

Abstract
Background
Constitutional deficiency in Factor XI (FXI) is a rare bleeding disorder in the general population, with the exception of Ashkenazi Jews. During the last decade, the detection of FXI-deficient patients has shifted from clinical screening identifying mostly severe bleeders to biological screening combining findings of prolonged activated partial thromboplastin time and FXI coagulation activity (FXI:C) below 50 U/dL.
Objectives
The goal of this study was to determine the molecular basis of FXI deficiency in western Brittany, France.
Patients/Methods
Over the course of 4 years, we detected 98 FXI-deficient patients through biological screening, and 44 patients agreed to participate in this study corresponding to 25 index cases. We developed an efficient mutation detection strategy (combining direct sequencing and QFM-PCR to search for heterozygous rearrangements in a routine setting) that detected F11 mutations in 24 out of the 25 index cases.
Results
An unexpected allelic heterogeneity was found, with 14 different single point mutations being detected, among which 9 are new. Moreover, a large heterozygous deletion of the entire F11 gene was detected, then further defined using a CGH array as a 4q34.2 telomeric deletion of 7 Mb containing 77 genes.
Conclusion
We propose that the observed recurrent mutations may be considered as genetic tags of a population. This study highlights the importance of screening for large deletions in molecular studies of F11.
Keywords: Factor XI deficiency, genetic analysis, mutation, deletion, F11 gene
INTRODUCTION
Factor XI (FXI) is a serine protease zymogen in the intrinsic pathway related to blood coagulation (1). The activated FXI glycoprotein (FXIa) has a two-chain structure, with a heavy chain composed of four apple domains in tandem (2) and a light chain harboring a serine protease domain (3). FXI deficiency is defined as severe when FXI Coagulation activity (FXI:C) is less than 15 U/dL and as moderate when it is between 15 and 50 U/dL. The detection of patients with FXI deficiency has shifted during the last decade from clinical screening mostly identifying severe bleeders to biological screening that detects a wide range of bleeding phenotypes (4). Patients with FXI deficiency are screened following a finding of prolonged activated partial thromboplastin time (aPTT), and the diagnosis of FXI deficiency is established when FXI:C is below 50 U/dL. The value of the FXI antigen (FXI:Ag) discriminates between two phenotypes of FXI deficiency: CRM− exhibits reduced levels of FXI:Ag associated with the low FXI:C, whereas CRM+ shows only reduced FXI:C with normal levels of FXI:Ag, indicating that there is a normal amount of FXI protein in the plasma, but with a loss of function (5). According to the FXI mutations database (http://www.factorxi.com/, version 2.2, updated in 2009), most of the mutations that have been reported are CRM− (6).
The gene encoding FXI (the F11 gene) is located on the long arm of chromosome 4 (4q35) (7) and is 50 kilobases in length, with 15 exons. Exon 1 is non-coding; exon 2 encodes the signal peptide; exons 3 to 10 code for the four apple domains; and exons 11 to 15 encode the carboxy-terminal serine protease (SP) domain, which contains the active site.
FXI deficiency is a rare bleeding disorder that is reported in all human populations, but it is particularly common in Ashkenazi Jews (estimated carrier rate of 9.0%) (8), with two major mutations occurring with equal frequencies: c.403G>T (p.E135X) in exon 5, and c.901T>C (p.F301L) in exon 9 (9,10). Two mutations appear to have a higher prevalence in France: c.316C>T (p.Q106X) in the western part of France (Nantes area) (11) and c.166T>C (p.C56R) in Basque families in the southwest of France (12). As of the last update of the database, 191 mutations causing FXI deficiency had been reported, all of which are single point mutations, except for two large deletions: a 31.5 kb deletion that encompasses the entire F11 gene (13) and a smaller deletion that extends from exon 11 to exon 15 (14).
In this study, we determined the molecular basis of FXI deficiency in 25 index patients detected based on exhibiting a FXI activity below 50 U/dL at Brest University Hospital, located in western Brittany (northwest of France). We developed, for the first time, a technique to systematically screen for rearrangements at the FXI locus in a routine setting to carry out complete direct sequencing analysis of this genomic region.
PATIENTS, MATERIALS, AND METHODS
PATIENTS
Over the course of 4 years (Jan. 2003 to Dec. 2006), 98 constitutional FXI deficiency patients were detected through a two-step biological screening strategy that combined the determination of a prolonged activated partial thromboplastin time (aPTT) with a ratio >1.2 (normal prothrombin time, normal fibrinogen and the absence of interfering heparin) followed by the detection of FXI coagulant activity (FXI:C) below 50 U/dL in the laboratory of Hematology at Brest University Hospital. The abnormal FXI:C values were confirmed in a second sample collected at a different time, and the two measurements were averaged.
Patients were recalled and forty-four patients (participation rate: 45%) agreed to participate in this study, belonging to 25 families living in the western Brittany area. A family tree was established for all families, and individuals were questioned about the origin of their ancestry based on the origins of their grandparents.
All 44 patients were questioned about their own bleeding history (spontaneous like epistaxis, gum bleeding, ecchymosis or related to surgical procedures) that was obtained verbally. To minimize the subjectivity of the record, 2 hematologists with expertise in hemostasis (JFA and BPP) met the patients and classified them as either “bleeder” or “non-bleeder” by each physician and blinded from the other physician’s assessment. In case of discordance the case was classified by a third clinician (PG).
Informed consent for molecular genetic analysis was obtained for these individuals by the referring clinician as part of our study protocol, which was approved by the local research ethics committee (CPP Ouest 6-485 and DGS 2007-0288).
Blood was obtained by venipuncture for coagulation testing and genetic analysis.
COAGULATION TEST
FXI:C was determined with an STA-evolution (Stago, Asnieres, France), using an activated partial thromboplastin time-based clotting test incorporating FXI-deficient plasma (Ref 00723;Stago). FXI antigen levels (FXI:Ag) were evaluated by an immunosorbent assay using paired polyclonal antibodies (Ref FXI-EIA; Kordia).
POINT MUTATION DETECTION
After DNA extraction from whole blood of patient’s leukocytes by standard procedures (15), screening for single point variations was performed by Sanger sequencing with the Dye terminator kit v3.1 (Appliedbiosystems). Fifteen primer pairs were designed for PCR amplification of the fifteen exons of the F11 gene and their immediate flanking sequences (16).
MUTATION NOMENCLATURE
To avoid confusion related to the variants described in this study, we provide both the commonly used name found in the published literature based on the active peptide (7) and the international nomenclature name, which is indicated between brackets in the text or tables. The international mutation nomenclature is based on the guidelines of the Human Genome Variation Society (http://www.hgvs.org/mutnomen/). cDNA-based numbering was assigned with the A of the ATG translational initiation codon being designated as +1 in the context of the reference sequence NM_000128.
POPULATION SCREENING OF THE 7 NOVEL MISSENSE MUTATIONS
To estimate the frequency of the new mutations in the general population, we set up an allele specific PCR with melting curve detection; briefly, seven primers pairs with one primer specific to each of the seven new missense mutations of the F11 gene were mixed with an internal control fragment (promotor of the HAMP gene) with the presence in the PCR of Syto9 (Invitrogen) as a fluorescent dye. The melt curve analysis (Lightscanner, Idaho Tech) allowed to discriminate between the control amplicon and the mutation specific ones. Five hundreds controls (relatives of cystic fibrosis patients recruited through cascade screening and representing the general population of our geographic area) were screened.
SCREENING OF REARRANGEMENTS BY QUANTITATIVE FLUORESCENT MULTIPLEX PCR (QFM-PCR)
Seventeen primer pairs targeting each of the 15 exons of the F11 gene (available upon request) and 2 amplicons upstream of the first exon (promoter region), together with two control regions (exon 14 of the F2 gene located on chromosome 11 as a diploid reference and exon 8 of the F9 gene located on the X chromosome as a quantitation control) were designed and simultaneously amplified in a single reaction for systematic screening of gross rearrangements by mean of quantitative fluorescent multiplex PCR (QFM-PCR) (17). The fluorescent profiles (electropherograms) were normalized based on the peak heights of the F2 amplicon, and the samples were then superimposed upon those of a normal diploid sample. A two-fold reduction in peak height was taken as being indicative of a deletion, while a 50% increase suggested a duplication.
REFINEMENT OF REARRANGEMENT BOUNDARIES BY A COMBINATION OF OLIGO-ARRAY CGH AND “WALKING” QFM-PCR
Whole-Genome Oligoarray comparative genomic hybridization (CGH) was performed using the Human Genome CGH Microarray kit 244A and a DNA microarray scanner (Agilent Technologies, Santa Clara, CA-USA). The obtained data were analyzed with Agilent Feature Extraction v.9.5.1 and Agilent CGH Analytics v.3.5.14 software (Agilent Technologies, Santa Clara, CA-USA). Hybridization was conducted with a pool of DNA samples from 10 control males (Human Genomic DNA: Male, Promega #G1471) as the reference. The nucleotide positions are indicated according to the hg18 build.
Based on the aCGH results, to narrow down the breakpoints, several sets of primer pairs located both in the upstream and downstream regions of the deletion were designed in the gaps between the aCGH probes to carry out a ‘walking QFM-PCR’ strategy with the aim of precisely localizing the centromeric breakpoint of the large telomeric deletion.
RESULTS
MOLECULAR EPIDEMIOLOGY: ALLELIC HETEROGENEITY
The strategy used in this study was to select patients based on their biological phenotypes with a prolonged aPTT and at least a 50 U/dL reduction of their FXI:C activity. Among the 25 index cases that participated in our study, thorough screening for both point mutations and rearrangements successfully identified an F11 gene mutation in 24 individuals. As anticipated based on their FXI:C phenotype (Table 1), 22 patients are heterozygous carriers, except for two patients who are homozygous for 2 mutations and one patient with no mutation detected. Most of the investigated families are native of western Brittany (northwest of France), except for 5 families either of different European ancestry (from France, Germany, Italy, and Russia) or from the Middle East (Lebanon).
Table 1.
Origin, mutations and phenotype findings of 25 index cases with FXI deficiency
| Family/Patient | Gender/Age | Geographic origin | Nucleotide change | Exon | Amino acid | HGV protein nomenclature | Domain | FXI:C (UI/dL)‡ | FXI:Ag (UI/dL) | Bleeding classification | References |
|---|---|---|---|---|---|---|---|---|---|---|---|
| XII/22 | F/19 | Western Brittany | c.151A>C | 3 | Thr33Pro | p.Thr51Pro | Apple 1 | 24 | 22 | Bleeder | Fard-Esfahani & al, 2008 |
| XIII/23 | M/16 | Western Brittany | c.151A>C | ” | ” | ” | ” | 22 | 18 | Nonbleeder | ” |
|
| |||||||||||
| XVIII/31 | F/34 | Western Brittany | c.166T>C | 3 | Cys38Arg | p.Cys56Arg | Apple 1 | 23 | 20 | Bleeder | Zivelin & al, 2002 |
| XIX/33 | F/39 | Western Brittany | c.166T>C | ” | ” | ” | ” | 33 | 30 | Nonbleeder | ” |
| XX/37 | M/79 | Western Brittany | c.166T>C | ” | ” | ” | ” | 25 | 23 | Bleeder | ” |
|
| |||||||||||
| XIV/24 | F/53 | Western Brittany | c.188C>T | 3 | Ala45Val | p.Ala63Val | Apple 1 | 42 | 56 | Bleeder | This study |
|
| |||||||||||
| XXII/39 | M/39 | Italy | c.403G>T * | 5 | Glu117X | p.Glu135X | Apple 2 | 1 | 2 | Bleeder | Asakai & al, 1989 |
|
| |||||||||||
| XV/25 | M/17 | Western Brittany | c.595+3A>G | Intron 6 | Splicing | Splicing | Introgenic region | 40 | 32 | Bleeder | Zivelin & al, 2002 |
|
| |||||||||||
| XVI/26 | F/11 | Western Brittany | c.664G>T | 7 | Asp204Tyr | p.Asp222Tyr | Apple 3 | 29 | 29 | Bleeder | This study |
|
| |||||||||||
| XXI/38 | F/33 | Germany | c.683G>A | 7 | Arg210Gln | p.Arg228Gln | Apple 3 | 50 | 56 | Bleeder | This study |
|
| |||||||||||
| XXIII/40 | F/30 | Lebanon | c.827C>G | 8 | Ser258Cys | p.Ser276Cys | Apple 3 | 40 | 105 | Nonbleeder | This study |
|
| |||||||||||
| XVII/27 | M/44 | Western Brittany | c.830G>A | 8 | Gly259Asp | p.Gly277Asp | Apple 3 | 33 | 25 | Nonbleeder | This study |
|
| |||||||||||
| IX/18 | F/21 | Western Brittany | c.1313C>A | 12 | Ser420X | p.Ser438X | Serine Protease Domain | 33 | 31 | Bleeder | This study |
|
| |||||||||||
| XI/20 | F/32 | Russia | c.1541G>T | 13 | Cys496Phe | p.Cys514Phe | Serine Protease Domain | 35 | 41 | Bleeder | This study |
|
| |||||||||||
| X/19 | F/31 | France (Picardie) | c.1565delG | 13 | Arg504fs | p.Arg522fs-Frame Shift | Serine Protease Domain | 49 | 37 | Nonbleeder | This study |
|
| |||||||||||
| VI/12 | F/60 | Western Brittany | c.1724C>T * | 15 | Ser557Leu | p.Ser575Leu | Active site | 1 | 60 | Nonbleeder | This study |
| VII/16 | M/78 | Western Brittany | c.1724C>T | ” | ” | ” | ” | 28 | 75 | Nonbleeder | ” |
| VIII/17 | F/17 | Western Brittany | c.1724C>T | ” | ” | ” | ” | 38 | 86 | Bleeder | ” |
|
| |||||||||||
| I/1 | F/28 | Western Brittany | c.1789G>A | 15 | Glu579Lys | p.Glu597Lys | Serine Protease Domain | 21 | 22 | Nonbleeder | Quelin & al, 2006 |
| II/5 | M/49 | Western Brittany | c.1789G>A | ” | ” | ” | ” | 36 | 35 | Bleeder | ” |
| III/8 | F/48 | Western Brittany | c.1789G>A | ” | ” | ” | ” | 25 | 22 | Bleeder | ” |
| IV/10 | F/51 | Western Brittany | c.1789G>A | ” | ” | ” | ” | 41 | 38 | Nonbleeder | ” |
| V/11 | M/67 | Western Brittany | c.1789G>A | ” | ” | ” | ” | 22 | 20 | Nonbleeder | ” |
|
| |||||||||||
| XXIV/41 | F/43 | Western Brittany | 4q telomeric del | / | / | / | / | 44 | 38 | Bleeder | This study |
|
| |||||||||||
| XXV/44 | M/19 | Western Brittany | ND | / | / | / | / | 50 | 41 | Nonbleeder | - |
ND : Not determinated
Homozygote
Average of 2 measurements from 2 samples
We observed allelic heterogeneity illustrated by 14 different single base changes identified by direct sequencing in the 23 index cases with a detected point mutation (Table 1). Ten of the mutations are missense mutations affecting conserved amino-acids, as determined through multiple protein sequence alignment against animal sequences. Only two mutations are nonsense mutations, one of which is located at the consensus splice site of intron 6, while one patient is a carrier of a one base deletion at position c.1565 in exon 13 resulting in a frameshift after amino acid p.Arg522. Among the 14 different mutations detected, 12 are CRM− mutations (quantitative), whereas two mutations are CRM+ (qualitative) (Table 1). Five mutations (identified in 12 families) were previously reported, whereas 9 point mutations (identified in 11 families) were novel. Among these 9 novel mutations, 4 are located in the serine protease domain or the active site (c.1313C>A, c.1541G>T, c.1565delG and c.1724C>T), and 5 are found within apple domains 1 to 3 (c.188C>T, c.664G>T, c.683G>A, c.827C>G and c.830G>A). The 7 novel missense mutations were not detected in the general population (500 controls) which rules out the hypothesis of frequent polymorphisms.
Four mutations were recurrent (c.151A>C, c.166T>C, c.1724C>T and c.1789G>A), the most frequent of which was the mutation c.1789G>A, which was encountered in 5 patients whose families are native to a small area in northwestern Brittany, suggesting a founder effect. Most of the carriers in these families presented a moderate bleeding tendency, usually associated with a cutaneo-mucous hemorrhagic symptoms.
Only 5 of the mutations found here were previously described (c.151A>C, c.166T>C, c.403G>T, c.595+3A>G, c.1789G>A) in different populations in Europe or the Middle East, and 2 patients (22 and 23) with familial origins in northwestern Brittany present a mutation in exon 3 (c.151A>C) that was recently described in Iran (18).
IDENTIFICATION of a LARGE 4q Ter DELETION
After sequencing the complete coding and flanking sequences of the F11 gene, there were two patients remaining for whom no mutation had been detected. Therefore, we developed an efficient, rapid QFM-PCR technique that included designing tag amplicons located in each of the 15 exons, allowing us, for the first time, to systematically and routinely screen the heterozygous rearrangements at the F11 locus. This strategy was found to be a success, as we were able to detect a large deletion in one patient (XXIV/41) that unexpectedly encompassed the entire F11 gene (Figure 1a). To refine the breakpoints of the deletion, we combined array CGH (Figure 1b) and “walking QFM-PCR” analyses, which indicated a loss of 4q telomeric sequences and located the centromeric breakpoint between nucleotide positions 183720115 and 183722461 (according to hg19 updated in February 2009), resulting in an estimation of the size of this new large deletion of 7 Mb. The deleted sequences contain 77 known genes, stretching from the DCTD gene on the centromeric side of the deletion to LOC728410 before the telomeric repeated sequences. The familial molecular analysis confirmed that the deleted allele was transmitted (mother and 2 children) with a CRM− biological phenotype and a variable bleeding phenotype.
Figure 1.
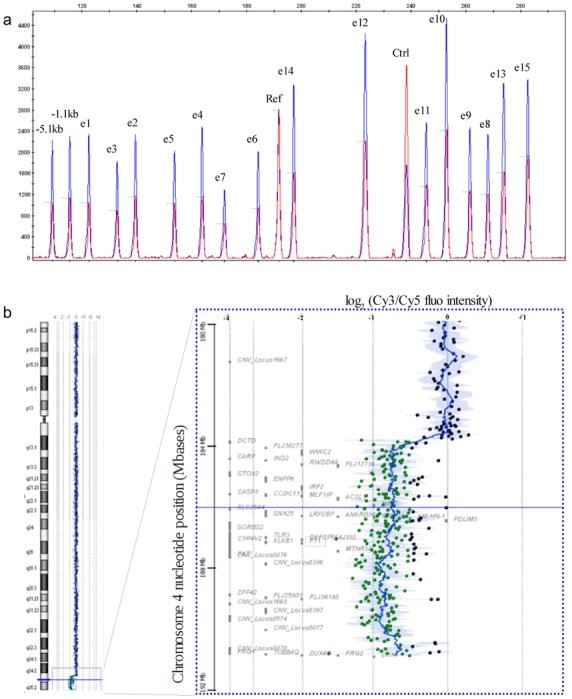
Delineation of the large heterozygous 4qTer deletion. (a) Systematic screening of heterozygous rearrangements at the F11 locus by QFM-PCR. The QFM-PCR electropherogram of patient XXIV/41 (red) is overlaid with that of a normal control (green) following normalization against the reference diploid F2 amplicon located on chromosome 11 (Ref). Another control amplicon located in the F9 gene on chromosome X (control) was used to validate the quantitative analysis by determining the sex based on chromosome X dosage. All peaks corresponding to sequences in the F11 gene show a two-fold reduction of their height, indicating a heterozygous deletion of the whole gene. (b) The large deletion of 4qTer revealed by arrayCGH. The log2 of the Cy3/Cy5 fluorescence intensity ratio is plotted against the chromosome 4 nucleotide position (hg18). Green data points indicate copy number losses, while black dots indicate no copy number change. More than 7 Mb of sequences are involved in this deletion, and the F11 gene (outlined in red) is located in the middle.
DISCUSSION
We investigated the molecular basis of FXI-deficient patients from western Brittany (northwest region of France). This study confirms the ethnic and molecular heterogeneity of the F11 even if we recruited patients in a limited geographical area where ~1 million people are living. As previously reported (see review (19)), only 5 clusters of patients have been identified to date with recurrent mutations, and these clusters are found among the Ashkenazi Jewish population (mutations c.403G>T and c.901T>C), Caucasians in UK (c.438C>A) and in France among Basques (c.166T>C) and in western France (c.316C>T). Among the 25 families investigated in this study, 4 mutations were found to be recurrent. Based on information about the geographic origins of the ancestors of our study population (grandparents of the index cases), we observed that the carriers of each recurrent mutation were clustered in small areas and shared the same haplotype (2 intragenic STRs in introns 2-B and 13-M), indicating a probable founder effect (data not shown). We propose that two of these (c.1789G>A and c.1724C>T) may be considered as tags of the population of the far west of France because they are described for the first time as recurrent in a population with non-consanguineous families, all the carriers sharing the same haplotype indicating a probable founder effect.
The most frequent mutation observed in our study (c.1789G>A, p.E597K), which was found in 5 index cases, is the first tag of our population, though it was previously reported (20) in a single patient living in France (Paris) who does not know if his ancestors lived in Brittany according to the team who reported it. The second tag, the c.1724C>T (p.S575L) mutation, is of particular interest because in addition to being original, it is the first time that a CRM+ mutation has been found to be recurrent (3 index cases), including one homozygous case being observed. This study confirms the usefulness of a rare, but not private, mutation as a tag of a population in a gene involved in a rare Mendelian disease.
The Basque mutation (c.166T>C) was also found in 3 index cases, (same haplotype with the original 12 repeats at marker CAi3(B) rs72428153, data not shown), representing a tag mutation shared between our study population and the Basques. The economic links (fishermen) between the Basques and Britons during the last centuries are documented in French religious records (available upon request). However “classical” population genetics studies using mtDNA (21) have failed to connect these two populations at the genetic level. Interestingly, the identification of the rare variant c.166T>C in the F11 gene relates the Brittany and Basque populations at the genetic level and strengthens the interest in the investigation of such “rare variants” in population genetics. The population of origin of the c.166T>C mutation remains unknown because, as in the Basque population (12), we did not find CAi3-12 repeats allele among 100 healthy controls, which may indicate that the mutation originated in another population that migrated to the western part of continental Europe. After the first step of the strategy for mutation detection developed in this study, consisting of screening for point mutations in the 15 exons of the F11 gene, no mutation had been detected in 2 patients among the 25 index cases.
Based on the accumulation of data during the last decade, it is now possible to systematically complete the mutation detection process by screening for large rearrangements. In one of the two patients lacking mutations, we suspected the occurrence of this type of large deletion based on a transmission discrepancy at rs5969 in exon 7, suggesting hemizygosity at this location. The second justification for screening for genomic events was that two F11 gene deletions had been previously reported: a 31.5 kb deletion was found using an EBV- transformed cell assay converting to haploid chromosomes (13), and the exons 11-15 del was detected in a homozygous carrier from Tunisia through the failure of PCR amplification of these five exons (22). Based on this information, we decided to develop, for the first time, a systematic strategy for screening for heterozygous rearrangements at the F11 locus by quantitative multiplex PCR. This method is inexpensive and robust, and thus, can be performed routinely. Using this strategy, we successfully identified a 7 Mb heterozygous deletion at 4qter.
We postulate that our method of patient selection based on aPTT, which corresponds to a mild phenotype, explains why we mostly found heterozygous carriers (21/23). The exhaustive mutation detection strategy used in this study (for point mutations and rearrangements) failed to identify the mutational status in only one patient among the 25 individuals studied, which is concordant with the results of previous studies (16). Our analyses were focused on the exonic and flanking sequences of the F11 gene, thus missing any nucleotide variation located in intron or promoter sequences or even in other putative genes resulting in FXI deficiency. Some types of mutations, such as large inversions, remain undetected by the techniques used. It can be observed that the FXI levels of patient XXV/44 are only at the threshold for FXI deficiency, but they were confirmed in two samples, and thus, we kept him in the cohort. In the perspective of a routine diagnostic analysis, describing the molecular epidemiology allows to optimize the strategy by targeting the exons carrying most of the mutations : in our population, 50% of the mutated alleles (12/24) are located in only 2 exons (i.e. exons 3 and 15).
The combination of arrayCGH (Figure 1b) and “walking QFM-PCR” was very efficient for delineating the loss of 4q telomeric sequences. However, based on the pauci-symptomatic phenotype of the patient and her children, the discovery of such a large deletion (~7 Mb) was unexpected. In 1999, Tsai & al (23) reported a 4q34.2 telomeric deletion in a child, identified by FISH, associated with velocardiofacial syndrome. Other cases of 4q deletion are described in the literature, with a phenotypic spectrum ranging from asymptomatic (24) to a polymalformative syndrome (25) or mental retardation (26). Based on physical examination, patients from family XXIV (a mother and two children) do not exhibit any cardiac abnormalities, facial dysmorphy or evident mental retardation; their only particular phenotype appears to be a FXI deficiency.
These findings highlight the importance of screening for large heterozygous deletions in molecular studies of F11.
What is known on this topic.
Near 200 mutations have been published in the literature (according to the FXI mutation database V2.2 updated in 2009)
Two large deletions causing FXI deficiency are reported (a 31.5 kb deletion that encompasses the entire F11 gene detected by EBV-transformed cell assay and a smaller deletion that extends from exon 11 to exon 15 detected in a homozygous carrier)
What this paper adds.
We set up an original and robust multiplex technique to routinely screen for heterozygous carriers of large rearrangements. A large heterozygous deletion of the entire F11 gene was detected, that was further delineated using CGH array as a 4q34.2 telomeric deletion of 7 Mb. These findings highlight the importance of screening for large heterozygous deletions in molecular studies of F11
We highlight 9 novel point mutations in our cohort from Western Brittany (France), including a new mutation in the active site (c.1724C>T) classified as CRM+
We propose that recurrent variants, that are rare in the general population, may be considered as tags of a population, or more interestingly, the variants shared between two populations, such as c.166T>C, may indicate that exchanges have occurred between populations
Acknowledgments
We thank V Scotet for her advices in population genetic, F Bridey and LFB for their financial support.
Footnotes
Disclosure of Conflicts of Interest
The authors state that they have no conflict of interest.
References
- 1.Emsley J, McEwan PA, Gailani D. Structure and function of factor XI. Blood. 2010;115:2569–2577. doi: 10.1182/blood-2009-09-199182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McMullen BA, Fujikawa K, Davie EW. Location of the disulfide bonds in human coagulation factor XI: the presence of tandem apple domains. Biochemistry. 1991;30:2056–2060. doi: 10.1021/bi00222a008. [DOI] [PubMed] [Google Scholar]
- 3.van der Graaf F, Greengard JS, Bouma BN, Kerbiriou DM, Griffin JH. Isolation and functional characterization of the active light chain of activated human blood coagulation factor XI. J Biol Chem. 1983;258:9669–9675. [PubMed] [Google Scholar]
- 4.Gomez K, Bolton-Maggs P. Factor XI deficiency. Haemophilia. 2008;14(6):1183–1189. doi: 10.1111/j.1365-2516.2008.01667.x. [DOI] [PubMed] [Google Scholar]
- 5.Saunders RE, Shiltagh N, Gomez K, Mellars G, Cooper C, Perry DJ, et al. Structural analysis of eight novel and 112 previously reported missense mutations in the interactive FXI mutation database reveals new insight on FXI deficiency. Thromb Haemost. 2009;102:287–301. doi: 10.1160/TH09-01-0044. [DOI] [PubMed] [Google Scholar]
- 6.Saunders RE, O’Connell NM, Lee CA, Perry DJ, Perkins SJ. Factor XI deficiency database: an interactive web database of mutations, phenotypes, and structural analysis tools. Hum Mutat. 2005;26:192–198. doi: 10.1002/humu.20214. [DOI] [PubMed] [Google Scholar]
- 7.Asakai R, Davie EW, Chung DW. Organization of the gene for human factor XI. Biochemistry. 1987;26:7221–7228. doi: 10.1021/bi00397a004. [DOI] [PubMed] [Google Scholar]
- 8.Shpilberg O, Peretz H, Zivelin A, Yatuv R, Chetrit A, Kulka T, et al. One of the two common mutations causing factor XI deficiency in Ashkenazi Jews (type II) is also prevalent in Iraqi Jews, who represent the ancient gene pool of Jews. Blood. 1995;85:429–432. [PubMed] [Google Scholar]
- 9.Asakai R, Chung DW, Davie EW, Seligsohn U. Factor XI deficiency in Ashkenazi Jews in Israel. N Engl J Med. 1991;325:153–158. doi: 10.1056/NEJM199107183250303. [DOI] [PubMed] [Google Scholar]
- 10.Hancock JF, Wieland K, Pugh RE, Martinowitz U, Schulman S, Kakkar VV, et al. A molecular genetic study of factor XI deficiency. Blood. 1991;77:1942–1948. [PubMed] [Google Scholar]
- 11.Quélin F, Trossaërt M, Sigaud M, Mazancourt PDE, Fressinaud E. Molecular basis of severe factor XI deficiency in seven families from the west of France. Seven novel mutations, including an ancient Q88X mutation. J Thromb Haemost. 2004;2:71–76. doi: 10.1111/j.1538-7836.2004.00554.x. [DOI] [PubMed] [Google Scholar]
- 12.Zivelin A, Bauduer F, Ducout L, Peretz H, Rosenberg N, Yatuv R, et al. Factor XI deficiency in French Basques is caused predominantly by an ancestral Cys38Arg mutation in the factor XI gene. Blood. 2002;99:2448–2454. doi: 10.1182/blood.v99.7.2448. [DOI] [PubMed] [Google Scholar]
- 13.Mitchell M, Dai L, Savidge G, Alhaq A. An Alu-mediated 31.5-kb deletion as the cause of factor XI deficiency in 2 unrelated patients. Blood. 2004;104:2394–2396. doi: 10.1182/blood-2004-04-1318. [DOI] [PubMed] [Google Scholar]
- 14.Zucker M, Zivelin A, Landau M, Salomon O, Kenet G, Bauduer F, et al. Characterization of seven novel mutations causing factor XI deficiency. Haematologica. 2007;92:1375–1380. doi: 10.3324/haematol.11526. [DOI] [PubMed] [Google Scholar]
- 15.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mitchell M, Mountford R, Butler R, Alhaq A, Dai L, Savidge G, et al. Spectrum of factor XI (F11) mutations in the UK population--116 index cases and 140 mutations. Hum Mutat. 2006;27:829. doi: 10.1002/humu.9439. [DOI] [PubMed] [Google Scholar]
- 17.Le Maréchal C, Masson E, Chen J-M, Morel F, Ruszniewski P, Levy P, et al. Hereditary pancreatitis caused by triplication of the trypsinogen locus. Nat Genet. 2006;38:1372–1374. doi: 10.1038/ng1904. [DOI] [PubMed] [Google Scholar]
- 18.Fard-Esfahani P, Lari GR, Ravanbod S, Mirkhani F, Allahyari M, Rassoulzadegan M, et al. Seven novel point mutations in the F11 gene in Iranian FXI-deficient patients. Haemophilia. 2008;14:91–95. doi: 10.1111/j.1365-2516.2007.01593.x. [DOI] [PubMed] [Google Scholar]
- 19.Seligsohn U. Factor XI deficiency in humans. J Thromb Haemost. 2009;7 (Suppl 1):84–87. doi: 10.1111/j.1538-7836.2009.03395.x. [DOI] [PubMed] [Google Scholar]
- 20.Quélin F, Mathonnet F, Potentini-Esnault C, Trigui N, Peynet J, Bastenaire B, et al. Identification of five novel mutations in the factor XI gene (F11) of patients with factor XI deficiency. Blood Coagul Fibrinolysis. 2006;17:69–73. doi: 10.1097/01.mbc.0000198054.50257.96. [DOI] [PubMed] [Google Scholar]
- 21.Richard C, Pennarun E, Kivisild T, Tambets K, Tolk H-V, Metspalu E, et al. An mtDNA perspective of French genetic variation. Ann Hum Biol. 2007;34:68–79. doi: 10.1080/03014460601076098. [DOI] [PubMed] [Google Scholar]
- 22.Hill M, McLeod F, Franks H, Gordon B, Dolan G. Genetic analysis in FXI deficiency: six novel mutations and the use of a polymerase chain reaction-based test to define a whole gene deletion. Br J Haematol. 2005;129:825–829. doi: 10.1111/j.1365-2141.2005.05536.x. [DOI] [PubMed] [Google Scholar]
- 23.Tsai CH, Van Dyke DL, Feldman GL. Child with velocardiofacial syndrome and del (4) (q34.2): another critical region associated with a velocardiofacial syndrome-like phenotype. Am J Med Genet. 1999;82:336–339. [PubMed] [Google Scholar]
- 24.Tupler R, Berardinelli A, Barbierato L, Frants R, Hewitt JE, Lanzi G, et al. Monosomy of distal 4q does not cause facioscapulohumeral muscular dystrophy. J Med Genet. 1996;33:366–370. doi: 10.1136/jmg.33.5.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kitsiou-Tzeli S, Sismani C, Koumbaris G, Ioannides M, Kanavakis E, Kolialexi A, et al. Distal del(4) (q33) syndrome: detailed clinical presentation and molecular description with array-CGH. Eur J Med Genet. 2008;51:61–67. doi: 10.1016/j.ejmg.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 26.Pickard BS, Hollox EJ, Malloy MP, Porteous DJ, Blackwood DHR, Armour JAL, et al. A 4q35.2 subtelomeric deletion identified in a screen of patients with co-morbid psychiatric illness and mental retardation. BMC Med Genet. 2004;5:21. doi: 10.1186/1471-2350-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]