

Abstract
Hydrogen sulfide, as a novel gaseous mediator, has been suggested to play a key role in atherogenesis. However, the precise mechanisms by which H2S affects atherosclerosis remain unclear. Therefore, the present study aimed to investigate the potential role of H2S in atherosclerosis and the underlying mechanism with respect to chemokines (CCL2, CCL5 and CX3CL1) and chemokine receptors (CCR2, CCR5, and CX3CR1) in macrophages. Mouse macrophage cell line RAW 264.7 or mouse peritoneal macrophages were pre-incubated with saline or NaHS (50 µM, 100 µM, 200 µM), an H2S donor, and then stimulated with interferon-γ (IFN-γ) or lipopolysaccharide (LPS). It was found that NaHS dose-dependently inhibited IFN-γ or LPS-induced CX3CR1 and CX3CL1 expression, as well as CX3CR1-mediated chemotaxis in macrophages. Overexpression of cystathionine γ-lyase (CSE), an enzyme that catalyzes H2S biosynthesis resulted in a significant reduction in CX3CR1 and CX3CL1 expression as well as CX3CR1-mediated chemotaxis in stimulated macrophages. The inhibitory effect of H2S on CX3CR1 and CX3CL1 expression was mediated by modulation of proliferators-activated receptor-γ (PPAR-γ) and NF-κB pathway. Furthermore, male apoE−/− mice were fed a high-fat diet and then randomly given NaHS (1 mg/kg, i.p., daily) or DL-propargylglycine (PAG, 10 mg/kg, i.p., daily). NaHS significantly inhibited aortic CX3CR1 and CX3CL1 expression and impeded aortic plaque development. NaHS had a better anti-atherogenic benefit when it was applied at the early stage of atherosclerosis. However, inhibition of H2S formation by PAG increased aortic CX3CR1 and CX3CL1 expression and exacerbated the extent of atherosclerosis. In addition, H2S had minimal effect on the expression of CCL2, CCL5, CCR2 and CCR5 in vitro and in vivo. In conclusion, these data indicate that H2S hampers the progression of atherosclerosis in fat-fed apoE−/− mice and downregulates CX3CR1 and CX3CL1 expression on macrophages and in lesion plaques.
Introduction
Hydrogen sulfide is commonly considered to be a toxic gas with the smell of rotten eggs. However, it is generated endogenously during cysteine metabolism in a reaction catalyzed by two pyridoxal phosphate-dependent enzymes, cystathionine β-synthase (CBS, EC4.2.1.22) and cystathionine γ-lyase (CSE, EC4.4.1.1) [1]. CSE is the major H2S-producing enzyme in the cardiovascular system, while CBS is the main H2S-forming enzyme in the central nervous system. It has become clear that H2S fulfills a wide range of physiological functions and plays important roles in the pathogenesis of various cardiovascular diseases, such as hypertension, pulmonary hypertension, and myocardial injury [2]–[3]. Furthermore, recent advances in the understanding of the biological importance of endogenous H2S has shed light on the potential role of the gas in atherosclerosis. Wang et al. first reported a direct correlation between endogenous H2S and atherosclerosis in apoE−/− mice [4]. Some studies have suggested that H2S may hinder the development of atherosclerosis by inhibiting vascular smooth muscle cell proliferation, adhesion molecules expression in endothelial cells and foam cell formation [4]–[6]. However, because of the complexity of the atherogenic process, the anti-atherogenic mechanisms of H2S are still far from clear. The present study seeks to investigate whether H2S also reduced the expression of chemokines and their receptors, which have been shown to play a key role in inflammatory conditions and atherosclerotic lesion development [7], [8].
In atherogenesis, chemokines and chemokine receptors may coordinate communication between inflammatory cellular components of the peripheral blood and cellular components of the arterial wall, thereby regulating leukocyte influx, capture, efflux and activation, as well as proliferation and/or apoptosis of reside cells in the plaque [7], [8]. The significance of chemokines (CCL2 [monocyte chemotactic protein-1], CCL5 [RANTES] and CX3CL1 [fractalkine]) and their receptors (CCR2, CCR5 and CX3CR1) in atherosclerosis has been demonstrated in animal models and clinical studies [8]–[10]. For instance, CCL2, CCL5, CX3CL1 and CX3CR1 have been identified in human atherosclerotic plaques [10], [11]. Targeting CCR2-CCL2 axis [12], [13], CCR5-CCL5 axis [14]–[16] or CX3CR1-CX3CL1 [17]–[20] axis with genetic and pharmacologic interventions reduced aortic lesion size, decreased macrophage infiltration and increased plaque stability. Because chemokines and chemokine receptors are important in the development of atherosclerosis, identifying the regulation of their expression by H2S may contribute to a better understanding of the precise mechanisms by which H2S hinders the progression of atherosclerosis.
Accordingly, in the present study, we examined whether H2S could regulate the expression of chemokines (CCL2, CCL5, CX3CL1) and chemokine receptors (CCR2, CCR5, CX3CR1) in vivo and in vitro. We found that H2S inhibited CX3CR1 expression on stimulated macrophages and reduced aortic CX3CR1 expression in fat-fed apoE−/− mice. The reduction in CX3CR1 expression induced by H2S was mediated by peroxisome proliferators-activated receptor-γ pathway. Moreover, H2S downregulated CX3CL1 expression in stimulated macrophages and decreased aortic CX3CL1 expression in vivo by suppressing the activation of nuclear factor (NF-κB). However, H2S had negligible effect on CCL2-CCR2 axis and CCL5-CCR5 axis in vitro and in vivo. Our findings suggest that H2S protects against the formation of atherosclerotic lesions in mice, along with downregulating macrophage CX3CR1 and CX3CL1 expression by a PPAR-γ and NF-κB dependent mechanism.
Results
H2S inhibits CX3CR1 Expression and CX3CR1-mediated Chemotaxis in IFN-γ or LPS-stimulated Macrophages
Using RT-PCR and western blot analysis, we found that NaHS dose-dependently decreased the mRNA and protein expression of CX3CR1 in RAW264.7 cells stimulated with either IFN-γ (500 U/ml) or LPS (10 µg/ml) (Figure 1A and 1B). Similar effects of NaHS on CX3CR1 expression were observed in IFN-γ or LPS stimulated mouse peritoneal macrophages (Figure S1). Furthermore, NaHS inhibited the chemotaxis of stimulated RAW264.7 towards soluble CX3CL1 (50 ng/ml) in a dose-dependent manner (Figure 1C and 1D). Treatment with NaHS at a concentration of 100 µM significantly inhibited the migration of stimulated RAW264.7 cells in response to increasing doses of CX3CL1 (Figure S2).
Figure 1. Effect of NaHS on CX3CR1 expression and CX3CR1-mediated chemotaxis in RAW264.7 cells stimulated with IFN-γ or LPS. RT-PCR analysis for CX3CR1 mRNA (A), western blot analysis for CX3CR1 protein expression (B) and chemotaxis towards CX3CL1 (50 ng/ml) (C, D) were carried out as described in Materials and Methods.

The data are means ± SEM of at least three independent experiments. *P<0.05, compared with unstimulated cells (control). ‡P<0.05, compared with stimulated cell pretreated with NaHS (50 µM). & P<0.05, compared with unstimulated cells pretreated with NaHS at the same concentration. #P<0.05, compared with stimulated cells pretreated with NaHS at a concentration of 50 µM.
Suppression of Macrophage CX3CR1 Expression by H2S is PPAR-γ Dependent
The nuclear receptor PPAR-γ has the inherent capacity to regulate expression of macrophage chemokine receptors (CX3CR1 and CCR2) in lipid-mediated inflammation in atherosclerosis lesions [21], [22]. Therefore, we examined the involvement of PPAR-γ in H2S-induced downregulation of CX3CR1 in stimulated macrophages. It was found that unstimulated RAW264.7 cells exhibited a very low level of DNA binding activity for PPAR-γ, whereas a relatively high DNA binding activity for PPAR-γ was observed in mouse peritoneal macrophages (Figure 2A and 2B). This is consistent with a previous report that the basal expression levels of PPAR-γ vary in different macrophages populations [23]. Because the basal expression levels of PPAR-γ differ between RAW264.7 and peritoneal macrophages, the DNA binding activity of PPAR-γ is diverse in these two types of macrophages after exposure to stimulators. Both IFN-γ and LPS resulted in an obvious reduction in the activation of PPAR-γ in mouse peritoneal macrophages, whereas they only slightly decreased the DNA binding activity of PPAR-γ in RAW264.7 cells (Figure 2A and 2B). Regardless of the disparate activation of PPAR-γ between RAW264.7 and peritoneal macrophages after IFN-γ or LPS exposure, NaHS dose-dependently enhanced the DNA binding activity of PPAR-γ in stimulated macrophages (Figure 2A and 2B).
Figure 2. Effect of NaHS on PPAR-γ activation in IFN-γ or LPS-stimulated macrophages.

(A, B) RAW264.7 cells (A) or mouse peritoneal macrophages (B) were pre-incubated with saline or NaHS (50 µM, 100 µM, 200 µM) for 6 hours and then stimulated with IFN-γ or LPS for 12 hours in the continuous presence of NaHS or saline. The DNA binding activity of PPAR-γ in nuclear extracts was carried out as described in Materials and Methods. (C, D) RAW264.7 cells were incubated with GW9662 (10 µM) or vehicle for 1 hour, further incubated with NaHS (100 µM) or saline for 6 hours and then stimulated with IFN-γ or LPS for 12 hours in the continuous presence of NaHS or saline. Western blot analysis for CX3CR1 expression (C) and chemotaxis towards CX3CL1 (50 ng/ml) (D) were assayed as described in Materials and Methods. The data are means ± SEM of at least three independent experiments. *P<0.05, compared with unstimulated cells (control). #P<0.05, compared with stimulated cells pretreated with saline. **P<0.05, compared with stimulated cell pretreated with NaHS (50 µM). †P<0.05, compared with stimulated cells treated with saline and vehicle. ‡P<0.05, compared with stimulated cell treated with NaHS and vehicle.
To further test whether PPAR-γ signaling was necessary for the inhibitory effect of H2S on CX3CR1 expression in stimulated macrophages, GW9662, a selective PPAR-γ antagonist, was utilized. As shown in Figure 2C, blocking PPAR-γ activity by pretreatment with GW9662 significantly reduced the downregulation of CX3CR1 induced by NaHS in IFN-γ or LPS stimulated RAW264.7 cells. Consequently, NaHS-induced inhibition of CX3CR1-mediated chemotaxis in stimulated RAW264.7 cells was considerably reduced by GW9662 pretreatment (Figure 2D). In addition, GW9662 also abolished NaHS-induced suppression of CX3CR1 expression and CX3CR1-mediated chemotaxis in stimulated peritoneal macrophages (Figure S3). Taken together, these data suggest that H2S may inhibit CX3CR1 expression and CX3CR1-mediated chemotaxis in stimulated macrophages in a PPAR-γ-dependent mechanism.
Overexpression of CSE Inhibits IFN-γ or LPS-induced CX3CR1 Expression by a PPAR-γ Dependent Mechanism
Overexpression of CSE was verified by western blot analysis and an H2S synthesis activity assay (Figure S4). As shown in Figure 3A, overexpression of CSE significantly inhibited IFN-γ or LPS-induced CX3CR1 expression in RAW264.7 cells. Furthermore, CSE overexpression significantly enhanced the activation of PPAR-γ in IFN-γ or LPS stimulated RAW264.7 cells (Figure 3B), while mock transfection had a negligible effect on CX3CR1 expression and the nuclear activity of PPAR-γ (Figure 3A and 3B). In agreement with this finding, CSE transfected cells exhibited reduced chemotactic response to CX3CL1 compared with mock-transfected cells after IFN-γ or LPS exposure (Figure 3D). In addition, blockage of PPAR-γ activation by GW9662 distinctly reduced the inhibition of CX3CR1 expression as well as the chemotactic response to CX3CL1 induced by CSE overexpression in stimulated RAW264.7 cells (Figure 3C and 3D). Taken together, these data suggest that CSE overexpression may downregulate CX3CR1 expression and CX3CR1-mediated chemotaxis in stimulated macrophages by activating the PPAR-γ pathway.
Figure 3. Effect of CSE overexpression on CX3CR1 expression and PPAR-γ activation in stimulated RAW264.7 cells.

Cells were transfected with CSE cDNA construct or empty vector and then were stimulated with IFN-γ (500 U/ml) or LPS (10 µg/ml) for 12 hours. Some transfected cells were pre-incubated with GW9962 (10 µM) for 1 hour before addition of IFN-γ or LPS. Western blot analysis for CX3CR1 expression (A, C), PPAR-γ activation (B) and CX3CR1-mediated chemotaxis towards CX3CL1 (50 ng/ml) (D) were assayed as described in Materials and methods. The data are means ± SEM of at least three independent experiments. *P<0.05, compared with unstimulated cells transfected with CSE construct or empty vector. †P<0.05, compared with stimulated cells transfected with empty vector. #P<0.05, compared with mock-transfected and unstimulated cells treated with vehicle. **P<0.05, compared with mock-transfected and stimulated cells treated with vehicle. ‡P<0.05, compared with CSE-transfected and stimulated cells treated with vehicle.
H2S Inhibits IFN-γ or LPS-induced CX3CL1 Expression in Stimulated Macrophages by a NF-κB Dependent Mechanism
RAW264.7 cell lysates were analyzed for changes in CX3CL1 mRNA and protein expression (Figure 4A and 4B). Incubation with IFN-γ or LPS significantly increased the concentration of CX3CL1 in RAW264.7 lysates. Preincubation with NaHS decreased lysate CX3CL1 mRNA and protein levels in a concentration dependent manner (Figure 4A and 4B). Stimulation with IFN-γ or LPS also significantly increased the CX3CL1 level in RAW264.7 cell culture media (Table S1). This increase was remarkably reduced by NaHS pretreatment in a dose-dependent manner (Table S1). Similarly, NaHS at a concentration of 100 µM significantly reduced lysate CX3CL1 mRNA and protein levels in mouse peritoneal macrophages stimulate with IFN-γ or LPS (Table S2). It also decreased the CX3CL1 concentration in culture media of stimulated peritoneal macrophages (Table S2). Furthermore, in agreement with these observations, CSE overexpression not only significantly inhibited lysate CX3CL1 mRNA and protein expression but also reduced the CX3CL1 level in culture media of RAW264.7 cells simulated with IFN-γ or LPS (Table S3).
Figure 4. Effect of NaHS on CX3CL1 expression, IκBα content and p65 DNA binding activity in RAW264.7 cells stimulated with IFN-γ or LPS.

RT-PCR analysis for CX3CL1 mRNA (A), ELISA for lysate CX3CL1 protein level (B), western blot analysis for IκBα content (C) and ELISA for p65 DNA binding activity in nuclear extracts (D) were carried out as described in Materials and Methods. The data are means ± SEM of at least three independent experiments. *P<0.05, compared with unstimulated cells (control). ‡P<0.05, compared with stimulated cell pretreated with saline.
CX3CL1 expression is regulated by the transcriptional activity of NF-κB [24]. Therefore, we examined the involvement of NF-κB in H2S-induced downregulation of CX3CL1 in stimulated macrophages. As shown in Figure 4C and Figure 4D, IFN-γ or LPS significantly decreased the IκBα content in RAW264.7 cells and increased the DNA binding activity of NF-κB in nuclear extracts. The nuclear activation of NF-κB induced by IFN-γ or LPS was remarkably suppressed by NaHS in a dose-dependent manner, as characterized by a significant elevation in IκBα content and an obvious reduction in nuclear NF-κB activity (Figure 4C and Figure 4D). Consistent with the findings observed in RAW264.7 cells, NaHS also inhibited nuclear activation of NF-κB in IFN-γ or LPS-stimulated mouse peritoneal macrophages (Figure S5A and Table S2). Moreover, CSE overexpression increased IκBα content and inhibited DNA binding activity of NF-κB in stimulated RAW264.7 cells (Figure S5B and Table S3).
In addition, IFN-γ or LPS significantly upregulated CCR2 and CCR5 mRNA expression and increased lysate CCL2 and CCL5 level in RAW264.7 cells (Figure S6 and Table S4). However, NaHS had negligible effect on CCR2, CCR5, CLL2 and CCL5 expression in stimulated RAW264.7 cells (Figure S6 and Table S4).
Changes Over Time of Endogenous H2S Synthesis During the Development of Atherosclerosis
In an attempt to assess the potential role of H2S in atherosclerosis, we observed the time-dependent alterations of H2S bio-synthesis in the aorta during the development of atherosclerosis. As shown in Figure 5A, the plasma H2S concentration decreased in a time-dependent fashion during the development of atherosclerosis, with a significant reduction after 8-week fat feeding. We also utilized sulfur-sensitive electrode assay to measure the plasma H2S level and similar time-dependent changes were obtained (Figure S7). Furthermore, H2S synthesizing activity and CSE expression in the aorta gradually reduced in a time-dependent manner (Figure 5B and 5C). These data indicate that a deficiency of endogenous H2S formation in the aorta accompanied the development and progression of atherosclerosis. The insufficiency of endogenous H2S in atherosclerosis may have been due to the downregulation of CSE expression and reduced CSE activity in the aorta.
Figure 5. Alterations in H2S biosynthesis during the development of atherosclerosis in fat-fed apoE−/− mice.

Plasma H2S level (A), H2S synthesizing activity (B) and CSE expression in aorta (C) were assayed at indicated time points (0, 4, 8, 12, 24 weeks after fat feeding). Results shown are the mean ± SEM (n = 6 animals in each group). *P<0.05, compared with the basal level at 0 weeks. **P<0.01, compared with the basal level at 0 weeks. †P<0.05, compared with mice sacrificed 4 weeks after fat feeding. ‡P<0.01, compared with mice sacrificed 4 weeks after fat feeding.
Effect of H2S on the Development of Atherosclerosis
NaHS, an H2S donor, was given to mice during the early stage of atherosclerosis (4 weeks after fat feeding, early NaHS treatment). 24 weeks after fat feeding, mice were sacrificed and the extent of atherosclerotic lesions was assessed by ultrasound biomicroscopy (UBM) and histological analysis. UBM assessment showed that early NaHS treatment significantly impeded plaque development in the aorta, as characterized by a significant decrease in the brachiocephalic artery (BCA) plaque size, common carotid artery (CCA) intima-media complex thickness (IMT) and the aortic arch IMT (Figure 6A, Figure 6B and table 1). It was also verified by histological analysis that early NaHS treatment significantly reduced the plaque area and lipid core in BCA (Figure 6C and table 2). On the other hand, we delayed the commencement of NaHS treatment until the plaques were fully established as identified by UBM (12 weeks after fat feeding, delayed NaHS treatment). We found that delayed NaHS treatment significantly inhibited the progression of atherosclerosis (Figure 6, Table 1 and Table 2). However, when compared to delayed NaHS treatment, early NaHS treatment induced a greater reduction in plaque size and IMT. All of these findings suggest that H2S may have an anti-atherogenic benefit and that this effect is more potent when applied during the early stage of atherosclerosis.
Figure 6. Effect of NaHS on the development of atherosclerosis in fat-fed apoE−/− mice.
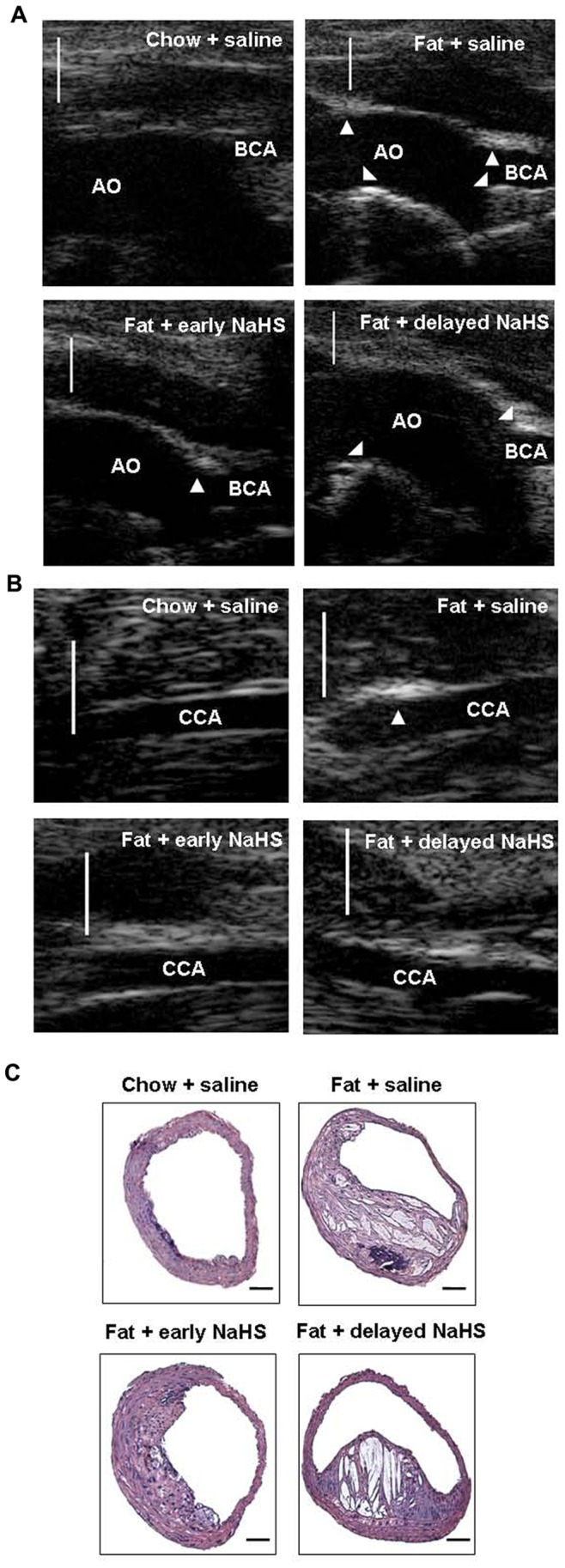
(A) Representative UBM images were taken from the aortic arch (AO) and BCA in mice with chow and saline, mice with fat feeding and saline, mice with fat feeding and early NaHS treatment or mice with fat feeding and delayed NaHS treatment. (B) Representative UBM images were taken from CCA in mice with chow and saline, mice with fat feeding and saline, mice with fat feeding and early NaHS treatment or mice with fat feeding and delayed NaHS treatment. Scale bar for UBM = 1 mm. Atherosclerotic plaques are identified by arrows. (C) Representative H&E staining images were taken from BCA in mice with chow and saline, mice with fat feeding and saline, mice with fat feeding and early NaHS treatment or mice with fat feeding and delayed NaHS treatment. Scale bar for histological images = 100 µm.
Table 1. Effect of treatment with NaHS on plaque development in aortic arch, brachiocephalic artery (BCA) and common carotid artery (CCA) by ultrasound biomicroscopy (UBM) technology.
| Aortic arch IMT | CCA IMT | Plaque area in BCA | |
| (mm) | (mm) | (mm2) | |
| chow + saline | 0.0537±0.0069 | 0.0415±0.0051 | 0.0511±0.0064 |
| Fat + saline | 0.1450±0.0076* | 0.1213±0.0115* | 0.1660±0.0139* |
| Fat + early NaHS treatment | 0.0879±0.0065** | 0.0621±0.0062** # | 0.0816±0.0137** # |
| Fat + delayed NaHS treatment | 0.0969±0.0074** | 0.0930±0.0044** | 0.1226±0.0064** |
P<0.05, vs. chow + saline group;
P<0.05, vs. fat + saline group;
P<0.05, vs. fat + delayed NaHS treatment group.
Table 2. Effect of treatment with NaHS on plaque development in brachiocephalic artery (BCA) by histology assessment.
| Plaque area (mm2) | Lipid core (%) | |
| chow + saline | 0.0358±0.0051 | 9.53±1.28 |
| Fat + saline | 0.1480±0.0152* | 39.63±4.25* |
| Fat + early NaHS treatment | 0.0641±0.0107** # | 15.81±1.87** # |
| Fat + delayed NaHS treatment | 0.1013±0.0052** | 25.72±1.96** |
P<0.05, vs. chow + saline group;
P<0.05, vs. fat + saline group;
P<0.05, vs. fat + delayed NaHS treatment group.
Moreover, administration with PAG significantly depleted endogenous H2S in fat –fed apoE−/− mice (plasma H2S concentration in fat+saline and fat+PAG group: 16.43±2.43 µmol/L vs. 5.32±0.87 µmol/L, P<0.05). As shown in Figure 7 and table 3, PAG treatment significantly exacerbated atherosclerosis in aorta, as characterized by a significant increase in the BCA plaque size, CCA IMT and aortic arch IMT assessed by UBM, as well as an obvious rise in BCA plaque area and lipid core evaluated by histology analysis.
Figure 7. Effect of PAG on the development of atherosclerosis in fat-fed apoE−/− mice.

(A) Representative UBM images were taken from the aortic arch (AO) and BCA in mice with fat feeding and saline, mice with fat feeding and PAG treatment. (B) Representative UBM images were taken from CCA in mice with fat feeding and saline, mice with fat feeding and PAG treatment. Scale bar for UBM = 1 mm. Atherosclerotic plaques are identified by arrows. (C) Representative H&E staining images were taken from BCA in mice with fat feeding and saline, mice with fat feeding and PAG treatment. Scale bar for histological images = 100 µm.
Table 3. Effect of PAG on plaque development in aorta by UBM and histology assessment.
| UBM | Histology | ||||
| Aortic arch IMT | CCA IMT | Plaque area in BCA | Plaque area | Lipid core | |
| (mm) | (mm) | (mm2) | (mm2) | (%) | |
| Fat + saline | 0.1039±0.0087 | 0.0946±0.0060 | 0.1164±0.0086 | 0.1073±0.0092 | 28.65±3.06 |
| Fat + PAG | 0.1343±0.0092* | 0.1236±0.0074* | 0.1470±0.0087* | 0.1368±0.0099* | 37.48±2.02* |
P<0.05, vs. fat + saline group;
In addition, treatment with NaHS or PAG did not affect body weight or the concentrations of plasma lipids but resulted in a slight reduction in blood pressure (Table S5 and S6).
Effect of H2S on CX3CR1 and CX3CL1 Expression in vivo
Immunohistochemistry revealed that CX3CR1 was widely expressed in macrophages and foam cells in atherosclerotic plaques (Figure 8A). Both early and delayed treatment with NaHS suppressed the expression of CX3CR1 in atherosclerotic plaques (Figure 8). Early NaHS treatment induced a greater reduction in the CX3CR1 expression in atherosclerotic plaques compared with delayed NaHS treatment (Figure 8). However, PAG treatment upregulated CX3CR1 expression in plaques (Figure 9). Furthermore, as shown in Figure 10 and 11, CX3CR1 staining predominantly colocalized in macrophages within plaques, suggesting that alterations in CX3CR1 expression in plaque lesions are mainly caused by macrophages and that H2S may directly act on plaque macrophages to lower CX3CR1 expression in vivo (Figure 8S, negative control for immunofluorescent staining).
Figure 8. Effect of NaHS on the expression of CX3CR1 in atherosclerotic plaques in fat-fed apoE−/− mice.

(A) Representative immunohistochemical images were taken from BCA in mice with chow and saline, mice with fat feeding and saline, mice with fat feeding and early NaHS treatment or mice with fat feeding and delayed NaHS treatment. Scale bar for histological images = 100 µm. (B) BCA sections from mice with chow and saline, mice with fat feeding and saline, mice with fat feeding and early NaHS treatment or mice with fat feeding and delayed NaHS treatment, were quantified immunohistochemically for CX3CR1 positive staining. Results shown are the mean ± SEM (n = 6–8 animals in each group). *P<0.05, compared with mice with chow and saline; **P<0.05, compared with mice with fat feeding and saline.
Figure 9. Effect of PAG on the expression of CX3CR1 in atherosclerotic plaques in fat-fed apoE−/− mice.

(A) Representative immunohistochemical images were taken from BCA in mice with fat feeding and saline, mice with fat feeding and PAG treatment. Scale bar for histological images = 100 µm. (B) BCA sections from mice with fat feeding and saline and mice with fat feeding and PAG treatment were quantified immunohistochemically for CX3CR1 positive staining. Results shown are the mean ± SEM (n = 6–8 animals in each group). *P<0.05, compared with mice with fat feeding and saline.
Figure 10. Effect of NaHS on the expression of CX3CR1 in macrophages in atherosclerotic plaques.

(A) Representative immunofluorescent stainging images for CX3CR1 and F4/80 colocalization were taken from BCA in mice with chow and saline, mice with fat feeding and saline, mice with fat feeding and early NaHS treatment or mice with fat feeding and delayed NaHS treatment. Scale bar for histological images = 20 µm. (B) BCA sections from mice with chow and saline, mice with fat feeding and saline, mice with fat feeding and early NaHS treatment or mice with fat feeding and delayed NaHS treatment, were quantified immunohistochemically for CX3CR1 positive staining. Results shown are the mean ± SEM (n = 6–8 animals in each group). *P<0.05, compared with mice with chow and saline; **P<0.05, compared with mice with fat feeding and saline.
Figure 11. Effect of PAG on the expression of CX3CR1 in macrophage in atherosclerotic plaques.

(A) Representative immunofluorescent staining images for CX3CR1 and F4/80 colocalization were taken from BCA in mice with fat feeding and saline, mice with fat feeding and PAG treatment. Scale bar for histological images = 20 µm. (B) BCA sections from mice with fat feeding and saline and mice with fat feeding and PAG treatment, were quantified immunohistochemically for CX3CR1 positive staining. Results shown are the mean ± SEM (n = 6–8 animals in each group). *P<0.05, compared with mice with fat feeding and saline.
Moreover, both early and delayed NaHS inhibited aortic CX3CL1 protein expression in fat-fed apoE−/− mice (Table 4). Early NaHS resulted in a greater reduction in aortic CX3CL1 expression in comparison to delayed NaHS treatment (Table 4). However, PAG significantly elevated aortic CX3CL1 expression (Table 5). Neither NaHS nor PAG had effect on plasma CX3CL1 level (Table 4 and 5).
Table 4. Effect of NaHS on CX3CL1 in vivo.
| Plasma CX3CL1 (ng/ml) | Aortic CX3CL1 (ng/ml) | |
| Aortic protein level (mg/ml) | ||
| chow + saline | 4.38±1.03 | 1.36±0.18 |
| Fat + saline | 10.32±1.08* | 2.82±0.31* |
| Fat + early NaHS treatment | 11.29±1.04 | 1.60±0.12**# |
| Fat + delayed NaHS treatment | 9.45±0.96 | 2.15±0.13** |
P<0.05, vs. chow + saline group;
P<0.05, vs. fat + saline group;
P<0.05, vs. fat+delayed NaHS treatment group.
Table 5. Effect of PAG on CX3CL1 in vivo.
| Plasma CX3CL1(ng/ml) | Aortic CX3CL1(ng/ml) | |
| Aortic protein level (mg/ml) | ||
| Fat + saline | 8.73±0.91 | 2.06±0.17 |
| Fat + PAG | 9.69±1.01 | 1.43±0.13* |
P<0.05, vs. fat + saline group.
In addition, we used flow cytometry to examine the effect of H2S on CX3CR1 expression on circulating mouse monocytes. Treatment with NaHS or PAG in fat-fed apoE−/− mice had negligible effect on CX3CR1 expression on CD11b+ mononuclear cells (Figure S9A and S9B). As a result, H2S did not impact CX3CR1-mediated chemotaxis of mononuclear cells isolated from peripheral blood of hyperlipidemic mice (Figure S9C).
Effect of H2S on CCR2, CCR5, CCL2 and CCL5 Expression in vivo
As shown in Table S7 and Table S8, both NaHS and PAG had minor effect on aortic CCR2, CCR5 mRNA expression and plasma CCL2 and CCL5 concentration in fat-fed apoE−/− mice.
Discussion
We have shown here that H2S synthesis was decreased in a time-dependent manner during the development of atherosclerosis. In fat-fed apoE−/− mice, H2S synthesis activity and CSE expression in the aorta gradually decreased and remained extremely low during the late stage of atherosclerosis. As a result, plasma H2S concentration began to decline as early as four weeks after high-fat feeding, followed by a significant decrease 8 weeks after fat feeding, and thereafter remained at a low level. The disturbance of the CSE/H2S pathway in atherosclerosis was also reported in other studies. Wang et al. first found that the plasma H2S level and H2S production rate in the aorta were significantly decreased in apoE−/− mice [4]. CSE expression and H2S production were reduced during the development of balloon injury-induced neointimal hyperplasia [6]. Taken together, these findings demonstrate that endogenous H2S deficiency in the aorta may accompany the pathogenesis of atherosclerosis. These data provide a solid basis for the use of H2S donor drugs in the treatment of atherosclerosis. Although the present study is not the first to address the direct correlation between H2S and atherosclerosis, our observation on the dynamic alterations of H2S metabolism during atherosclerosis may be helpful to identify the right time point for starting preventive or therapeutic interventions for atherosclerosis.
The critical role of H2S in the pathogenesis of atherosclerosis is further highlighted by experiments in which insufficiency of endogenous H2S formation was manipulated by NaHS, an H2S donor. NaHS treatment was given to mice either at the early stage of atherosclerosis or was delayed until the plaques were fully established. Aortic plaque burden was evaluated by both UBM technology and histological analysis. It was found that NaHS significantly reduced plaque size and IMT in the aortic arch and the main branches of the aortic arch. The anti-atherogenic effect of NaHS was more potent when the treatment was commenced at the early stage of atherosclerosis rather than at the advanced stage. Furthermore, depletion of endogenous H2S formation by PAG treatment significantly aggravated atherosclerotic lesions in aorta. These interesting observations reinforce the view that H2S is involved in the initiation and progression of atherosclerosis. They also clearly suggest that prevention of atherosclerosis is correlated with a supplement of exogenous H2S and that H2S may exert a more prominent anti-atherosclerotic effect when it is applied during the initial stage of lesion formation.
H2S has been shown to be endogenously generated in monocytes and macrophages and in turn plays an important role in regulating the biological functions of these cells [25], [26]. H2S inhibited LPS-induced NO production and alpha-tumor necrosis factor (TNF-α) secretion in microglia, a specialized form of macrophages residing in central nervous systems [27]. Zhi et al. reported that H2S stimulated the activation of human monocytes with the generation of cytokines [25]. However, to date, the effect of H2S on macrophage recruitment and hemostasis in atherosclerotic lesions has not been determined. Recently, chemokines (CCL2, CCL5 and CX3CL1) and chemokine receptors (CCR2, CCR5 and CX3CR1) have been strongly implicated to play critical roles in mediating inflammation, macrophage recruitment and atherosclerosis progression by multiple lines of evidence, including expression, functional and epidemiologic data [8]–[20]. Therefore, we performed a series of in vitro experiments to investigate the role of H2S in regulating CCL2-CCR2, CCL5-CCR5 and CX3CL1-CX3CL1 expression in RAW264.7 cells and mouse peritoeal macrophages. Activation of macrophages was achieved by classic stimulators, IFN-γ and LPS. Cell activation resulted in enhanced CX3CL1 and CX3CR1 expression and chemotaxis towards CX3CL1. Supplement of exogenous H2S dose-dependently reduced the LPS or IFN-γ induced-upregulation of CX3CL1 and CX3CR1 in macrophages. Similarly, overproduction of endogenous H2S via CSE overexpression also inhibited CX3CL and CX3CR1 expression in stimulated macrophages. As a result, either exogenous applied H2S or overexpression of CSE reduced macrophage migration in response to CX3CL1 after stimulation with IFN-γ or LPS. Collectively, our findings demonstrate that H2S plays a crucial role in regulating CX3CL1 and CX3CR1 expression and CX3CR1-mediated chemotaxis in stimulated macrophages.
To further demonstrate the involvement of H2S in regulating CX3CL1 and CX3CR1 expression during atherosclerosis and to establish its pathophysiological relevance in vivo, we examined the effect of H2S on CX3CL1 and CX3CR1 expression in fat-fed apoE−/− mice. Administration of NaHS at either the early or the advanced stage of atherosclerosis suppressed the aortic expression of CX3CL1 and CX3CR1, together with reduced plaque size and IMT thickness in the main branches of the aortic arch. In contrast, inhibition of endogenous H2S formation upregulated aortic expression of CX3CL1 and CX3CR1, thus exacerbating atherosclerosis in aorta. Furthermore, immunofluorescent staining revealed that CX3CR1 colocalized with macrophages, suggesting that the reduction in plaque CX3CR1 expression is predominantly by macrophages and that other cell types are likely to play insignificant roles. These findings indicated that H2S may directly act on plaque macrophages and inhibit CX3CR1 expression in vivo. These results are consistent with the findings of in vitro studies with mouse macrophage cell line or isolated mouse peritoneal macrophages. However, flow cytometry analysis found that injection of NaHS or PAG did not alter CX3CR1 expression on circulating monocytes isolated from fat-fed aopE−/− mice. This lack of change may be due to the short half-life (approximately 6 hours) of circulating monocytes, such that their exposure time to H2S is inadequate to inhibit CX3CR1 expression. This result also supports the concept that H2S induced reduction in CX3CR1 expression occurs in macrophages within plaques but not in the circulating monocytes. A decline in CX3CR1 expression in macrophages at the site of lesions may be beneficial, eg suppressing the recruitment and retention of macrophages in blood vessel walls and thereby retarding the development and progression of atherosclerosis.
The present study found that H2S inhibited CX3CL1 expression in stimulated macrophages and in aorta tissues. However, it had no effect on plasma CX3CL1 level in vivo. It may be attributed to the fact that CX3CL1 is predominantly a membrane-bond chemokine. Furthermore, H2S did not affect CCL2-CCR2 and CCL5-CCR5 expression both in vitro and in vivo. These observations support the notion that CCL2-CCR2, CCL5-CCR5 and CX3CL1-CX3CR1 axis impact the pathogenesis of atherosclerosis independently.
Recent studies have shown that PPARγ has the inherent capacity to regulate expression of macrophage chemokine receptors CX3CR1 in the lipid-mediated inflammation process of atherosclerosis lesions [21], [22]. Wan et al. identified a direct link between PPARγ and the CX3CL1/CX3CR1 signaling [28]. Activation of PPARγ not only downregulated CX3CR1 gene transcription but also prevented plasma membrane translocation of CX3CR1 protein [28]. Therefore, we investigated the underlying mechanism by which H2S regulates CX3CR1 expression and CX3CR1-mediated chemotaxis in macrophages with respect to the PPARγ pathway. Here, we showed that H2S-induced inhibition of CX3CR1 expression in stimulated macrophages is dependent on PPAR-γ activation. Support for this concept comes from the observation that NaHS dose-dependently increased the DNA binding activity of PPAR-γ in RAW264.7 cells and mouse peritoneal macrophages stimulated with IFN-γ or LPS. CSE overexpression also enhanced the activation of PPAR-γ in IFN-γ or LPS stimulated RAW264.7 cells. Moreover, our notion regarding the involvement of PPAR-γ was further strengthened by the finding that blockage of PPAR-γ activation by GW9662 markedly reduced the inhibition of CX3CR1 expression induced by NaHS or CSE overexpression in stimulated macrophages. Consequently, GW9662 eliminated exogenous or endogenous H2S-induced inhibition of CX3CR1-mediated chemotaxis in activated macrophages. Together, these results indicate that PPAR-γ is possibly involved in the signaling transduction of H2S-induced inhibition of CX3CR1 expression in macrophages.
H2S is a highly reactive molecule that readily reacts with free reactive oxygen species (ROS) and reactive nitrogen species (RNS) [29]–[31]. All these compounds react rapidly to form peroxynitrite, which then attacks tyrosine residues in proteins to form nitrotyrosine. Nitration of PPAR-γ following inflammatory stimulation in RAW 264.7 cells inhibited its ligand-dependent translocation into the nucleus [32]. These findings raise the possibility that H2S may suppress the nitration of PPAR-γ by directly scavenging ROS and/or RNS, thus provoking the activation of PPAR-γ and downregulating the expression of CX3CR1 in macrophages. However, the precise mechanism by which H2S regulates the activation of PPAR-γ in stimulated macrophages is still far from clear. Further investigations are required to address this and to resolve the detailed structural basis for PPAR-γ induction of CX3CR1 expression.
CX3CL1 expression has been shown to be modulated by NF-κB activation [24]. Therefore, we investigated the involvement of NF-κB pathway in regulating CX3CL1 expression by H2S. The present study found that H2S inhibited IκB degradation, the key step to NF-κB nuclear translocation and activation, and decreased the DNA binding activity of NF-κB p65 in macrophage nuclear extracts. This suggests that H2S may downregulate CX3CL1 expression in macrophages by suppressing the activation of NF-κB pathway.
Finally, it is to be noted that our data are still insufficient to establish a clear causal link between the inhibitory effect of H2S on athoerslcerosis and macrophage CX3CR1-CX3CL1 expression. Further work is needed most likely in CX3CR1/CX3CL1 gene modified mice to probe the exact linkage involved. In addition to its effect on the macrophage CX3CR1-CX3CL1 expression, H2S has been shown to impede the pathogenesis of atherosclerosis in various ways [33]. For instance, H2S plays a role in regulating vascular smooth muscle cell (VSMC) proliferation and apoptosis. It also inhibits atherogenic modification of low-density lipoprotein (LDL), interferes with vascular calcification and is involved in platelet function.
In summary, the present study suggests that deficiency of endogenous H2S formation in the aorta accompanies the development and progression of atherosclerosis. H2S may have an anti-atherogenic benefit and this effect seems to be more potent when it is applied during the early stage of atherosclerosis. H2S may prevent the progression of atherosclerosis, along with downregulating macrophage CX3CR1 and CX3CL1 expression by a PPAR-γ and NF-κB dependent mechanism. Our study therefore provides evidence that H2S donors may be a possible new therapeutic strategy to counteract the development of atherosclerosis or other immune diseases.
Materials and Methods
Cell Culture
RAW 267.4 murine macrophage cells were obtained from ATCC. Cells were grown in Dubbelco’s Modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA, USA), 100 units/ml penicillin, and 100 mg/ml streptomycin and plated on 6-well plates or 100 mm tissue culture dishes 24 h before experiments. Near-confluent cultures were switched to medium containing 0.5% FBS for 24 h before NaHS treatment or stimulation with LPS or IFN-γ. No cells older than passage 10 were used, because RAW cells showed highly variable responses at higher passage numbers.
Macrophage Isolation
Peritoneal macrophages were isolated from 10-week, male C57BL/6J mice. A 3% thioglycolate solution (Sigma-Aldrich, St Louis, MO, USA) was injected intraperitoneally and, 4 days later, peritoneal macrophages were harvested through phosphate-buffered saline lavage. Ten milliliters of ice-cold phosphate-buffered saline was injected into the mouse peritoneal cavity and gently removed. Once collected, cells were seeded in RPMI medium 1640 supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin/streptomycin for 4 h and then rinsed and maintained in RPMI medium 1640 supplemented with 0.5% FBS for 24 h before NaHS treatment or stimulation with LPS or IFN-γ.
Cell Treatment
Cells were rinsed twice with serum-free culture medium before treatment and all treatments were carried out in serum-free culture medium containing penicillin and streptomycin. Cells were pre-incubated with saline or NaHS (50, 100 and 200 µM) for 6 hours and then stimulated with IFN-γ (500 U/ml) or LPS (10 µg/ml) for 12 hours in the continuous presence of NaHS. For experiments using inhibitors, cells were pre-incubated with GW9662 (10 µM) for 1 hour before NaHS addition.
Animal Experiments
The study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the Shanghai JiaoTong University School of Medicine. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Shanghai JiaoTong University School of Medicine (Permit Number: [2011]-23). C57BL/6J male apoE−/− mice were purchased from the Animal Center of the Beijing University, Beijing, China. At the age of 8 weeks, feeding of animals on a high-fat diet began that contained 21% fat from lard and was supplemented with 0.15% (w/w) cholesterol. At indicated time points (0, 4, 8, 12, 24 weeks after fat feeding), mice were sacrificed by an intraperitoneal (i.p.) injection of a lethal dose of pentobarbitone. To investigate the role of H2S in atherosclerosis, different drug administration schedules were used. In the early-NaHS treatment study, thirty mice began to receive NaHS (1 mg/kg, i.p., daily) 4 weeks after fat feeding and were sacrificed 24 weeks after fat feeding. In the delayed-NaHS treatment study, thirty mice began to receive NaHS (1 mg/kg, i.p., daily) 12 weeks after fat feeding and were sacrificed 24 weeks after fat feeding. In DL-propargylglycine (PAG) treatment study, eighteen mice began to receive PAG (10 mg/kg, i.p., daily) 4 weeks after fat feeding and were sacrificed 12 weeks after fat feeding. Samples of aortas and plasma were harvested and stored at –80°C for subsequent measurement.
Systolic blood pressure (SBP) was measured at the beginning and the end of the studies by using a non-invasive computerized tail cuff system (Blood Pressure Analysis System BP- 98AW monitor, Softron Co. Ltd., Tokyo, Japan). A series of readings was taken and an average of 3–5 BP readings was used. Blood samples were drawn from the left ventricle of mice at the end of the studies and plasma lipid concentrations were determined in heparinized plasma.
Cloning of CSE cDNA and Transient Transfection
PCR was used to amplify the open reading frame of CSE (GenBankTM accession number: NM_145953) from C57BL/6J mouse liver using the primers 5′-CAT GCA GAA GGA CGC CTC TTT GA-3′ and 5′-TTA AGG GTG CGC TGC CTT CAA AGC -3′. The amplified open reading frame of CSE was subcloned into the TA cloning vector (PCR®2.1-TOPO, Invitrogen). A positive clone containing CSE open reading frame insert was sequenced to confirm the accuracy of the inserted CSE sequence. The constructs containing CSE cDNA were cleaved and subcloned into the mammalian expression vector pcDNA3.1 (Invitrogen). The CSE expression vectors or the control vectors (pcDNA3.1 empty vectors) were transiently transfected into RAW264.7 cells using FuGENE HD Transfection Reagent (6 µl:2 µg reagent to plasmid DNA ratio, Roche, Switzerland), according to manufacturer’s instruction.
RT-PCR
Total RNA from cells or tissues was extracted with Trizol ® reagent (Invitrogen) according to the manufacturer’s protocol. The concentration of isolated nucleic acids was determined spectrophotometrically by measuring the absorbance at 260 nm and the integrity was verified by ethidium bromide staining of 18 S and 28 S rRNA bands on a denaturing agarose gel. All samples were thereafter stored at −80°C until required. 1 µg of RNA was reversely transcribed using an iScript™ cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA, USA) at 25°C for 5 minutes, 42°C for 30 minutes, followed by 85°C for 5 minutes. The cDNA was used as a template for PCR amplification by iQ™ Supermix (Bio-Rad Laboratories). The primer sequences, optimal annealing temperature, optimal cycles and product sizes are as shown in Table 6. PCR amplification was carried out in a MyCycler™ system (Bio-Rad Laboratories). The reaction mixture was first subjected to 95°C for 3 minutes, followed by an optimal cycle of amplifications, consisting of 95°C for 50 s, optimal annealing temperature for 45 s and 72°C for 1 minute. PCR products were analyzed on 1.5 % w/v agarose gels containing 0.5 µg/ml ethidium bromide.
Table 6. PCR primer sequences, optimal conditions and product sizes.
| Gene | Primer sequence | Optimal conditions | Size |
| CSE | Forward:5′-GAC CTC AAT AGT CGG CTT CGT TTC-3′Reverse:5′-CAG TTC TGC GTA TGC TCC GTA ATG-3′ | 34cyclesAnnealing: 61°C | 618 bp |
| CX3CR1 | Forward:5′-CCT GCC TCT GAG AAA TGG AG -3′Reverse: 5′- ATC TCT CCA GCC CCT GAA AT -3′ | 36 cyclesAnnealing: 58°C | 332 bp |
| CX3CL1 | Forward:5′-ATG ACC TCA CGA ATC CCA GTG -3Reverse: 5′-CCG CCT CAA AAC TTC CAA TGC -3′ | 36 cyclesAnnealing: 60°C | 453 |
| CCR2 | Forward: 5′-AGAGAGCTGCAGCAAAAAGG-3′Reverse: 5′-GGAAAGAGGCAGTTGCAAAG-3 | 35 cyclesAnnealing: 56°C | 185 bp |
| CCR5 | Forward: 5′-CGAAAACACATGGTCAAACG-3′Reverse: 5′- CATGGCACCTGCCTCAAGTCTC-3′ | 36 cyclesAnnealing: 52°C | 364 bp |
| GAPDH | Forward: 5′-GCA CAG TCA AGG CCG AGA AT -3′Reverse: 5′-GCC TTC TCC ATG GTG GTG AA -3′ | 22cyclesAnnealing: 54°C | 151 bp |
Quantification of Chemokine Concentrations by ELISA
CCL2, CCL5 and CX3CL1 protein levels were assayed in aorta and cell homogenates as well as mouse plasma and culture media using commercially available ELISA kits (Quantikine, R&D systems).
Western Immunoblot
Cultured cells (3×106) were harvested and lysed at 4°C in radioimmunoprecipitation assay lysis buffer. Aorta was homogenized at 4°C in radioimmunoprecipitation assay lysis buffer. The cell lysate or tissue homogenates were clarified by centrifugation at 14,000×g for 10 min at 4°C. Protein concentration in the soluble fraction was determined by the Bradford method. Protein samples (50–100 µg) were separated by a 12% SDS-polyacrylamide gel and then transferred onto polyvinylidene difluoride membranes. Membranes were then washed, blocked, and probed overnight at 4°C with rabbit polyclonal anti-CX3CR1 (1∶1000, Abcam, USA), IκBα (1∶1000, Cell Signaling Technology, USA), β-actin antibodies (1∶2000, Santa Cruz Biotechnology, USA) and mouse monoclonal anti-CSE antibody (1∶1000, Abnova, Taiwan) respectively, followed by secondary antibody for 2 h with a 1∶2000 dilution of HRP-conjugated, goat anti-rabbit IgG or goat anti-mouse IgG (Santa Cruz Biotechnology). Membranes were washed and then incubated in SuperSignal™ West Pico chemiluminescent substrate (Pierce, USA) before exposure to X-ray films (Pierce). The intensity of bands was quantified using LabWorks™ Image Analysis software (UVP).
Flow Cytometry
Male apoE−/− were given saline, NaHS (1 mg/kg, i.p., daily) or PAG (10 mg/kg, i.p., daily) at the same time as the commencement of fat feeding and were sacrificed 8 weeks after fat feeding. Mononuclear cell (MNC) fraction was isolated from 1 ml of whole mouse blood using centrifugation (400 g, 30 min) with Ficoll-Paque Plus (Amersham Biosciences, Sweden) as described previously [34]. After washing with PBS, monocytes (1×106/tube) were labelled with rabbit anti-mouse CX3CR1 antibody for 1 h at room temperature, followed by incubation for a further 45 min with secondary PE-labelled antibodies (BD Biosciences) at room temperature. A third 30 min incubation was then carried out with a FITC conjugated CD11b antibody (BD Pharmingen). Appropriate anti-rabbit IgG control (Abcam) and Rat IgG control (BD Pharmingen) were also tested for CX3CR1 and CD11b antibodies, respectively. CX3CR1+CD11b+ cells were identified by flow cytometry (BD FACSCalibur Flow Cytometer) using FlowJo 5.7.2 software. For each sample, 100 000 cells were acquired.
Chemotaxis Assay
Chemotaxis was evaluated with QCM™ Chemotaxis 5 µm 96-well Cell Migration Assay (Chemicon, US). Serum free medium or chemoattractant solution (150 µL) was added to the lower chamber and 100 µL of cell suspension (2×106/ml) was added to the upper compartment. The cell migration plate assembly was incubated at 37°C with 5% CO2 for 2 h. Migratory cells on the bottom of insert membrane were dissociated from the membrane when incubated with Cell Detachment Buffer. The cells that migrated into the medium in the lower chamber or detached from the membrane by Cell Detachment Buffer were subsequently lysed and detected by CyQuant GR dye. This green fluorescent dye exhibits strong fluorescence enhancement when bound to cellular nucleic acids. The intensity of fluorescence, which correlated with the number of migrated cells, was measured with a fluorescence plate reader using a 485/520 nm filter set. Results were expressed as fluorescence intensity, RFU (relative fluorescence unit), by subtracting background fluorescence in cells migrating to the medium without chemoattractant, from specific fluorescence in cells migrating to CX3CL1.
Transcription Activity of PPAR-γ and NF-κB
Nuclear extracts from RAW264.7 or peritoneal cells were prepared using a Nuclear Extraction Kit, as described by the manufacturer (Active Motif, Tokyo, Japan). Protein concentrations in nuclear extracts were determined using the Bradford assay (Bio-Rad Laboratories). PPAR-γ or NF-κB p65 DNA binding activity in nuclear extracts was assayed in triplicate with TransAM PPAR-γ Transcription Factor Assay Kits (Active Motif) according to manufacturer’s instructions. The OD450 was read on a 96-well microplate reader (Tecan Systems Inc.).
Measurement of Plasma H2S
Aliquots (120 µl) of plasma were mixed with distilled water (100 µl), trichloroacetic acid (10% w/v, 120 µl), zinc acetate (1% w/v, 60 µl), N, N-dimethyl-p-phenylenediamine sulfate (20 µM; 40 µl) in 7.2 M HCl and FeCl3 (30 µM; 40 µl) in 1.2 M HCl. The absorbance of the resulting solution (670 nm) was measured 10 min thereafter by spectrophotometry (Tecan Systems Inc.). H2S was calculated against a calibration curve of sodium hydrosulfide (NaHS; 3.125–100 µM). Results were shown as plasma H2S concentration in µM.
Measurement of Plasma H2S by Sulfur-sensitive Electrode
Plasma H2S was measured by a sulfur-sensitive electrode (PXS-270, Shanghai, China) [4]. Briefly, 0.2 mL of plasma was mixed with 0.2 mL of antioxidant buffer. After being rinsed with distilled water and dried, the electrode was immersed into the plasma. Electrode potential was recorded when the reading stabilized. H2S was calculated against a calibration curve of sodium hydrosulfide (NaHS; 3.125–100 µM). Results were shown as plasma H2S concentration in µM.
H2S Synthesizing Activity Assay
H2S synthesiszing activity in aorta homogenates was measured essentially as described elsewhere [35]. Briefly, the assay mixture contained 100 mM potassium phosphate buffer (pH 7.4), L-cysteine (20 µl, 20 mM), pyridoxal 5′-phosphate (20 µl, 2 mM), saline (30 µl) and 4.5% w/v tissue homogenate (430 µl). The reaction was performed in tightly sealed micro centrifuge tubes and initiated by transferring the tubes from ice to a water bath at 37°C. After incubation for 30 min, 1% w/v zinc acetate (250 µl) was added to trap evolved H2S, followed by 10% v/v trichloroacetic acid (250 µl) to denature the protein and stop the reaction. Subsequently, N, N-dimethyl-p-phenylenediamine sulfate (20 µM; 133 µl) in 7.2 M HCl was added, immediately followed by FeCl3 (30 µM; 133 µl) in 1.2 M HCl. The absorbance of the resulting solution at 670 nm was measured by spectrophotometry (Tecan Systems Inc). The H2S concentration was calculated against a calibration curve of NaHS. Results were then corrected for the DNA content of the tissue sample and expressed as nmoles H2S formed/mg DNA.
Ultrasound Biomicroscopy
Ultrasound biomicroscopy was performed as previously described [36], [37]. Briefly, mice were lightly anesthetized with isoflurane gas. A UBM system (Vevo 770, Visualsonics, Toronto, Canada) equipped with a 40 MHz mechanical transducer was used for all the examinations. A right parasternal long-axis view was employed to visualize the ascending aorta, aortic arch and the neck vessels in one plane. A parasternal short-axis view was taken to visualize the same arterial site in a cross-sectional view immediately proximal to the branch of the brachiocephalic artery. By adjusting the distance between the transducer and the target vascular site of interest, the intima-media complex thickness (IMT) and atherosclerotic lesions can be readily visualized. Thereafter, during diastole, an optimal freeze-frame image was taken manually to visualize the plaque area by using the leading-to-leading edge approach. IMT was measured from the leading echo edge (intima) closest to the blood stream to the next leading echo edge representing the adventitial layer. Plaque area was delineated in the cross-sectional view also by using the leading-to-leading edge approach, which also includes the underlying smooth muscle cell layers in the plaque area measurement. In vivo length of the internal elastic lamina was approximated by delineation of the lumen circumference. A 10 s CINE loop was stored digitally for off-line image analysis using an image analysis system (Visualsonics, Toronto, Canada). All measurements were repeated twice at the same site. All images were analyzed by an operator blinded to the identities of the animals.
Histological Examination
Mice were anesthetized, followed by left ventricle perfusion with PBS and 10% neutralized formalin at a constant pressure of 100 mmHg. Brachiocephalic arteries were removed with a piece of the aortic arch and the stump of the right subclavian artery still attached to aid orientation during histological processing. Morphometric studies were performed in brachiocephalic artery. Brachiocephalic arteries were embedded in paraffin. Five sections (5 µm) taken 30 µm apart from the brachiocephalic artery of every mouse (starting from the proximal end) were stained with hematoxylin and eosin and then used for histological analysis (n = 6 mice in each group). Estimation of plaque lipid content used a valid method as previously described [38]. The maximum plaque thickness was averaged from three lesion sites around the thickest part of a plaque, approximately 100 µm apart. Every morphological parameter was quantified using computer-assisted morphometry (Leica Q win, Heidelberg, Germany) and performed by an investigator blinded for the treatment.
Immunohistochemistry of CX3CR1
Brachiocephalic arteries were deparaffinized in xylene and rehydrated in aqueous solutions with decreasing alcohol content, followed by a wash in PBST (1×PBS with 0.5% Tween 20, pH 7.4). Antigen retrieval was achieved by heating the slides in 10 mM sodium citrate (pH 6.0) at 95°C for 20 min. Slides were cooled for 20 min and washed in H2O and PBST. Endogenous peroxidase activity was inactivated by treatment in 3% H2O2 for 15 min, followed by a wash in PBST. Nonspecific staining was blocked by incubation with Ultra-V block (Labvision) for 5 min. Slides were incubated with rabbit anti-mouse CX3CR1 antibody (Abcam) in 1∶100 dilution for 2 h at 37°C, followed by secondary antibody in a 1∶200 dilution of HRP-conjugated, goat anti-rabbit IgG (Santa Cruz Biotechnology) for 2 h at room temperature. Immunohistochemical staining was developed by exposure to 3, 3′-diaminobenzidine and counterstaining was performed with hematoxylin. The staining was examined by light microscopy using a light microscope (objective lens magnification of ×40; eyepiece magnification of ×10). For fluorescent immunohistochemical staining, sections were incubated with rabbit anti-mouse CX3CR1 antibody (Abcam, 1∶100) and rat anti mouse F4/80 antibody (Santa Cruz, 1∶100) overnight at 4°C, followed by incubation for 2 hour with fluorescently (Alexa Fluor 488 or Alexa Fluor 594) labeled secondary antibodies (Invitrogen, 1∶100). Sections were then mounted and visualized using a fluorescent microscope (Olympus).
Statistics
The data were expressed as mean ± SEM. The significance of differences among groups was evaluated by analysis of variance (ANOVA) with the post-hoc Tukey’s test when comparing three or more groups. The significance of differences between two groups was evaluated by the Student’s t-test. A P < 0.05 was regarded as statistically significant.
Supporting Information
Effect of NaHS on CX3CR1 expression in mouse peritoneal macrophages stimulated with IFN-γ or LPS. RT-PCR analysis for CX3CR1 mRNA (A) and western blot analysis (B) for CX3CR1 protein expression were carried out as described in Materials and Methods. The data are means ± SEM of at least three independent experiments. *P<0.05, compared with unstimulated cells (control). ‡P<0.05, compared with stimulated cell pretreated with saline.
(TIF)
Effect of NaHS (100 µM) on chemotactic response to increasing doses of CX3CL1 in RAW264.7 cells stimulated with IFN-γ- or LPS. Chemotaxis of RAW264.7 cells towards CX3CL1 was assayed as described in Materials and Methods. The data are means ± SEM of at least three independent experiments. *P<0.05, compared with saline-pretreated, unstimulated cells (CX3CL1 10 ng/ml). **P<0.05, compared with saline-pretreated, stimulated cells (CX3CL1 10 ng/ml). †P<0.05, compared with saline-pretreated, unstimulated cells (CX3CL1 at the same concentration). ‡P<0.05, saline-pretreated, stimulated cells (CX3CL1 at the same concentration).
(TIF)
Effect of pretreatment with GW9662 on NaHS-induced downregulation of CX3CR1 expression and NaHS-induced inhibition of CX3CR1-mediated chemotaxis in IFN-γ or LPS-stimulated mouse peritoneal macrophages. Cells were incubated with GW9662 (10 µM) or vehicle for 1 hour, further incubated with NaHS (100 µM) or saline for 6 hours and then stimulated with IFN-γ (500 U/ml) or LPS (10 µg/ml) for 12 hours in the continuous presence of NaHS or saline. Western blot analysis for CX3CR1 expression (A) and chemotaxis towards CX3CL1 (50 ng/ml) (B) were assayed as described in Materials and Methods. The data are means ± SEM of at least three independent experiments. *P<0.05, compared with unstimulated cells (control). †P<0.05, compared with stimulated cells treated with saline and vehicle. ‡P<0.05, compared with stimulated cell treated with NaHS and vehicle.
(TIF)
Transfection of CSE cDNA construct in RAW264.7 cell. Cells were transfected with CSE cDNA construct or an identical empty vector lacking a cDNA insert as a control. The expression of CSE was verified by western blot analysis (A) and the H2S synthesizing activity (B) was assayed as described in Materials and Methods. The data are means ± SEM of four independent experiments. *P<0.05, compared with control cells. †P<0.05, compared with cells transfected with empty vector.
(TIF)
Effect of NaHS or CSE overexpression on CX3CL1 mRNA expression and IκBα content in macrophages stimulated with IFN-γ or LPS. (A) Mouse peritoneal macrophages were pre-incubated with saline or NaHS (100 µM) for 6 hours and then stimulated with IFN-γ or LPS for 12 hours in the continuous presence of NaHS or saline. (B) RAW264.7 cells were transfected with CSE cDNA construct or empty vector and then were stimulated with IFN-γ or LPS for 12 hours. RT-PCR analysis for CX3CL1 mRNA and western blot analysis for IκBα content were carried out as described in Materials and Methods.
(TIF)
Effect of NaHS on CCR2 and CCR5 mRNA expression in macrophages stimulated with IFN-γ or LPS. RAW264.7 cells (A, B) or mouse peritoneal macrophages (C, D) were pre-incubated with saline or NaHS (50 µM, 100 µM, 200 µM) for 6 hours and then stimulated with IFN-γ or LPS for 12 hours in the continuous presence of NaHS or saline. RT-PCR analysis for CCR2 and CCR5 mRNA was carried out as described in Materials and Methods.
(TIF)
Alterations in plasma H2S level during the development of atherosclerosis in fat-fed apoE−/− mice. Plasma H2S levels were assayed at indicated time points (0, 4, 8, 12, 24 weeks after fat feeding) by sulfur-sensitive electrode method. Results shown are the mean ± SEM (n = 6 animals in each group). *P<0.05, compared with the basal level at 0 weeks. #P<0.05, compared with mice sacrificed 4 weeks after fat feeding. & P<0.05, compared with mice sacrificed 8 weeks after fat feeding.
(TIF)
Negative control immunofluorescent staining in BCA with mouse IgG1 isotype control and Alexa Fluor 488 or Alexa Fluor 594 labeled secondary antibodies. Scale bar for histological images = 20 µm.
(TIF)
Effect of H2S on CX3CR1 expression and CX3CL1-mediated chemotaxis of circulating monocytes isolated from fat-fed apoE−/− mice. ApoE−/− mice (n = 10 in each group) were fed a high-fat diet and at the same time received saline, NaHS (1 mg/kg, daily, i.p.), or PAG (10 mg/kg, daily, i.p.) and sacrificed 8 weeks after fat feeding. Whole blood was collected and the mononuclear cell fraction was isolated, and then incubated with antibodies to detect CD11b+CX3CR1+ cell populations using flow cytometry (A). CX3CR1 mean fluorescence intensity (B) and CX3CL1-induced chemotaxis (C) were determined among treatment groups. Results are expressed as mean±SEM. *P<0.05, compared with chow+saline group.
(TIF)
Effect of NaHS on CX3CL1 level in RAW264.7culture media stimulated with IFN-γ or LPS.
(DOC)
Effect of NaHS on CX3CL1 in stimulated mouse peritoneal macrophages.
(DOC)
Effect of CSE overexpression on CX3CL1 in stimulated RAW264.7 cells.
(DOC)
Effect of H2S on CCL2 and CCL5 in stimulated RAW264.7.
(DOC)
Effect of treatment with NaHS on blood pressure and plasma lipids.
(DOC)
Effect of treatment with PAG on blood pressure and plasma lipids.
(DOC)
Effect of H2S on CCL2, CCL5, CCR2 and CCR5 in vivo.
(DOC)
Effect of PAG on CCL2, CCL5, CCR2 and CCR5 in vivo.
(DOC)
Acknowledgments
We thank Jia Jianguo for his help with UBM technology.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported by the National Natural Science Foundation of China grant (Grant No. 30900522) www.nsfc.gov.cn, the Shanghai Pujiang Program (Grant No. 09PJ1407000) www.stcsm.gov.cn, and the Scientific Research Foundation for the Returned Overseas Chinese Scholars, Ministry of Education of China www.cscse.edu.cn. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Renga B. Hydrogen sulfide generation in mammals: the molecular biology of cystathionine-β-synthase (CBS) and cystathionine-γ-lyase (CSE). Inflamm Allergy Drug Targets. 2011;10:85–91. doi: 10.2174/187152811794776286. [DOI] [PubMed] [Google Scholar]
- 2.Lefer DJ. A new gaseous signaling molecule emerges: cardioprotective role of hydrogen sulfide. Proc Natl Acad Sci. 2007;104:17907–17908. doi: 10.1073/pnas.0709010104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elsey DJ, Fowkes RC, Baxter GF. Regulation of cardiovascular cell function by hydrogen sulfide (H2S). Cell Biochem Funct. 2010;28:95–106. doi: 10.1002/cbf.1618. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Zhao X, Jin H, Wei H, Li W, et al. Role of hydrogen sulfide in the development of atherosclerotic lesions in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2009;29:173–179. doi: 10.1161/ATVBAHA.108.179333. [DOI] [PubMed] [Google Scholar]
- 5.Zhao ZZ, Wang Z, Li GH, WanG R, Tan JM, et al. Hydrogen sulfide inhibits macrophage-derived foam cell formation. Exp Biol Med (Maywood) 2011;236:169–176. doi: 10.1258/ebm.2010.010308. [DOI] [PubMed] [Google Scholar]
- 6.Meng QH, Yang G, Yang W, Jiang B, Wu L, et al. Protective effect of hydrogen sulfide on balloon injury-induced neointima hyperplasia in rat carotid arteries. Am J Pathol. 2007;170:1406–1414. doi: 10.2353/ajpath.2007.060939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 8.Zernecke A, Shagdarsuren E, Weber C. Chemokine in atherosclerosis: an update. Arterioscler Thromb Vasc Biol. 2008;28:1897–1908. doi: 10.1161/ATVBAHA.107.161174. [DOI] [PubMed] [Google Scholar]
- 9.Tacke F, Alvarez D, Kaplan J, Jakubzick C, Spanbroek R, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–194. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao Q. Dual targeting of CCR2 and CCR5: therapeutic potential for immunologic and cardiovascular diseases. J Leukoc Biol. 2010;88:41–55. doi: 10.1189/jlb.1009671. [DOI] [PubMed] [Google Scholar]
- 11.Wong BW, Wong D, McManus BM. Characterization of fractalkine (CX3CL1) and CX3CR1 in human coronary arteries with native atherosclerosis, diabetes mellitus, and transplant vascular disease. Cardiovasc Pathol. 2002;11:332–338. doi: 10.1016/s1054-8807(02)00111-4. [DOI] [PubMed] [Google Scholar]
- 12.Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 13.Dawson TC, Kuziel WA, Osahar TA, Maeda N. Absence of CC chemokine receptor-2 reduces atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis. 1999;143:205–211. doi: 10.1016/s0021-9150(98)00318-9. [DOI] [PubMed] [Google Scholar]
- 14.Braunersreuther V, Zernecke A, Arnaud C, Liehn EA, Steffens S, et al. CCR5 but not CCR1 deficiency reduces development of diet-induced atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2007;27:373–379. doi: 10.1161/01.ATV.0000253886.44609.ae. [DOI] [PubMed] [Google Scholar]
- 15.Veillard NR, Kwak B, Pelli G, Mulhaupt F, James RW, et al. Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ Res. 2004;94:253–261. doi: 10.1161/01.RES.0000109793.17591.4E. [DOI] [PubMed] [Google Scholar]
- 16.Braunersreuther V, Steffens S, Arnaud C, Pelli G, Burger F, et al. A novel RANTES antagonist prevents progression of established atherosclerotic lesions in mice. Arterioscler Thromb Vasc Biol. 2008;28:1090–1096. doi: 10.1161/ATVBAHA.108.165423. [DOI] [PubMed] [Google Scholar]
- 17.Teupser D, Pavlides S, Tan M, Gutierrez-Ramos JC, Kolbeck R, et al. Major reduction of atherosclerosis in fractalkine (CX3CL1)-deficient mice is at the brachiocephalic artery, not the aortic root. Proc Natl Acad Sci USA. 2004;101:17795–17800. doi: 10.1073/pnas.0408096101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Combadiere C, Potteaux S, Gao JL, Esposito B, Casanova S, et al. Decreased atherosclerotic lesion formation in CX3CR1/apolipoprotein E double knockout mice. Circulation. 2003;107:1009–1016. doi: 10.1161/01.cir.0000057548.68243.42. [DOI] [PubMed] [Google Scholar]
- 19.Lesnik P, Haskell CA, Charo IF. Decreased atherosclerosis in CX3CR1−/− mice reveals a role for fractalkine in atherogenesis. J Clin Invest. 2003;111:333–340. doi: 10.1172/JCI15555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bursill CA, Castro ML, Beattie DT, Nakhla S, van der Vorst E, et al. High-density lipoproteins suppress chemokines and chemokine receptors in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2009;30:1773–1778. doi: 10.1161/ATVBAHA.110.211342. [DOI] [PubMed] [Google Scholar]
- 21.Barlic J, Zhang Y, Foley JF, Murphy PM. Oxidized lipid-driven chemokine receptor switch, CCR2 to CX3CR1, mediates adhesion of human macrophages to coronary artery smooth muscle cells through a peroxisome proliferator-activated receptor γ-dependent pathway. Circulation. 2006;114:807–819. doi: 10.1161/CIRCULATIONAHA.105.602359. [DOI] [PubMed] [Google Scholar]
- 22.Barlic J, Murphy PM. An oxidized lipid-PPARγ-chemokine pathway in the regulation of macrophage: vascular smooth muscle cell adhesion. Trends Cardiovasc Med. 2007;17:269–274. doi: 10.1016/j.tcm.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 24.Bhavsar PK, Sukkar MB, Khorasani N, Lee KY, Chung KF. Glucocorticoid suppression of CX3CL1 (fractalkine) by reduced gene promoter recruitment of NF-κB. FASEB J. 2008;22:1807–1816. doi: 10.1096/fj.07-094235. [DOI] [PubMed] [Google Scholar]
- 25.Zhi L, Ang AD, Zhang H, Moore PK, Bhatia M. Hydrogen sulfide induces the synthesis of proinflammatory cytokines in human monocyte cell line U937 via the ERK-NF-kappaB pathway. J Leuk Biol. 2007;81:1322–1332. doi: 10.1189/jlb.1006599. [DOI] [PubMed] [Google Scholar]
- 26.Oh GS, Pae HO, Lee BS, Kim BN, Kim JM, et al. Hydrogen sulfide inhibits nitric oxide production and nuclear factor-kappaB via heme oxygenase-1 expression in RAW264.7 macrophages stimulated with lipopolysaccharide. Free Radic Biol Med. 2006;41:106–119. doi: 10.1016/j.freeradbiomed.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 27.Hu LF, Wong PTH, Moore PK, Bian JS. Hydrogen sulfide attenuates lipopolysaccharide-induced inflammation by inhibition of p38 mitogenactivated protein kinase in microglia. J Neurochem. 2007;100:1121–1128. doi: 10.1111/j.1471-4159.2006.04283.x. [DOI] [PubMed] [Google Scholar]
- 28.Wan Y, Evans RM. Rosiglitazone activation of PPARγ suppresses fractalkine signaling. J Mol Endocrinol. 2010;44:135–142. doi: 10.1677/JME-09-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kimura Y, Kimura H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004;18:1165–1167. doi: 10.1096/fj.04-1815fje. [DOI] [PubMed] [Google Scholar]
- 30.Whiteman M, Armstrong JS, Chu SH, Jia-Ling S, Wong BS, et al. The novel neuromodulator hydrogen sulfide: an endogenous peroxynitrite ‘scavenger’? J Neurochem. 2004;90:765–768. doi: 10.1111/j.1471-4159.2004.02617.x. [DOI] [PubMed] [Google Scholar]
- 31.Whiteman M, Li L, Kostetski I, Chu SH, Siau JL, et al. Evidence for the formation of a novel nitrosothiol from the gaseous mediators nitric oxide and hydrogen sulphide. Biochem Biophys Res Commun. 2006;343:303–310. doi: 10.1016/j.bbrc.2006.02.154. [DOI] [PubMed] [Google Scholar]
- 32.Shibuya A, Wada K, Nakajima A, Saeki M, Katayama K, et al. Nitration of PPARγ inhibits ligand-dependent translocation into the nucleus in a macrophage-like cell line, RAW 264. FEBS Lett. 2002;525:43–47. doi: 10.1016/s0014-5793(02)03059-4. [DOI] [PubMed] [Google Scholar]
- 33.Wang Q, Tang C, Jin H, Du J. Endogenous hydrogen sulfide is involved in the pathogenesis of atherosclerosis. Biochem Biophys Res Commun. 2010;396:182–186. doi: 10.1016/j.bbrc.2010.04.061. [DOI] [PubMed] [Google Scholar]
- 34.Zhang H, Zhi L, Moochhala SM, Moore PK, Bhatia M. Endogenous hydrogen sulfide regulates leukocyte trafficking in cecal ligation and puncture-induced sepsis. J Leukoc Biol. 2007;82:894–905. doi: 10.1189/jlb.0407237. [DOI] [PubMed] [Google Scholar]
- 35.Zhang H, Zhi L, Moore PK, Bhatia M. Role of hydrogen sulfide in cecal ligation and puncture-induced sepsis in the mouse. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1193–L1201. doi: 10.1152/ajplung.00489.2005. [DOI] [PubMed] [Google Scholar]
- 36.Gan LM, Grönros J, Hägg U, Wikström J, Theodoropoulos C, et al. Non-invasive real-time imaging of atherosclerosis in mice using ultrasound biomicroscopy. Atherosclerosis. 2007;190:313–320. doi: 10.1016/j.atherosclerosis.2006.03.035. [DOI] [PubMed] [Google Scholar]
- 37.Wu DJ, Xu JZ, Wu YJ, Jean-Charles L, Xiao B, et al. Effects of fasudil on early atherosclerotic plaque formation and established lesion progression in apolipoprotein E-knockout mice. Atherosclerosis. 2009;207:68–73. doi: 10.1016/j.atherosclerosis.2009.04.025. [DOI] [PubMed] [Google Scholar]
- 38.Johnson J, Carson K, Williams H, Karanam S, Newby A, et al. Plaque rupture after short periods of fat feeding in the apolipoprotein E-knockout mouse: model characterization and effects of pravastatin treatment. Circulation. 2005;111:1422–1430. doi: 10.1161/01.CIR.0000158435.98035.8D. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect of NaHS on CX3CR1 expression in mouse peritoneal macrophages stimulated with IFN-γ or LPS. RT-PCR analysis for CX3CR1 mRNA (A) and western blot analysis (B) for CX3CR1 protein expression were carried out as described in Materials and Methods. The data are means ± SEM of at least three independent experiments. *P<0.05, compared with unstimulated cells (control). ‡P<0.05, compared with stimulated cell pretreated with saline.
(TIF)
Effect of NaHS (100 µM) on chemotactic response to increasing doses of CX3CL1 in RAW264.7 cells stimulated with IFN-γ- or LPS. Chemotaxis of RAW264.7 cells towards CX3CL1 was assayed as described in Materials and Methods. The data are means ± SEM of at least three independent experiments. *P<0.05, compared with saline-pretreated, unstimulated cells (CX3CL1 10 ng/ml). **P<0.05, compared with saline-pretreated, stimulated cells (CX3CL1 10 ng/ml). †P<0.05, compared with saline-pretreated, unstimulated cells (CX3CL1 at the same concentration). ‡P<0.05, saline-pretreated, stimulated cells (CX3CL1 at the same concentration).
(TIF)
Effect of pretreatment with GW9662 on NaHS-induced downregulation of CX3CR1 expression and NaHS-induced inhibition of CX3CR1-mediated chemotaxis in IFN-γ or LPS-stimulated mouse peritoneal macrophages. Cells were incubated with GW9662 (10 µM) or vehicle for 1 hour, further incubated with NaHS (100 µM) or saline for 6 hours and then stimulated with IFN-γ (500 U/ml) or LPS (10 µg/ml) for 12 hours in the continuous presence of NaHS or saline. Western blot analysis for CX3CR1 expression (A) and chemotaxis towards CX3CL1 (50 ng/ml) (B) were assayed as described in Materials and Methods. The data are means ± SEM of at least three independent experiments. *P<0.05, compared with unstimulated cells (control). †P<0.05, compared with stimulated cells treated with saline and vehicle. ‡P<0.05, compared with stimulated cell treated with NaHS and vehicle.
(TIF)
Transfection of CSE cDNA construct in RAW264.7 cell. Cells were transfected with CSE cDNA construct or an identical empty vector lacking a cDNA insert as a control. The expression of CSE was verified by western blot analysis (A) and the H2S synthesizing activity (B) was assayed as described in Materials and Methods. The data are means ± SEM of four independent experiments. *P<0.05, compared with control cells. †P<0.05, compared with cells transfected with empty vector.
(TIF)
Effect of NaHS or CSE overexpression on CX3CL1 mRNA expression and IκBα content in macrophages stimulated with IFN-γ or LPS. (A) Mouse peritoneal macrophages were pre-incubated with saline or NaHS (100 µM) for 6 hours and then stimulated with IFN-γ or LPS for 12 hours in the continuous presence of NaHS or saline. (B) RAW264.7 cells were transfected with CSE cDNA construct or empty vector and then were stimulated with IFN-γ or LPS for 12 hours. RT-PCR analysis for CX3CL1 mRNA and western blot analysis for IκBα content were carried out as described in Materials and Methods.
(TIF)
Effect of NaHS on CCR2 and CCR5 mRNA expression in macrophages stimulated with IFN-γ or LPS. RAW264.7 cells (A, B) or mouse peritoneal macrophages (C, D) were pre-incubated with saline or NaHS (50 µM, 100 µM, 200 µM) for 6 hours and then stimulated with IFN-γ or LPS for 12 hours in the continuous presence of NaHS or saline. RT-PCR analysis for CCR2 and CCR5 mRNA was carried out as described in Materials and Methods.
(TIF)
Alterations in plasma H2S level during the development of atherosclerosis in fat-fed apoE−/− mice. Plasma H2S levels were assayed at indicated time points (0, 4, 8, 12, 24 weeks after fat feeding) by sulfur-sensitive electrode method. Results shown are the mean ± SEM (n = 6 animals in each group). *P<0.05, compared with the basal level at 0 weeks. #P<0.05, compared with mice sacrificed 4 weeks after fat feeding. & P<0.05, compared with mice sacrificed 8 weeks after fat feeding.
(TIF)
Negative control immunofluorescent staining in BCA with mouse IgG1 isotype control and Alexa Fluor 488 or Alexa Fluor 594 labeled secondary antibodies. Scale bar for histological images = 20 µm.
(TIF)
Effect of H2S on CX3CR1 expression and CX3CL1-mediated chemotaxis of circulating monocytes isolated from fat-fed apoE−/− mice. ApoE−/− mice (n = 10 in each group) were fed a high-fat diet and at the same time received saline, NaHS (1 mg/kg, daily, i.p.), or PAG (10 mg/kg, daily, i.p.) and sacrificed 8 weeks after fat feeding. Whole blood was collected and the mononuclear cell fraction was isolated, and then incubated with antibodies to detect CD11b+CX3CR1+ cell populations using flow cytometry (A). CX3CR1 mean fluorescence intensity (B) and CX3CL1-induced chemotaxis (C) were determined among treatment groups. Results are expressed as mean±SEM. *P<0.05, compared with chow+saline group.
(TIF)
Effect of NaHS on CX3CL1 level in RAW264.7culture media stimulated with IFN-γ or LPS.
(DOC)
Effect of NaHS on CX3CL1 in stimulated mouse peritoneal macrophages.
(DOC)
Effect of CSE overexpression on CX3CL1 in stimulated RAW264.7 cells.
(DOC)
Effect of H2S on CCL2 and CCL5 in stimulated RAW264.7.
(DOC)
Effect of treatment with NaHS on blood pressure and plasma lipids.
(DOC)
Effect of treatment with PAG on blood pressure and plasma lipids.
(DOC)
Effect of H2S on CCL2, CCL5, CCR2 and CCR5 in vivo.
(DOC)
Effect of PAG on CCL2, CCL5, CCR2 and CCR5 in vivo.
(DOC)