptides were directly introduced into the nanospray source. Mass spectrometry experiments were performed on a hybrid quadrupole/linear ion trap 4000 QTRAP MS/MS system (ABSciex, Foster City, CA, USA) running with the Analyst 1.5.1 software package. The 4000 QTRAP equipped with a nanospray source was operated in positive ionization mode. LC-MS analyses for cyclotide quantification and identification by molecular weight were performed using enhanced multiple scan (EMS) acquisition with a scan speed of 1000 amu/s in the mass range from 400 to 1400 Da. LC-MS data were analyzed by “LC-MS reconstruct” in the MW range from 2700 to 3500 Da and by using several S/N filter settings to obtain the molecular weight and validity score of all peptide peaks. LC-MS/MS analyses were performed using information dependent acquisition (IDA). The acquisition protocol used to provide mass spectral data for database searching involved the following procedure: the HPLC eluant was mass profiled using EMS; ions over the background threshold were subjected to examination using the enhanced resolution (ER) scan to confirm charge states of the multiply charged molecular ions. The most and next most abundant ions in each of these scans with a charge state of +2 to +4 or with unknown charge were subjected to collision-induced dissociation (CID) using rolling collision energy. An enhanced product ion scan was used to collate fragment ions and present the product ion spectrum for subsequent database searches.
Enzymatic Digest and Peptide Sequencing Using Database Analysis
C18 prepurified O. affinis extract cyclotides were prepared for MS/MS sequencing as described earlier.58,59 The extract was reduced, alkylated with iodoacetamide, and enzymatically digested using trypsin or endo-GluC (Sigma-Aldrich, Austria). Digested peptide extracts were analyzed with nano LC-MS/MS as described above, and IDA data were used for further analysis. Database searching of LC-MS/MS data was carried out using the ProteinPilot software and the Paragon algorithm with the custom-made ERA database tool for the identification of cyclotides.42
Relative Quantification of Cyclotides Using Nano LC-MS Analysis
C18 prepurified O. affinis extract was separated by one-dimensional nano LC-MS as described above. Cyclotide peaks were quantified by relative area under the curve (all peaks at 214 nm absorbance from 15 to 55 min were processed) using the quantification wizard of Chromeleon 6.8 software. Peaks in the LC chromatogram were identified by molecular weight and retention time from corresponding LC-MS peaks. Quantification was performed on five independent LC-MS experiments, and relative cyclotide abundance is presented as mean ± SEM.
Preparation of Human Peripheral Blood Mononuclear Cells and Cell Culture
PBMC were isolated from the blood of healthy adult donors obtained from the Blood Transfusion Centre, University Medical Center, Freiburg, Germany. Venous blood was centrifuged on a LymphoPrep gradient (density: 1.077 g/cm3, 20 min, 500g, 20 °C; Progen, Heidelberg, Germany). Afterward cells were washed twice with medium, and cell viability and concentration were determined using the trypan blue exclusion test. PBMC were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum (PAA, Coelbe, Germany), 2 mM l-glutamine, 100 U/mL penicillin, and 100 U/mL streptomycin (all from Invitrogen, Karlsruhe, Germany). The cells were cultured at 37 °C in a humidified incubator with a 5% CO2/ 95% air atmosphere. All experiments conducted on human material were approved by the Ethics Committee of the University of Freiburg.
Activation and Treatment of Peripheral Blood Mononuclear Cells
PBMC (105) were stimulated with anti-human CD3 (clone OKT3) and anti-human CD28 (clone 28.2) mAbs (both from eBioscience, Frankfurt, Germany) for 72 h in the presence of medium, the control agents camptothecin (CPT; 30 μg/mL: Tocris, Eching, Germany), and Triton-X 100 (0.5%; Carl Roth, Karlsruhe, Germany) or different concentrations of O. affinis extract, melittin (PolyPeptide, Strasbourg, France), or kalata B1, respectively. After cultivation, the cells were assessed in bioassays as described in the text.
Determination of Cell Proliferation and Cell Division
For cell proliferation and cell division tracking analysis PBMC were harvested and washed twice in cold PBS and resuspended in PBS at a concentration of 5 × 106 cells/mL. Cells were incubated for 10 min at 37 °C with CFSE (5 μM; Sigma-Aldrich, Taufkirchen, Germany). The staining reaction was stopped by washing twice with complete medium. Afterwards, the cell division progress was analyzed using flow cytometric analysis.
Determination of PBMC Apoptosis and Necrosis Using Annexin V and Propidium Iodide Staining
The levels of apoptosis were determined using the annexin V-FITC apoptosis detection kit (eBioscience, Frankfurt, Germany) according to the manufacturer’s instructions.60 After annexin V staining, propidium iodide solution (eBioscience) was added, and the cells were incubated in the dark, followed by a flow cytometric analysis to determine the amount of apoptosis and necrosis. We used CPT (30 μg/mL) and Triton-X 100 (0.5%) as positive controls for apoptosis and necrosis, respectively.
Data Analysis
All graphs were prepared using GraphPad Prism software, and data are presented as mean ± standard error (SEM). Where applicable, data were statistically analyzed using the one-way ANOVA Kruskal–Wallis test and Dunn’s multiple comparison post analysis.
Supplementary Material
ACKNOWLEDGMENTS
Thanks to the individuals at the Department of Pharmacognosy, University of Vienna, Austria, who helped during the growth and harvest of O. affinis plants and to D. J. Craik, Institute for Molecular Bioscience, Australia, for the gift of seeds. MS data of this research have been obtained by access to the MS core facility of the Center for Physiology and Pharmacology, Medical University of Vienna. Work on bioactive peptides in the laboratory of C.W.G. is funded by the Austrian Science Fund (FWF, P22889-B11). C.G. and R.H. receive financial support from Software AG foundation and DAMUS eV.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Supplemental figures: Structural alignment of kalata B1 and B2 (Figure S1), determination of IC50 for antiproliferative effect of kalata B1 on PBMC (Figure S2), and effects of melittin on cell proliferation and cytotoxicity on PBMC (Figure S3). Supplemental table: Comparison of IC50 values for kalata B1 on various cellular test systems (Table S1). Supplemental files: LC-MS reconstruct of O. affinis cyclotides (file S1), O. affinis database search results following digests and LC-MS/MS analysis (file S2), and cyclotide quantification data (file S3). This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Matsuda S, Koyasu S. Immunopharmacology. 2000;47:119–125. doi: 10.1016/s0162-3109(00)00192-2. [DOI] [PubMed] [Google Scholar]
- (2).Schreiber SL. Science. 1991;251:283–287. doi: 10.1126/science.1702904. [DOI] [PubMed] [Google Scholar]
- (3).de Mattos AM, Olyaei AJ, Bennett WM. Am. J. Kidney Dis. 2000;35:333–346. doi: 10.1016/s0272-6386(00)70348-9. [DOI] [PubMed] [Google Scholar]
- (4).Craik DJ. Science. 2006;311:1563–1564. doi: 10.1126/science.1125248. [DOI] [PubMed] [Google Scholar]
- (5).Martinez-Bueno M, Maqueda M, Galvez A, Samyn B, Van Beeumen J, Coyette J, Valdivia E. J. Bacteriol. 1994;176:6334–6339. doi: 10.1128/jb.176.20.6334-6339.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Rosengren KJ, Clark RJ, Daly NL, Göransson U, Jones A, Craik DJ. J. Am. Chem. Soc. 2003;125:12464–12474. doi: 10.1021/ja0367703. [DOI] [PubMed] [Google Scholar]
- (7).Luckett S, Garcia RS, Barker JJ, Konarev AV, Shewry PR, Clarke AR, Brady RL. J. Mol. Biol. 1999;290:525–533. doi: 10.1006/jmbi.1999.2891. [DOI] [PubMed] [Google Scholar]
- (8).Mylne JS, Colgrave ML, Daly NL, Chanson AH, Elliott AG, McCallum EJ, Jones A, Craik DJ. Nat. Chem. Biol. 2011;7:257–259. doi: 10.1038/nchembio.542. [DOI] [PubMed] [Google Scholar]
- (9).Tang Y-Q, Yuan J, Ösapay G, Ösapay K, Tran D, Miller CJ, Ouellette AJ, Selsted ME. Science. 1999;286:498–502. doi: 10.1126/science.286.5439.498. [DOI] [PubMed] [Google Scholar]
- (10).Gruber CW, Muttenthaler M, Freissmuth M. Curr. Pharm. Des. 2010;16:3071–3088. doi: 10.2174/138161210793292474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Craik DJ, Daly NL, Bond T, Waine C. J. Mol. Biol. 1999;294:1327–1336. doi: 10.1006/jmbi.1999.3383. [DOI] [PubMed] [Google Scholar]
- (12).Clark RJ, Daly NL, Craik D. J. Biochem. J. 2006;394:85–93. doi: 10.1042/BJ20051691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Colgrave ML, Craik DJ. Biochemistry. 2004;43:5965–5975. doi: 10.1021/bi049711q. [DOI] [PubMed] [Google Scholar]
- (14).Gillon AD, Saska I, Jennings CV, Guarino RF, Craik DJ, Anderson MA. Plant J. 2008;53:505–515. doi: 10.1111/j.1365-313X.2007.03357.x. [DOI] [PubMed] [Google Scholar]
- (15).Saska I, Gillon AD, Hatsugai N, Dietzgen RG, Hara-Nishimura I, Anderson MA, Craik DJ. J. Biol. Chem. 2007;282:29721–29728. doi: 10.1074/jbc.M705185200. [DOI] [PubMed] [Google Scholar]
- (16).Gruber CW, Cemazar M, Clark RJ, Horibe T, Renda RF, Anderson MA, Craik DJ. J. Biol. Chem. 2007;282:20435–20446. doi: 10.1074/jbc.M700018200. [DOI] [PubMed] [Google Scholar]
- (17).Poth AG, Colgrave ML, Lyons RE, Daly NL, Craik D. J. Proc. Natl. Acad. Sci. U. S. A. 2011;108:10127–10132. doi: 10.1073/pnas.1103660108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Poth AG, Colgrave ML, Philip R, Kerenga B, Daly NL, Anderson MA, Craik DJ. ACS Chem. Biol. 2011;6:345–355. doi: 10.1021/cb100388j. [DOI] [PubMed] [Google Scholar]
- (19).Nguyen GK, Zhang S, Nguyen NT, Nguyen PQ, Chiu MS, Hardjojo A, Tam JP. J. Biol. Chem. 2011;286:24275–24287. doi: 10.1074/jbc.M111.229922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Gruber CW, O’Brien M. Planta Med. 2011;77:207–220. doi: 10.1055/s-0030-1250317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Barbeta BL, Marshall AT, Gillon AD, Craik DJ, Anderson MA. Proc. Natl. Acad. Sci. U. S. A. 2008;105:1221–1225. doi: 10.1073/pnas.0710338104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Gruber CW, Cemazar M, Anderson MA, Craik DJ. Toxicon. 2007;49:561–575. doi: 10.1016/j.toxicon.2006.11.018. [DOI] [PubMed] [Google Scholar]
- (23).Colgrave ML, Kotze AC, Huang YH, O’Grady J, Simonsen SM, Craik DJ. Biochemistry. 2008;47:5581–5589. doi: 10.1021/bi800223y. [DOI] [PubMed] [Google Scholar]
- (24).Colgrave ML, Kotze AC, Kopp S, McCarthy JS, Coleman GT, Craik DJ. Acta Trop. 2009;109:163–166. doi: 10.1016/j.actatropica.2008.11.003. [DOI] [PubMed] [Google Scholar]
- (25).Göransson U, Sjogren M, Svangard E, Claeson P, Bohlin L. J. Nat. Prod. 2004;67:1287–1290. doi: 10.1021/np0499719. [DOI] [PubMed] [Google Scholar]
- (26).Wang CK, Colgrave ML, Gustafson KR, Ireland DC, Goransson U, Craik DJ. J. Nat. Prod. 2008;71:47–52. doi: 10.1021/np070393g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ireland DC, Wang CK, Wilson JA, Gustafson KR, Craik DJ. Biopolymers. 2008;90:51–60. doi: 10.1002/bip.20886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Svangard E, Goransson U, Hocaoglu Z, Gullbo J, Larsson R, Claeson P, Bohlin L. J. Nat. Prod. 2004;67:144–147. doi: 10.1021/np030101l. [DOI] [PubMed] [Google Scholar]
- (29).Lindholm P, Göransson U, Johansson S, Claeson P, Gulbo J, Larsson R, Bohlin L, Backlund A. Mol. Cancer Ther. 2002;1:365–369. [PubMed] [Google Scholar]
- (30).Gran L. Medd. Nor. Farm. Selsk. 1970;12:173–180. [Google Scholar]
- (31).Gran L. Acta Pharmacol. Toxicol. (Copenh.) 1973;33:400–408. doi: 10.1111/j.1600-0773.1973.tb01541.x. [DOI] [PubMed] [Google Scholar]
- (32).Gruber CW, Elliott AG, Ireland DC, Delprete PG, Dessein S, Goransson U, Trabi M, Wang CK, Kinghorn AB, Robbrecht E, Craik DJ. Plant Cell. 2008;20:2471–2483. doi: 10.1105/tpc.108.062331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Simonsen SM, Sando L, Ireland DC, Colgrave ML, Bharathi R, Goransson U, Craik DJ. Plant Cell. 2005;17:3176–3189. doi: 10.1105/tpc.105.034678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Trabi M, Svangard E, Herrmann A, Goransson U, Claeson P, Craik DJ, Bohlin L. J. Nat. Prod. 2004;67:806–810. doi: 10.1021/np034068e. [DOI] [PubMed] [Google Scholar]
- (35).Gruber CW. Biopolymers. 2010;94:565–572. doi: 10.1002/bip.21414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Gran L, Sletten K, Skjeldal L. Chem. Biodiversity. 2008;5:2014–2022. doi: 10.1002/cbdv.200890184. [DOI] [PubMed] [Google Scholar]
- (37).Craik DJ. Toxicon. 2001;39:1809–1813. doi: 10.1016/s0041-0101(01)00129-5. [DOI] [PubMed] [Google Scholar]
- (38).Craik DJ, Cemazar M, Daly NL. Curr. Opin. Drug Discovery Dev. 2007;10:176–184. [PubMed] [Google Scholar]
- (39).Craik DJ, Cemazar M, Wang CK, Daly NL. Biopolymers. 2006;84:250–266. doi: 10.1002/bip.20451. [DOI] [PubMed] [Google Scholar]
- (40).Craik DJ, Daly NL, Waine C. Toxicon. 2001;39:43–60. doi: 10.1016/s0041-0101(00)00160-4. [DOI] [PubMed] [Google Scholar]
- (41).Seydel P, Gruber CW, Craik DJ, Dörnenburg H. Appl. Microb. Biotechnol. 2007;77:275–284. doi: 10.1007/s00253-007-1159-6. [DOI] [PubMed] [Google Scholar]
- (42).Colgrave ML, Poth AG, Kaas Q, Craik DJ. Biopolymers. 2010;94:592–601. doi: 10.1002/bip.21400. [DOI] [PubMed] [Google Scholar]
- (43).Wang CK, Kaas Q, Chiche L, Craik DJ. Nucleic Acids Res. 2008;36:D206–210. doi: 10.1093/nar/gkm953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Plan MRR, Göransson U, Clark RJ, Daly NL, Colgrave ML, Craik DJ. ChemBioChem. 2007;8:1001–1011. doi: 10.1002/cbic.200700097. [DOI] [PubMed] [Google Scholar]
- (45).Huang YH, Colgrave ML, Clark RJ, Kotze AC, Craik DJ. J. Biol. Chem. 2010;285:10797–10805. doi: 10.1074/jbc.M109.089854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Daly NL, Gustafson KR, Craik DJ. FEBS Lett. 2004;574:69–72. doi: 10.1016/j.febslet.2004.08.007. [DOI] [PubMed] [Google Scholar]
- (47).Barry DG, Daly NL, Clark RJ, Sando L, Craik DJ. Biochemistry. 2003;42:6688–6695. doi: 10.1021/bi027323n. [DOI] [PubMed] [Google Scholar]
- (48).Henriques ST, Huang YH, Rosengren KJ, Franquelim HG, Carvalho FA, Johnson A, Sonza S, Tachedjian G, Castanho MA, Daly NL, Craik DJ. J. Biol. Chem. 2011;286:24231–24241. doi: 10.1074/jbc.M111.253393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Pratt JP, Ravnic DJ, Huss HT, Jiang X, Orozco BS, Mentzer SJ. In Vitro Cell. Dev. Biol. Anim. 2005;41:349–355. doi: 10.1007/s11626-005-0007-1. [DOI] [PubMed] [Google Scholar]
- (50).Colgrave ML, Jones A, Craik DJ. J. Chromatogr. A. 2005;1091:187–193. doi: 10.1016/j.chroma.2005.07.094. [DOI] [PubMed] [Google Scholar]
- (51).Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Drug Discovery Today. 2010;15:40–56. doi: 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]
- (52).Werle M, Kafedjiiski K, Kolmar H, Bernkop-Schnurch A. Int. J. Pharm. 2007;332:72–79. doi: 10.1016/j.ijpharm.2006.09.028. [DOI] [PubMed] [Google Scholar]
- (53).Kolmar H. Curr. Opin. Pharmacol. 2009;9:608–614. doi: 10.1016/j.coph.2009.05.004. [DOI] [PubMed] [Google Scholar]
- (54).Clark RJ, Jensen J, Nevin ST, Callaghan BP, Adams DJ, Craik DJ. Angew. Chem., Int. Ed. 2010;49:6545–6548. doi: 10.1002/anie.201000620. [DOI] [PubMed] [Google Scholar]
- (55).Jennings C, West J, Waine C, Craik D, Anderson M. Proc. Natl. Acad. Sci. U. S. A. 2001;98:10614–10619. doi: 10.1073/pnas.191366898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Kimura RH, Tran AT, Camarero JA. Angew. Chem., Int. Ed. 2006;45:973–976. doi: 10.1002/anie.200503882. [DOI] [PubMed] [Google Scholar]
- (57).Clark RJ, Craik DJ. Biopolymers. 2010;94:414–422. doi: 10.1002/bip.21372. [DOI] [PubMed] [Google Scholar]
- (58).Chen B, Colgrave ML, Daly NL, Rosengren KJ, Gustafson KR, Craik DJ. J. Biol. Chem. 2005;280:22395–22405. doi: 10.1074/jbc.M501737200. [DOI] [PubMed] [Google Scholar]
- (59).Ireland DC, Colgrave ML, Craik DJ. Biochem. J. 2006;400:1–12. doi: 10.1042/BJ20060627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Grundemann C, Gruber CW, Hertrampf A, Zehl M, Kopp B, Huber R. J. Ethnopharmacol. 2011;136:444–451. doi: 10.1016/j.jep.2011.05.018. [DOI] [PubMed] [Google Scholar]
- (61).Ireland DC, Clark RJ, Daly NL, Craik DJ. J. Nat. Prod. 2010;73:1610–1622. doi: 10.1021/np1000413. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.