

Abstract
Introduction
Alzheimer’s disease (AD) is a common, progressive neurological disorder whose incidence is reaching epidemic proportions. The prevailing ‘amyloid cascade hypothesis’, which maintains that the aberrant proteolysis of beta-amyloid precursor protein (βAPP) into neurotoxic amyloid beta (Aβ)-peptides is central to the etiopathology of AD, continues to dominate pharmacological approaches to the clinical management of this insidious disorder. This review is a compilation and update on current pharmacological strategies designed to down-regulate Aβ42-peptide generation in an effort to ameliorate the tragedy of AD.
Areas Covered
This review utilized on-line data searches at various open online-access websites including the Alzheimer Association, Alzheimer Research Forum; individual drug company databases; the National Institutes of Health (NIH) Medline; Pharmaprojects database; Scopus; inter-University research communications and unpublished research data.
Expert Opinion
Aβ immunization-, anti-acetylcholinesterase-, β-secretase-, chelation-, γ-secretase-, N-methyl D-aspartate (NMDA) receptor antagonist-, statin-based and other strategies to modulate βAPP processing have dominated pharmacological approaches directed against AD-type neurodegenerative pathology. Cumulative clinical results of these efforts remain extremely disappointing, and have had little overall impact on the clinical management of AD. While a number of novel approaches are in consideration and development, to date there is still no effective treatment or cure for this expanding healthcare concern.
Keywords: Alzheimer’s disease (AD), amyloid beta (Aβ) peptides, amyloidosis, beta-amyloid cleavage enzyme 1 (BACE-1), beta-amyloid precursor protein (βAPP), chelation therapy, γ-secretase, immunological strategies, neurodegenerative disease, presenilin-1 (PS1), secretase, statins
1. Background
1.1 The Looming Alzheimer’s Disease (AD) Healthcare Problem
Alzheimer’s disease (AD) is a tragic, age-related neurological disease, and represents the most prevalent neurodegenerative disorder in industrialized societies. AD is the most common type of cognitive impairment and memory loss of the aged, characterized by the progressive erosion of cognition, functional ability, mood, behavior and memory. An estimated 5.4 million people in the United States have AD, and healthcare treatment for AD patients in the United States currently involves a staggering 15 million unpaid caregivers and 183 billion dollars in annual costs. The projected yearly expense of AD healthcare is estimated to soar to 1.1 trillion dollars by the year 2050 [1,2]. Currently, this places a tremendous socioeconomic burden on both AD caregivers and an already strained healthcare system. In the foreseeable future the prognosis for AD incidence and soaring medical costs become even more stark and overwhelming. Globally, 5 million new cases of AD are diagnosed annually, with one new AD case being reported every 7 seconds [1–5]. Importantly, our increasing life expectancy and the demographics of our aging population on a global scale cast significant concerns over our medical and socioeconomic capability to manage this rapidly expanding brain disease. Currently, there are no adequate preventive or curative treatments for this leading cause of senile dementia, and pharmacological strategies and treatments directed at AD symptoms, and specifically targeted to neurotransmitter deficits and the progressively amyloidogenic and inflammatory nature of this brain degeneration, have collectively met with extremely disappointing results [1–7].
1.2 The Neuropathology of Alzhemier’s Disease (AD)
Neuropathologically, AD is characterized (i) by a progressive, age-related generation, aggregation and accumulation of amyloid-beta (Aβ) peptides as dense, insoluble, pro-inflammatory and pathogeic deposits of senile plaque (SP), (ii) the appearance of tau-protein containing intracellular neurofibrillary tangles (NFTs), (iii) reduced synaptic densities, and (iv) neuronal loss in temporal lobe and hippocampal structures that, as AD progresses, radiates into the more distal parietal, frontal and occipital poles of the brain [11–16]. In the recent past, the density of SP and NFT lesions required extensive post-mortem histopathological confirmation for an AD diagnosis, however current autoradiographic, tomographic, magnetic resonance and related electronic digitization and quantification technologies are capable of effectively resolving SP, NFT or Aβ peptide load both in aging brains and in patients with AD, and in transgenic animal models of AD [16–20]. The initial generation of Aβ peptides, their aggregation from soluble monomers into insoluble deposits, and their unusual biophysical properties have been suggested by many to be the earliest markers for cognitive disturbances and AD onset that precedes by decades the appearance of mature SP and NFT lesions [19–23].
1.3 The ‘Amyloid Cascade Hypothesis’
The essentials of the ‘amyloid cascade hypothesis’ are that proteolysis of the ~770 amino acid, polytopic, transmembrane β-amyloid precursor protein (βAPP; chr 21q21.1; GenBank: BAA22264.1), though interaction with associated membrane proteins such as nicastrin, and tandem beta (β)- and gamma (γ)-secretase cleavage yields a series of ragged Aβ peptide monomers Figure 1). The (γ)-secretase is a rather unusual aspartic protease that cleaves βAPP within the hydrophobic transmembrane domain, and its catalytic activity is thought to be the rate-limiting enzyme in Aβ peptide production [24–26]. Because a water molecule is absolutely required for βAPP cleavage, cleavage within the hydrophobic transmembrane domain of βAPP may be accelerated by membrane disruption or dynamic ‘vibrational’ movement of βAPP within the membrane (Figure 1; see below). The (γ)-secretase complex is actually composed of several high molecular weight integral proteins including presenilin 1 (PS1; PSEN1), nicastrin, Aph-1 and Pen-2 [12–15,23–27]. While each of these four components are required for γ-secretase activity, the precise mechanism of how this membrane-associated complex recognizes and cleaves its βAPP substrate is incompletely understood, and remains a critical issue in the development of compounds that specifically regulate β- and γ-secretase activity. Importantly, Aβ peptides generated include primarily, two noteworthy isoforms, including a 40 amino acid Aβ40 peptide that associates with the endothelium of the cerebral vasculature, and the more neurotoxic, albeit less abundant, Aβ42 species, containing 42 amino acids that is strongly self-associating and forms the essential nucleus of the SP lesion. Interestingly, the extra two amino acids in Aβ42 (versus Aβ40) appear to convey many of the toxic biophysical attributes of this slightly larger molecule [25–29]. The recognition of these end-stage Aβ peptides by the brain’s microglial surveillance system, and the inability of the microglial cell to deal with these toxic, insoluble pro-inflammatory inclusions are thought to form the basis for the elevated inflammatory and oxidative stress signaling that is characteristic of the AD process [9–16,30]. One major ongoing pharmacological strategy for the treatment of AD has been the targeting of the specific secretases responsible for Aβ40 and Aβ42 production. A highly informative website that includes video animation of the currently understood βAPP cleavage mechanism and how the modulation of Aβ42 abundance may be of use in down-regulating these AD-related insoluble deposits is freely available online [31].
Figure 1.
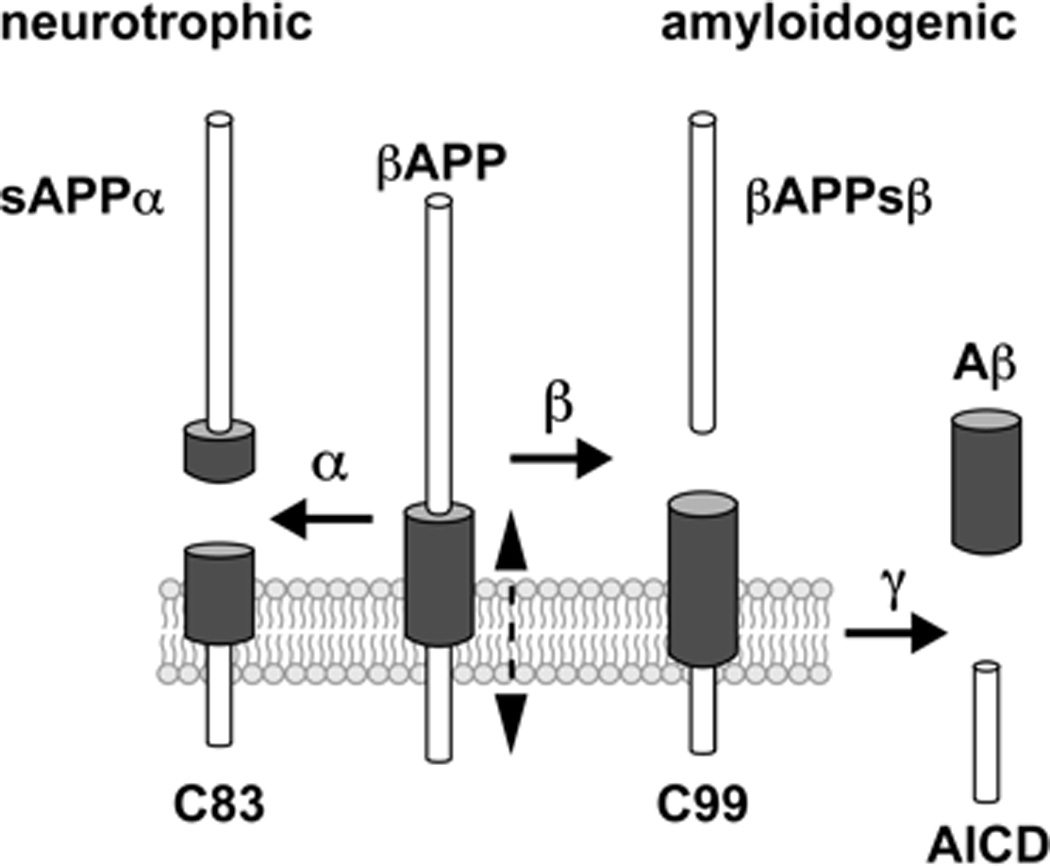
This diagram depicts the essentials of beta-amyloid precursor protein (βAPP) proteolytic cleavage system [64]. The 770 amino acid βAPP holoprotein is an extremely abundant polytopic type 1 neuronal membrane-spanning glycoprotein that can be processed via a neurotrophic (non-amyloidogenic) or amyloidogenic signaling pathway. In the neurotrophic (non-amyloidogenic) pathway, βAPP is first cleaved by a distintegrin and metallo-protease (ADAM) class of enzymes possessing α-secretase activity. This occurs 12 amino acids from the transmembrane domain of βAPP. This cleavage generates a soluble βAPP ectodomain called sAPPα and a C-terminal fragment (C83; which is further processed by γ-secretase, leading to a secreted p3-peptide). The transmembrane portion of βAPP that gives rise to sAPPα via the α-secretase-mediated pathway supports non-amyloidogenic, neuritogenic and neurotrophic signaling. Proteases similar to α-secretase are essential for a wide range of biological processes, such as cell adhesion and embryonic development. In the alternate amyloidogenic pathway βAPP is sequentially cleaved, first by β-secretase (beta-amyloid cleavage enzyme 1, or BACE1) within the ectodomain of βAPP close to the transmembrane domain, and then by γ-secretase, resulting in the generation of the soluble βAPP ectodomain (βAPPsβ), and the formation of the membrane-bound C-terminal fragment C99 (the C-terminal 99 amino acid peptide of βAPP). The γ-secretase next cleaves C99 giving rise to soluble Aβ-peptides (Aβ), chiefly Aβ40 and Aβ42 peptides, and secretion and the formation of the βAPP intracellular domain AICD which may function as a gene expression regulator. Over time soluble Aβ-peptides aggregate to form insoluble, pathogenic senile plaques (SP); trace metals appear to aid in this aggregation (see text) [98–103]. ‘Vibrational’ molecular movement of portions of the βAPP transmembrane polypeptide chain within the hydrophobic membrane (movement depicted by a hatched vertical line next to βAPP) may transiently expose the γ-secretase cleavage site to the aqueous cytoplasmic environment (see text). Notably, the membrane depicted in this drawing may be that of the endoplasmic reticulum, Golgi apparatus or neuronal plasma membrane, so amyloidogenic cleavage of βAPP into Aβ40 or Aβ42 peptides may culminate in either intracellular or extracellular Aβ peptide evolution with ensuing neurotoxic events in that compartment [1–8]. Aβ peptides are intensely hydrophobic, and these self-aggregating 40 (Aβ40) and 42 (Aβ42) amino acid peptides accumulate preferentially within, respectively, the microvasculature and senile plaque (SP) cores in AD brain [1–13,17–23]. The activity of the γ-secretase complex (containing βAPP, PS1 and nicastrin and associated peptides) is thought to be rate limiting in Aβ40 and Aβ42 peptide generation. As previously reviewed, the membrane-integral protein nicastrin and the βAPP sorting receptor sortilin-1 (SORL1) also direct βAPP trafficking, and down-regulation of SORL1 may lead to over activation of the amyloidogenic β-γ secretase axis and increased generation of Aβ peptides, both in AD brain and in stressed human brain cell models in vitro [24–28,64]. SORL-1 is known to interact with apoE which functions in part as a major cholesterol transporter [25–29,64,94]. Selective Aβ42-lowering agents or secretase inhibitors deigned to target β-secretase (BACE1) and in particular γ-secretase (PS1)-mediated Aβ peptide production are currently a principal research and development area, and their successful implementation are an important pharmacotherapeutic strategic goal for future AD treatment. Interactions between lipids, cholesterol, and the various βAPP-processing secretases and βAPP-associated proteins are highly complex. It appears that Aβ peptide production is favored in cholesterol-rich lipid raft domains [65,68,101–103]. While βAPP neurobiology is one of the most intensively studied areas of contemporary neurodegenerative disease research, many of the mechanistic details of βAPP proteolysis, including βAPP-membrane and βAPP-secretase interactions, remain incompletely understood.
In contrast to these smaller, intensely hydrophobic, pathogenic Aβ40 and Aβ42 peptides, a non-amyloidogenic, ~621 amino acid soluble amyloid precursor protein alpha (sAPPα) may be generated from βAPP through alternate α-secretase cleavage, and this relatively large extracellular peptide possesses neuritogenic, neurotrophic, neuroprotective and growth-promoting properties (Figure 1) [28–30]. sAPPα has been further shown to regulate neural cell excitability, synaptic plasticity, and has been demonstrated to be useful in promoting brain cell regeneration after acute brain injury after cerebral ischemia and stroke [32,33]. While the neurotoxic actions of Aβ40 and Aβ42 engender pathology by inducing oxidative stress, neural inflammation, neuronal dysfunction, apoptosis and brain cell death through the neurotoxic β-γ-secretase pathway, sAPPα production via the alternate α-secretase pathway both shunts production of pathogenic Aβ peptides while promoting the generation of the more neurotrophic sAPPα protein [32,33]. The single-transmembrane lipoprotein receptor SorLA/LR11, that normally regulates βAPP trafficking from the cell surface via the endocytic pathways, appears to play a determining role in the neuron’s decision of whether neurotoxic or neurotrophic forms of βAPP fragments are generated [34–36]. Indeed deficits in SORL1 abundance are associated with AD and in cytokine and Aβ peptide-stressed neuro-inflammatory models of AD using cultured primary human neural cells and inherited genetic variants in this membrane sorting receptor are associated with late-onset forms of AD [34,35]. It should be pointed out that there are several alternate hypotheses to the ‘amyloid cascade hypothesis’, including the idea that amyloid deposition is a neuro protective adaptation to AD, that Aβ42 peptide increases may be a response to pathogenic infection, and that AD is a pathogenetic autoimmune disorder caused by herpes simplex virus (HSV-1) or other brain pathogens in a gene-dependent manner [153–161]. Interestingly antiviral drugs such as acyclovir (1) have been shown to reduce the HSV-1 mediated up-regulation of a pro-inflammatory micro RNA known as miRNA-146a in human brain cells [159], (2) have been shown to reduce Aβ peptide accumulation in AD model systems, and (3) have been proposed to offer some therapeutic benefit towards the AD condition [159,164].
2. Medical need
The immense socioeconomic burden of AD is an expanding healthcare concern as demographic analyses indicate that the elderly currently constitute the fastest growing segment of Westernized societies [1,2,37–39]. Our deeper understanding of the basic molecular-genetic mechanisms involved in healthy aging versus AD remains a primary medical research area, and effective Aβ-peptide targeted drugs and strategies to treat AD are an urgent pharmacological goal. Unfortunately, while many pharmacological strategies and drug formulations have been advanced, and many primary clinical trials for AD are ongoing, no primary pharmacological-based prevention trial has yet successfully delayed the development or effectively cured this prevalent neurological disorder [37–40]. As is further discussed below, the strategic targeting of pharmaceuticals to the α-β-γ-secretase-SORL1-nicastrin axis of βAPP processing to traffic and compartmentalize βAPP-derived peptides away from the more neurotoxic into the more neurotrophic species is an obvious, and highly sought after, drug development strategy (Figure 1). Indeed elucidation of the fine details of amyloidogenic and pro-inflammatory signaling pathways continue to provide an abundance of disease markers and multiple biological targets useful for the future development of novel pharmaceuticals to retard AD progression. Clearly the refinement of our understanding on AD disease mechanisms, new secretase intervention strategies, novel pharmaceuticals and clinical treatments are essential to more effectively address this expanding health care problem.
3. Existing treatment
Due to the immense socioeconomic and health care concern of AD, the search for effective ways to prevent, alleviate and slow down AD progression is a paramount goal of contemporary neurodegenerative disease research. While not further discussed here, a wide range of antipsychotic drugs are currently used for reducing the severity and frequency of the often bizarre perceptual and behavioral symptoms of AD patients [41–43]. Currently there are no drugs available to cure, to effectively halt the progression, or to arrest further development of the AD, i.e., all current treatment approaches have little or no effect in altering the proliferative pathogenic mechanism of AD. Currently, in the AD clinical setting, suitable combinatorial drug treatment therapies have been suggested to be an effective way to address AD management and the quality of life for patients with AD. For example the AChE inhibitor donepezil hydrochloride [(RS)-2-[(1-benzyl-4-piperidyl)methyl]-5,6-dimethoxy-2,3-dihydroinden-1-one; Aricept®; Eisai, Pfizer] and more recently the glutamatergic antagonist memantine hydrochloride [3,5-dimethyltricyclo(3.3.1.1)decan-1amine; Namenda®; Forest; Merz; a moderate affinity NMDA-receptor antagonist], for the treatment of moderate to severe AD are currently the two most widely prescribed drugs in North America [43–58]. However, as with Aβ peptide immunization strategies, elaborated further upon below, unexpected complications such as acute hepatotoxicity have arisen, the cost-to-benefit ratios have been seriously questioned, and their general implementation against AD treatment have been fraught with difficulty. The hepatoxocity problem may be more acute and exacerbated in aged AD patients whose liver functions may already be compromised by age or by other medications. Drugs currently in use and under development further include compounds directed at γ-secretase, β-secretase, metal ion chelators, statins and related cholesterol lowering medications, other drugs which target the mechanism of Aβ peptide generation, and synthetic pharmaceutical and plant extracts and their derivatives which have been shown to modulate excess production of Aβ peptides in various ways [44–63]. The information in this review is essentially an update of a previous paper in Current Opinion on Emerging Drugs [64]. The use and potential benefits of these and several other categories of AD medications and pharmacological treatment strategies are further discussed in the updates and material presented below.
4. Therapeutic class review
4.1. Inhibitors of Acetylcholinesterase (AChE)
The inhibition of AChE is currently the most common (non-Aβ targeting) pharmacological approach for the clinical treatment of mild-to-moderate AD. As AChE is the enzyme that inactivates acetylcholine (Ach), an abundant brain neurotransmitter, especially in AD-targeted brain regions, the proposed mechanism is that AChE inhibitors can transiently increase acetylcholine concentration, and hence support cholinergic neurotransmission in ACh-depleted brain regions. Hence, the rate at which acetylcholine is degraded is decreased, thus compensating for AD-related ACh deficits due to cholinergic neuron loss (43–50). In the United States the Food and Drug Administration (FDA) has four approved AchE inhibitors as cholinergic drugs for AD. Donepezil (Aricept®) and galantamine (Razadyne®, Reminyl®, Nivalin®) are selective AchE inhibitors and rivastigmine (Exelon®) inhibits both acetylcholinesterase and buturylcholinesterase activities[43–47]. Tacrine is no longer used clinically because of its acute hepatotoxicity, its use being the leading cause of acute liver failure in the US, and the most common adverse event-causing drug non-approval and/or drug withdrawal by the FDA [39–42]. In fact, donepezil hydrochloride (Aricept®) is still the most widely prescribed single AChE inhibitor drug for AD in the world, however independent studies in Canada, the US and the UK have shown that its overall efficacy is rather poor, and that other anti-cholinesterase agents may soon overtake Donepezil as the current drug-of-choice for anti-AD therapy [45–47]. Since the inception of AChE inhibitors almost 25 years ago, there have been 850 reviews on the pharmacology, pharmacokinetics and efficacy of AD treatments using cholinergic approaches [43–50]. Besides considerable problems with hepatotoxicity, particularly in aged AD patients who are often taking multiple hepatotoxic medications, the use of AChE inhibitors has multiple and adverse off-target effects in normal cholinergic signaling. These include cardiovascular, cerebrovascular, cognitive, extrapyramidal, gastrointestinal, respiratory urinary, diurnal (i.e. wake-sleep cycle) and other neurological complications [42–46]. For example, increased cererbovascular and cardiovascular events have been observed in several placebo-controlled trials of galantamine [49]. In a recent review analyzing 22 published, double blind, randomized trials of 1 to 36 months duration, examining pharmaceutical efficacy on the basis of clinical outcomes, in which treatment with donepezil, rivastigmine, or galantamine were compared with placebo in AD concluded that the scientific basis for the use of AChE inhibitors for AD treatment was at best questionable [50–55]. Such pharmacological deficiencies underscore the need for (i) quantifying the clinical benefits of these medicines, (ii) clarifying the costs of the medication and (iii) who bears this cost, (iv) analyzing the statistical methods of weighing benefit against cost, (v) evaluating the consequences of using different approaches to cost-benefit analysis and the benefit-risk balance in individual AD patients, and (vi) ongoing re-evaluation of the efficacy of these medicines for the AD patient.
4.2. N-methyl D-aspartate (NMDA) receptor antagonists
The over-stimulation of N-methyl D-aspartate (NMDA) receptors by glutamate up-regulates excitatory signal transduction resulting in excitotoxicity, neural dysfunction and excitatory over-stimulation, and resulting in hyperstimulation, signaling dysfunction and progressive neurodegeneration [47,48]. Dysfunction of glutamatergic neurotransmission that manifests as a form of neuronal excitotoxicity has been repeatedly implicated in the etiopathogenesis of AD [47–49]. Aβ-peptides and SP lesions have been shown to inhibit glial-mediated glutamate reuptake and recycling, and with excess glutamate masking signal transmission, NMDA receptor antagonists have been strategically formulated to block the effects of excess glutamate and thereby restore physiological signal neurotransmission. Glutamate excitotoxic mechanisms have thereby become another focus for AD treatment with the moderate-affinity NMDA receptor antagonist memantine hydrochloride (3,5-dimethyltricyclo[3.3.1.13,7]decan-1amine; Namenda®), an anti-glutamatergic that modulates pathological over-stimulation of NMDA channels [51–55].
Interestingly, the drug memantine, first synthesized and patented by Eli Lilly in 1968, has been shown to possess atropinic, cholinergic, dopaminergic and serotonergic pharmacological effects, and has had a long and remarkable usage in neuropharmacological medicine. Memantine has been used clinically as, has been suggested to be used for, or is currently being tested as, a treatment for autism, nystagmus, ethanol addiction, as an anticonvulsant, as an antispasmodic, as an anti-Parkinsonian drug with neuroprotective properties, as a medication for opioid dependence, for systemic lupus erythematosis, multiple sclerosis, depression, obsessive compulsive disorder, glaucoma, tinnitus, Tourette syndrome, problem gambling, neuropathic pain and neurological developmental and neurodegenerative disorders including HIV-associated dementia [50–58]. As longer term clinical data become available, the efficacy of memantine hydrochloride for AD management is becoming increasingly questionable [55]. While both AChE inhibitors and NMDA antagonists have been reported to possess both neuroprotective and neurotoxic properties depending on dose, patient age and AD severity and other confounding factors, one recent clinical study reports that for patients not receiving a cholinesterase inhibitor and randomized to receive memantine (20 mg/d) or placebo during a 24-week, double-blind, placebo-controlled trial, that while short-term evaluation showed a minimal improvement, prospectively defined analyses failed to demonstrate a statistically significant benefit of memantine treatment compared with placebo [51–58]. Using the severe impairment battery analysis and a 24 week end point, the AD cooperative study-activities of daily living scale (ADCS-ADLS) did not differ significantly between groups in any statistical analysis [53]. Combinatorial AChE-glutamatergic drug strategies, such as the use of memantine with cholinergic inhibitors in ‘anti-AD’ AChE inhibitor-glutamatergic-cocktails showed that memantine considered to be in the therapeutic or neuroprotective range for rats, induced neurotoxicity in the adult rat brain, and co-administration of memantine with the cholinesterase inhibitor donepezil markedly potentiated cerebrotoxic reactions, causing brain cell injury at significantly lower doses of memantine [54,55]. Approximately 5 years ago, due to memantine’s limited clinical effectiveness and high cost-to-benefit ratios, the National Institute for Health and Clinical Excellence (NICE) in the UK made the substantial decision to the National Health Board against the continued use of memantine as a medication for AD, although more recently this decision has been reversed [56]. Understandably, these decisions have generated much discussion amongst pharmaceutical companies, healthcare providers, AD researchers and within the UK health care system as a whole [55,56]. Another low-affinity NMDA receptor antagonist Dimebon (latrepirdine; dimebolin; 2,3,4,5-tetrahydro-2,8-dimethyl-5-(2-(6-methyl-3-pyridyl)ethyl)-1H-pyrido(4,3-b)indole; Pfizer, Medivation), originally developed in Russia as an over-the-counter oral antihistamine, has shown equally disappointing results in Phase III CONNECTION clinical trials of patients with mild-to-moderate AD [58,145,146,149]. Clinically, potential interactions, therapeutic failures and unexpected clinical outcomes using AChE inhibitor and glutamatergic antagonists, either alone or in combination have prompted the redesign of clinical strategies and hopefully a more efficacious series of medical formulations. Some novel and alternate pharmacological strategies for AD management are further discussed below. Importantly, while some moderate benefit has been realized in drugs that may help in the early or ‘pre-clinical’ stages of AD, there is a growing skepticism on the ability of any current drug formulation to slow the rate of decline in patients with more advanced stages of this progressively incapacitating disease.
4.3. Immunization against Aβ and tau peptides
An intuitive intervention strategy was proposed that immunization against Aβ-peptides would be highly effective in reducing excessive amounts of Aβ-peptides in the AD brain, and hence be useful in lowering Aβ-peptide-triggered pathogenic change. While early studies in βAPP over-expressing transgenic mice showed a significant reduction in the extent and progression of AD-like neuropathology, early therapeutic trials of immunization against Aβ42 in humans were discontinued due to the development of significant meningo-encephalitic cellular inflammatory reactions and related life-threatening complications [58–62]. While active immunization against Aβ peptides, has received extensive pre-clinical validation in AD mouse models, effective strategies free of significant side effects in humans, while problematic, are still under active development. The novel AAB-001 (Bapineuzumab; Elan, Pfizer and Wyeth) administered via intravenous injection, is a humanized monoclonal anti-Aβ antibody for mild to moderate AD that received fast track designation from the US FDA. One unexpected side effect of AAB-001 was vasogenic cerebral edema, and those taking the largest doses had to be been removed from clinical trials [64]. In general the results have again been disappointing, however a second generation anti-Aβ monoclonal antibody solanezumab (LY2062430; a humanized monoclonal antibody that binds to the central region of β-amyloid) is currently in phase III clinical trials [62,63,144,149]. Further investigation on the usefulness of the immunization against Aβ peptides awaits critical refinements in our understanding of the physiological complexities of γ-secretase-substrate recognition, γ-secretase mediated βAPP cleavage and the kinetics of Aβ peptide metabolism, translocation, proteolysis and clearance, and off-target effects [20–23,61–63]. Interestingly, antibodies directed against small regions of Aβ peptides (i.e Aβ peptide residues 4–10) have been shown to inhibit cytotoxicity and fibrillogenesis [59]. More recently DNA immunization with HBsAg-based particles expressing a B cell epitope of Aβ-peptide have been shown to attenuate disease progression that prolongs survival in AD mouse models [60]. Further, recent studies have shown that tau-targeted immunization slows the progression of NFT histopathology and pathology in aged P301L Tau transgenic mice [61,62]. Even more recently, APP-C99 substrate-targeting monoclonal antibodies appear to have properties as novel immunotherapeutic and Notch-sparing agents to lower the levels of Aβ peptides in mice, but whether the same effects can be duplicated in humans in currently not known [154].
4.4. Targeting α-secretase, β-secretase and γ-secretase Aβ42- peptide lowering agents
Stimulation of α-secretase cleavage of the βAPP holoprotein into the neurotrophic sAPPα has been widely considered a therapeutic approach for AD, but the molecular mechanisms regulating α-secretase substrate targeting and cleavage are not clearly understood. Protein kinase C and mitogen-activated protein kinases constitute complex central signaling hubs for the regulation of α-secretase cleavage. Additionally, recent studies increasingly demonstrate that a favorable spatial and temporal localization of the two membrane integral proteins βAPP and α-secretase is essential for efficient α-secretase cleavage and sAPPα generation [35–40,64].
To date a number of selective γ-secretase inhibitors have been identified that inhibit the processing of βAPP into pathogenic Aβ peptides, their implementation into clinical trials have so far been fraught with difficulty due to multiple off-target effects and associated gastrointestinal tract bleeding, hyperplasia, immunological and neurological disturbances, including severe headache [18–21]. Typically large single drug doses of greater than 100 mg are required to obtain significant anti-amyloidogenic effects [18–23]. LY450139 is one γ-secretase inhibitor currently in clinical development, with doses being optimized through the use of post-treatment biomarkers such as Aβ levels in the plasma and in the cerebrospinal fluid [20–22]. In one preliminary study using LY450139 a proportional drug dose and Aβ peptide lowering response was apparent in plasma and in CSF markers [19–21]. Still in development are compounds such as JLK6, a selective inhibitor of γ-secretase that inhibits βAPP cleavage with minimized off target effects on other γ-secretase-mediated pathways. For example JLK6 has been shown to inhibit Aβ peptide shedding from HEK293 cells over-expressing wild-type or Swedish-mutated βAPP (IC50 ~ 30 µM), but displays no apparent activity on BACE, α-secretase or the proteosome clearance system [Tocris Bioscience; Eli Lily; 20–25]. One concern is that the Aβ peptide generating γ-secretase is a rather unusual aspartic protease that cleaves within the hydrophobic plasma transmembrane domain, and the specific targeting of enzymes integral to the lipid bilayer is a relatively new area of pharmaceutical design [18,20]. An additional concern is that the γ-secretase is actually a multi-protein complex with multiple molecular substrates. How each of the proteins of this γ-secretase complex recognizes and cleaves target substrates is of paramount importance in the development of more precise inhibitors that more specifically target and preferentially impair Aβ peptide generating activities [19–22,117]. Viable targets for anti-amyloidogenic drugs is the β-amyloid cleavage enzyme (BACE; β-secretase) and its active site. The US Biotech company CoMentis initiated a phase I study of its orally bioavailable small molecule CTS-2166, purportedly a highly potent, highly selective and efficacious brain-penetrating β-secretase inhibitor but preliminary clinical results were disappointing [56,62,64]. More recently, oral administration the retinoid X receptor (RXR) agonist bexarotene (used as treatment for skin cancer for 13 years) has been shown to rapidly clear Aβ42 deposits from the brain and improve cognitive deficits in mouse models, but whether this might aid in clearance of Aβ-enriched amyloid lesions and Aβ-induced deficits in the human brain is again, currently not known [151].
4.5. Anti-cholesterol drugs: statins
4.5.1
A class of well-tolerated drugs known as statins are currently the most widely prescribed drugs in this country. Statins are inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase which normally catalyzes the conversion of HMG-CoA into mevalonic acid in the cholesterol biosynthesis pathway. Statins have been widely shown to reduce plasma cholesterol and hence the risk of development of cerebrovascular and cardiovascular disease and stroke [65–69]. Interestingly, both cerebrovascular and cardiovascular disease are often present in aged AD patients and appear to be weighty contributors to the development of AD [36–40,66–68]. Both experimental and clinical studies have indicated a significant inter-relationship between elevated serum cholesterol, cholesterol metabolism and trafficking, and the enhanced processing of βAPP into neurotoxic Aβ40-42 peptides, a process that appears to preferentially occur within cholesterol-enriched lipid-raft domains of plasma membranes [68–75]. In early clinical trials, statins have been shown to increase βAPP/Aβ ratios, while reducing cholesterol and blood plasma lipids in AD patients [71,74,75]. The lipophilic lovastatin and simvastatin, which readily cross the blood brain barrier, and the more hydrophilic statins atorvastatin, pravastatin and fluvastatin, that do not easily transit the blood brain barrier, are currently the most widely prescribed group of oral cholesterol-lowering drugs and have been shown to have multiple additional or off-target ‘pleiotrophic’ effects. For example, statin-mediated inhibition of mevalonic acid not only reduces cholesterol biosynthesis but also inhibits the biosynthesis of isoprenoids, resulting in a decrease in protein isoprenylation. This is thought to contribute substantially to the cell- and tissue-specific anti-inflammatory and immuno-modulatory effects of statin activity [66–71]. Other "pleiotropic" effects of statins relate to endothelial-vascular function, increased myocardial and microcapillary perfusion, and enhancement of the bioavailability of nitric oxide; the mechanism of these effects remain poorly understood [67–75]. It is abundantly clear that additional mechanistic studies, modifications in statin dose, speciation and dosing parameters, and longer carefully controlled clinical trials are essential to more fully evaluate the usefulness of statins as anti-Aβ peptide modulators in the treatment of AD (see further discussion in Section 7 below). Recently two excellent reviews on the role of statins in AD research and pharmacotherapy have appeared in the literature [75,83].
4.5.2. Other cholesterol-lowering strategies and AD-relevant pharmaceuticals
Cholesterol reduction represents an important pharmacological insight to Aβ peptide modulation via simultaneous effects on Aβ peptide production and cholesterol lowering effects. Benefits from non-statin, lipid-and-cholesterol lowering therapies include inhibitors of the intracellular enzyme acyl-coenzyme A cholesterol acyltransferase (ACAT) that catalyzes the formation of cholesterol esters from cholesterol and fatty acyl-coenzyme A. These appear to reduce systemic cholesterol levels by suppressing absorption of dietary cholesterol and suppressing the assembly and secretion of apolipoprotein-containing lipoproteins [75,76]. Another ACAT inhibitor CI-976 (2,2-methyl-N-(2,4,6,-trimethoxyphenyl dodecanamide) has beneficial effects on multiple membrane trafficking pathways in eukaryotic cells, including the stimulation of retrograde trafficking of Golgi membranes to the endoplasmic reticulum wherein βAPP holoprotein resides [76]. The ACAT inhibitor avasimibe has been shown to decrease excessive hepatic apolipoprotein secretion into plasma while significantly increasing clearance of triglyceride-rich lipoprotein from the systemic circulation [77]. The novel selective cholesterol transporter inhibitor ezetimibe was demonstrated to reduce plasma low density lipoproteins, either alone or in combination with statins [78–80]. Even more recently, selective estrogen receptor modulators have shown potential to reduce serum cholesterol in postmenopausal women who are at increased AD risk [81,82]. For clinical application of cholesterol absorption and transport inhibitors, ACAT inhibitors and estrogen modulators the development of more potent compounds and improvements in the methods to evaluate their clinical efficacy are strongly needed. Concerns about clinical outcomes, safety, and efficacy in various combinations still remain; their mechanistic effects on βAPP processing and Aβ generation remain poorly understood [81–84].
4.6. Other AD drugs and formulations targeted against the generation of Aβ peptide
Several additional compounds that are currently being evaluated may provide potential alternative treatments for AD in the future in part due to their actions on βAPP processing. Tramiprosate (honotaurine; Alzhemed®; NC-531) is a glycosaminoglycan-mimetic molecule that interferes with soluble Aβ-peptide accumulation and prevents the formation of toxic SP, however European trials for Alzhemed were halted in November 2007 [80–83]. Another novel compound scyllo-cyclohexanehexol (AZD-103, ELND-005), a blood-brain barrier permeable stereoisomer of inositol which binds and arrests Aβ aggregation has displayed promising activity in animal studies in reducing Aβ40 and Aβ42 load, inflammation and restoring synaptic loss and improving cognition [84]. Non-steroidal anti-inflammatory drugs (NSAIDs) that possess anti-amyloidogenic, anti-inflammatory, and anti-angiogenic properties [(R)-flurbiprofen; Tarenflurbil; MPC-7869; Flurizan®] has been of particular interest among the NSAID group because they are also γ-secretase inhibitors and have been reported to reduce Aβ42 peptide load [78,83,84].
Medicinal plant extracts that include ginkgo biloba and ginseng saponins, also known as ginsenosides, also have been shown to possess selective anti-oxidant, anti-inflammatory, anti-apoptotic, anti-angiogenic and immune-stimulatory activities, and activity comparisons with donepezil and tacrine have been reported [85–88]. Further, the ginsenosides Rb1, Rg3 and RE have been found to significantly lower Aβ peptide abundance in AD animal models [81,89]. Other novel preventive drugs and pharmacological strategies that may have potential for Aβ peptide lowering effects are the Chinese medicinal herb Uncararia rhynchophylla, reported to inhibit Aβ peptide aggregation and stabilization [82], and 9-tetrahydrocannabinol (THC), the active compound in marijuana that acts as both a competitive AChE inhibitor that prevents Aβ aggregation [90]. Most recently, two herbal formulations have been described to have therapeutic effects on the treatment of AD associated Aβ peptide generation and they are medicinal extracts from the plants Melissa officinalis, Salvia officinalis, and the Chinese concoctions Yi-Gan San and Ba Wei Di Huang Wan [78,91]. Each have been shown to have some beneficial functions on improving cognitive impairment of mild-to-moderate AD, larger sample, double blind, multi-centered clinical studies remain to be carried out [91]. Two current excellent reviews on neutraceuticals, herbal medicines and the role of phytochemicals and ethnobotanicals in the treatment and prevention of dementia including AD have recently appeared in the literature [93,94]. Importantly, there are published reports to the contrary of the beneficial effects of neutraceuticals and medicinal plant extracts in Ad treatment [144].
4.7. Chelation of neurotoxic trace metal ions inhibit of Aβ peptide aggregation
There has been recently a resurgence of interest in the role of neurotoxic trace metals on Aβ peptide aggregation and hence their neurotoxicity and contributory role to AD, and the possibility of using chelation of these trace metals to inhibit Aβ aggregation ino SP and Aβ-triggered neurotoxicity [98–103]. Both the βAPP holoprotein and secretase-generated Aβ peptides bind divalent [copper, iron (Fe2+), zinc] and trivalent [aluminum, iron (Fe3+)] metal ions under physiological conditions. Abundant evidence continues to support the idea that these metals play a role in Aβ crosslinking, aggregation, inflammation, oxidative stress and neurotoxicity of different Aβ-peptide species [98–103]. In the presence of nanomolar amounts of these metals, soluble Aβ peptide monomers nucleate into higher order structures yielding dense, insoluble SP cores. Aggregation of Aβ peptides in the presence of trace amounts of aluminum and iron is remarkable in its biophysical efficiency [100–103]. As Aβ peptides coalesce into SP, self-reinforcing and self-perpetuating episodes of oxidative stress and neural inflammation occur. In the early and preclinical stages of AD, when SP and NFT lesions first appear it has been suggested that it may already be too late to utilize chelation strategies, due to chelator access to the SP core and other accessibility and solubility problems [33,37]. Because metal-ion catalyzed oxidative stress and inflammatory signaling precede the appearance of aggregated Aβ peptides and SP early on in the AD process, combined or integrated anti-oxidant and metal-ion chelation strategies for neurotoxic metals may hold promise for the management of AD in addition to strategies using cholinergic enhancers, glutamate antagonists, statins and NSAID-based strategies [37,98–101]. At present, metal ion chelators including desferrioxamine, clioquinol, Feralex-G and silicon, including combinatorial approaches and mixed antioxidant-chelation strategies such as ‘molecular shuttle chelation’ are highly active areas of research investigation [98–101]. Interestingly, more than 20 years ago low dose treatment with intramuscular desferrioxamine, an antioxidant and trivalent ion chelator that can remove excessive iron and aluminum from the body, was reported to slow the progression of AD by a factor of two, but why this treatment worked still is not clear [100]. While a growing body of evidence supports the idea that trace amounts of neurotoxic metal ions are essential for Aβ peptide aggregation, virtually nothing is known about the interaction of these toxic trace metals with βAPP-processing secretases, statins, or other kinds of Aβ peptide modulators and other AD-relevant drug compounds.
4.8. Supplementation with Essential Fatty acids (docosahexaenoic acid; DHA) and neuroprotectin D1 (NPD1)
Both in vitro and in vivo studies strongly suggest that docosahexaenoic acid (DHA) and DHA derivatives such as neuroprotectin D1 (NPD1) may slow or prevent age-related cognitive dysfunction, memory loss and dementia [104–106,142,145–147], however there are important reports to the contrary [143,144]. Strategies employing omega-3 fatty acids, and most recently in co-administered with antioxidants, appear to be the most efficacious in the rescue of altered βAPP processing and in preserving cognition, especially in the early stages of AD [104–106, 146]. Both esterified and free DHA and NPD1 are seriously depleted in AD brain [104]. In human brain primary tissue culture cell models and in transgenic AD animal models, DHA depletion dramatically enhances oxidative damage. The recently described DHA-derivative and endogenous docosanoid NPD1 has been shown to lower Aβ42 peptide secretion from pro-inflammatory cytokine-stressed human neural cells, to up-regulate the neurotrophic sAPPα, to induce the expression of anti-apoptotic members of the Bcl-2 gene family, and to quench pro-inflammatory signaling and innate immune system markers [104,107]. However, while DHA and NPD1 activities are present in human brain cells, what secretase and accessory enzyme pathways are targeted by these are currently under active investigation [104–106]. Additional pharmacological regimens using a combinatorial approach, may be useful in the pharmacotherapy of AD and diseases with both a neurovascular and cardio-vascular component [36,106–110]. Individually tailored mixtures of eicosapentaenoic (EPA) and DHA or other fatty acids may be the most beneficial in the treatment of moderate AD; the optimal formulations for DHA and EPA for example still remain unclear [142,149]. Lifelong enrichment of DHA in the diet may provide the best DHA supplementation strategy in AD prevention [146]. Interestingly, in transgenic animal models of AD (Tg-AD) endogenous conversion of omega-6 into omega-3 fatty acids shows significant improvements AD-type neuropathology [147]. It should be again mentioned that there are potential toxic effects of omega-3 fatty acid supplementation in the elderly – for example there are reports that elevated omega-3 fatty acid levels predict the risk of ventricular arrhythmias [148] but there are again reports to the contrary [149].
4.9. Non-pharmaceutical based strategies
Long-standing attitudes of Westernized medicine to complex, age-related diseases such as AD is more often than not a ‘quick-fix’, pharmacological-based approach to deal with chronic human diseases that take many decades to develop [101–105]. Recently, significant non-pharmaceutical-based protection against diseases of the elderly such as AD, cardiovascular, neurovascular disease and diabetes can clearly be obtained from nutritional and dietary education and brain- and heart-healthy lifestyles. Diets for both adults and the elderly focusing on high fiber intake, restricted intake of trans- and saturated-animal fat and cholesterol reduction, dietary enrichment in antioxidant, DHA and omega-3-fatty acid intake, the maintenance of appropriate body weight, regular physical activity, the cessation of smoking and excessive alcohol consumption, introspective health care education and ethnobotanical approaches collectively represent a realistic, statistically significant, and cost-effective therapeutic strategy for the management of not only AD but other age-related disorders [104,105,111]. In addition to non-pharmacological approaches, psychotherapy involving cognitive and behavioral interventions and rehabilitation strategies that include nutritional intervention appear to be highly effective adjunctive treatments directed at the clinical management of AD, especially in the earlier stages of this disease [36–42,111–122]. Recent, goal-oriented cognitive rehabilitation approaches for early-stage AD patients has shown a very positive clinical benefit [123–125].
5. Current research goals
The current research goals for the clinical management of AD are summarized as follows:
to more completely understand the molecular, genetic and epigenetic mechanism of Aβ40 and Aβ42 peptide evolution, and limiting the off-target effects of β-secretase (BACE) and γ-secretase,
pharmacologically, to strategically design the neurotrophic processing of βAPP into sAPPα and to limit neurotoxic Aβ40 and Aβ42 peptide generation,
modulation of nicastrin, SORL1, BACE, PS1, Aph-1, Pen-2 and other effectors of βAPP activity with βAPP, secretase inhibitors and Aβ peptide-modulators to achieve these goals
to advance the thoughtful design and study of transgenic animal models which will provide valuable in vivo models of AD, and on which the actions of novel secretase inhibitors and Aβ peptide-modulators can be further evaluated
to further evaluate statin and related therapies that support a multi-targeted and multi-modal approach to the clinical management of AD
prophylaxis to carry out highly rigorous studies and clinical trials in large, statistically significant populations of early, mild and moderate AD
to explore other treatment approaches that lie outside of the ‘amyloid cascade hypothesis’
Indeed, the ideal AD drug might be an Aβ-peptide modulator that would attenuate the pathogenic production of Aβ40 and Aβ42 peptides while up-regulating the production of sAPPα, be effective in all stages (early, moderate and severe) of AD, be useful in the reduction of oxidative stress and inflammatory signaling, and have negligible hepatic, gastrointestinal, neurological, respiratory or other serious off-target effects. Put another way the ideal Aβ peptide modulator would target multiple AD pathogenic mechanisms and have pleiotropic beneficial off-target effects. Unfortunately, the probability of developing such an efficacious drug is remote. It may however be possible to develop pharmaceuticals which target not only the major pathways but also ancillary mechanisms that contribute to amyloidogenic signaling and AD progression.
6. Scientific rationale
While the progressive generation of Aβ peptides from βAPP holoprotein via the actions of various secretases appears to be a major contributory factor to the development of Aβ-peptide-triggered neurodegeneration, these membrane-associated secretase-mediated proteolytic mechanisms have a considerable number of ancillary regulatory controls. For example βAPP may also be degraded by other families of proteases in addition to the α-, β-, or γ-secretase system and these include brain abundant proteolytic enzymes such as neprilysin [101–105]. The amyloidogenesis of AD can be considered as a fundamental biomembrane-derived neuropathology that involves a membrane-spanning substrate, membrane-associated secretases and accessory proteins and cellular trafficking factors such as SORL1 ad nicastrin. For example, the βAPP processing mechanism and Aβ peptide generation appears to be up-regulated in lipid raft domains of brain cell membranes that are enriched in cholesterol, and cholesterol modifying medications such as statins have attractive interventional potential especially over the long term [71]. In addition, statins are of effective use in hypercholesterolaemia, hyper-lipidemic cardiovascular and neurovascular diseases, common disorders known to be contributory to for the development of AD [80–83,112,141,142].
7. Competitive environment
As has been previously discussed, AChE and glutamatergic medications for AD treatment are generally not highly effective, especially in advanced AD, however they may help some AD patients at certain stages of AD under certain treatment regimens. Although stains are not the ‘magic bullet’ for AD, generally, most recent data both in the lab and in the clinical setting is that the use of statin pharmacotherapy continues to looks more promising as treatment regimens become more refined [67–75,83]. Currently AstraZeneca (Crestor®), Bristol-Myers Squibb Co (Pravachol®), Glaxo-Smith-Kline and Merck & Co (Zocor®; Mevacor®), Novartis (Lescol®), and Pfizer (Lipitor®) are the 6 major manufacturers of currently marketed statins with combined sales in excess of 30 billion dollars per year. Clinical studies for statins for use in AD management are on the rise; as of 2005, 22 ongoing trials from these manufacturers evaluated the non-lipid lowering ‘pleotropic’ effects of statins; the number of clinical trials involving statins had increased to 106 by the year 2010 [114]. These multiple clinical drug trials are currently examining overlapping end-point assessments assessing markers for inflammation and atherosclerotic plaque stabilization, oxidized vascular oxidant stress and low density lipoprotein abundance, end-stage renal disease, serum viscosity and fibrinogen status, endothelial function, aortic stenosis progression, acute coronary syndrome, and pleiotrophic effects on hypertension, osteoporosis, ischemic burden, multiple sclerosis, and stroke. Given the generally well tolerated aspects of statins, the evaluation of their ‘pleiotropic’ effects has considerable potential to provide additional benefits to patients suffering from a number of overlapping diseases related to aging in addition to AD [80,111–113].
Secretase-directed inhibitor strategies are ongoing, however the development of non-selective protease inhibitors for γ-secretase has been difficult due in part to cleavage of related substrates such as the Notch receptor and resulting impairment of Notch-related signaling; there may 50 or more natural endogenous substrates for γ-secretase [11,12,19]. For example, gastrointestinal irregularities after high dose LY-450139 treatment is also an ongoing concern as is with all current γ-secretase inhibitors [19–21]. Unfortunately, there are scarce data available on the effects of γ-secretase inhibitors on brain Aβ peptide accumulation, aggregation and deposition after prolonged administration. A large study with (R)-flurbiprofen (Flurizan®), a selective Aβ42 lowering agent, that showed little effectiveness has clouded the perception that modulation of γ-secretase activity is a clinically effective approach for AD [17,18,78,83,132,134]. Finalized commentaries on the Flurizan® trials are available on the website (Myriad Pharmaceuticals) [88]. According to a recent study from Pharmacor, part of the PharmaCare group of companies, a dramatic change in market dynamics will begin when the first disease-modifying therapies, Flurizan® (R-flurbiprofen), Alzhemed® (NC-531) enter the markets of the United States, Western Europe and Japan and when sufficient time has elapsed to evaluate their overall efficacy [87–90]. Lastly, and as outlined above, targeting γ-secretase, β-secretase with statins hold promise for the future of AD therapy as combinatorial drug treatment and improvements in pharmacotherapeutic strategy and design are further advanced.
8. Potential development issues
8.1. Statins
While current agents for anti-amyloid treatments provide limited benefits on the various target symptoms and despite some rather limited positive outcomes, potentially harmful side effects of statin-based therapies include hepatotoxicity, skeletal muscle pain and inflammation, (sometimes referred to as statin-induced myopathy or rhabdomyloysis), and symptoms similar to muscular dystrophy such as difficulty in initiating walking, rising from a seated position, gait, coordination, tremor, droopy eye, dry eye, memory loss, personality disturbances and irritability, and depletion of coenzyme Q10, which is, ironically, an essential nutrient for cardiovascular and neurovascular response, strength and function. There are additional emerging controversies concerning the suitability and effectives of statins for AD therapy. A recent animal study showed that in female Tg-AD (Tg-2576; transgenic, amyloid overexpressing) mice, lovastatin indeed lowered plasma cholesterol concentrations, but actually enhanced the rate of Aβ production and senile plaque deposition in the brain [109]. In one prospective, randomized, 36-week clinical treatment trial, no significant change in the level of Aβ peptides was found after statin therapy in hypercholesterolemic patients [110]. In another recent AD cholesterol-lowering treatment (ADCLT) double-blind, placebo-controlled, randomized (1:1) trial with a 1-year exposure to atorvastatin involving 166 individiuals, 98 of whom had mild-to-moderate AD (MMSE score of 12–28) circulating cholesterol levels, apoE genotype and initial dementia severity influenced the benefit of atorvastatin treatment [74,107]. Clearly these early studies should be interpreted with caution, and further careful experimentation and refinement in dosage, regimen, patient selection and characteristics etc., are required. Larger, well controlled human clinical trials using statins as a potential treatment for AD are now in progress. As of the writing of this paper 83 clinical drug trials for mild cognitive impairment, dementia and AD are now in progress and the Alzheimer’s Research Forum ‘Drugs in Clinical trials’ webpage lists another 61 drug studies in progress, many of which quantify serum or CSF Aβ peptide load as an outcome measure [108].
8.2. Secretase inhibitors
Clinical trials employing γ-secretase inhibitors are in still in their early stages, and there is a both a need for larger clinical trials and a close monitoring of patients for adverse gastrointestinal, hepatocytic, and immune system effects. Importantly, the neural membrane bound γ-secretase complex has at least 40 endogenous substrates that play critical roles in the adult human and γ-secretase inhibitor off target effects are a common clinical problem. For the same reasons strategies directed at β-secretase inhibition have met with equal difficulty [14–17]. Increasing evidence shows γ-secretase-mediated disruption of the neural net, especially during development, and another concern is that short term lowering of Aβ peptides in plasma are often followed by a rebound to significantly elevated blood serum levels of Aβ peptides [14–17].
9. Conclusion and Perspectives
While current academic and pharmaceutical research and pharmacological efforts to address the AD problem are immense, the prognosis for the AD patient remains dismal - pharmaceutical intervention strategies to date have been rather disappointing. One hundred and six years since Alois Alzheimer’s first description of this neurological affliction, a tremendous amount of scientific insight into the etiological, molecular-genetic, and pathological mechanisms of AD have been obtained, however, many appreciable gaps in our knowledge still remain [32–36]. While the neurotoxic Aβ peptides generated via the ‘amyloid cascade hypothesis’ and the secretase systems which generate them are an obvious therapeutic target, their management in the AD patient still remains an elusive, and to date no pharmaceutical-based clinical trial has yet successfully stabilized or reversed the course of this devastating neurological affliction. Currently only 4 therapeutics are being investigated in phase III clinical trials for AD; the γ-secretase inhibitor Tarenflurbil, the immunotherapeutics Bapineuzumab and solanezumab, and the low-affinity NMDA receptor antagonist and anti-acetylcholinesterase Dimebon (latrepirdine) [88,90,114,147, 148]. To date the results of clinical trials for all 4 drugs have been extremely disappointing [64,132–134,152–154].
Many researchers and clinicians agree that a cure for AD is probably not an achievable goal. The more realistic primary pharmacological strategy is to slow down the rate of cognitive decline in AD patients so that the disease can be more effectively managed by family care-givers and by our health care system. Familial and sporadic forms of AD account for 5% and 95% of all AD cases, respectively. Both forms of AD appear to be the consequence of composite age-related interactions between complex combinations of CNS genes, environmental and epigenetic factors. Environmental factors include lifestyle adjustable parameters such as tobacco and alcohol consumption, cholesterol and dietary saturated fat intake, environmental exposure to neurotoxic trace metals, previous viral infection, familial genetic history, predisposition to cardiovascular, neurovascular and neurological disease, educational background, drug medication history, etc., hence the etiopathogenesis of AD remains highly complicated and interactive both with both genetic and environmental components. There is clear room for improvement in the rather limited pharmaceutical arsenal we now possess to combat AD onset or delay AD development. While some drugs help some AD patients some of the time, it could still be easily argued that in the second hundred years of AD research that we are still in the infancy in our basic understanding of the AD process and in the development of suitable drugs and pharmaceutical strategies to arrest or if possible, reverse AD’s devastating neurological effects.
On a personal note I was once in the audience of a lecture given by Linus Pauling who, during an after-session question period, was asked about the intra-membrane γ-secretase cleavage site for beta-amyloid precursor protein (βAPP), a cleavage site that, as reviewed above, lies within the intensely hydrophobic domain of the plasma membrane lipid bilayer. As Pauling was the person who wrote the book on the peptide bond, amino acid hydrolysis, and the very nature of the peptide and hydrogen bonds, his first response was that this could not possibly happen, as you need an aqueous environment for any type of peptide hydrolysis and polypeptide chain scission. This was followed by a question by Pauling if the γ-secretase cleavage site and resultant Aβ peptides were discrete or ragged? The answer to this is of course, ragged – and it is indeed observed that many kinds of βAPP-excised Aβ peptides –Aβ40, Aβ42, and sometimes Aβ37 and Aβ43 peptides, are in fact generated. Pauling went on to interestingly comment that (i) either the lipid bilayer plasma membrane must be damaged or distorted to enable such polypeptide scission to occur, or (ii) equally likely that there must be some ‘vibrational’ movement of that segment of the βAPP polypeptide to transiently expose the γ-secretase cleavage site into an aqueous internal or external environment, where cleavage could rapidly occur. Hence, more research attention might be given to the basic in vivo biology of βAPP as an entity within various limiting and plasma membranes, and how membrane biophysics affects the generation of amyloidogenic Aβ peptides (Figure 1).
Indeed, novel research approaches, strategies, methodologies and clear outcome-defined clinical treatments based on the rapidly evolving AD research area are urgently required to more effectively contain the current AD situation. Over the long term there is clear benefit to be obtained in the prevention of AD by educating our aging population on the basic mechanisms of AD and the genetic, environmental and lifestyle factors associated with increasing the risk for this disease, and related to this, the diversion of our aging populations to more brain- and heart-healthy lifestyles [121–127]. Combinatorial studies involving pharmaceutical therapy and lifestyle modifications, or combinations of pharmaceuticals and traditional plant-derived natural medicines, have not been adequately addressed for use in AD treatment and these may ultimately prove to be beneficial. Hopefully, four dimensional quantitative analysis of the recognized pathological markers for AD onset and progression using novel imaging techniques should further facilitate the implementation of more efficacious pharmacological strategies for modulating Aβ peptide directed neuropathology and the advancement more effective AD treatment.
Lastly, the recent discoveries of docosahexaenoic acid (DHA) derivatives such as the brain’s endogenous neuroprotection system involving neuroprotectin D1 (NPD1) [104,105], and the novel manipulation of anti-micro RNA (anti-miRNA; antagomir) and antagomir-based strategies directed against degenerating human brain cells [128–131] have provided highly novel therapeutic applications and AD treatment strategies that have not previously been considered. Indeed such novel alternative, and perhaps combinatorial treatment strategies, are currently receiving a considerable amount of research attention as they are scrutinized by both academia and the pharmacology industry. These emerging AD treatment strategies include:
education of our aging population on the basic mechanisms of AD and the genetic, environmental and lifestyle factors associated with increasing the risk for this disease;
a daily exercise regimen in adults and the aged, including a heart- and brain-healthy lifestyle;
multiple γ-secretase and β-secretase inhibitors designed to reduce Aβ-peptide generation, and minimize both off-target effects and hepatotoxicity;
Aβ-peptide immunization strategies;
omega-3 fatty acid dietary (fish oil) supplementation such as DHA and DHA derivatives, such as neuroprotectin D1 (NPD1); most recently in combination with antioxidants such as vitamin E; combinations of EPA and DHA
various cholesterol-reducing 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-coA) reductase inhibitors (statins);
recently characterized lipid- and water-soluble antioxidants including chelators for neurotoxic metals such as aluminum and mercury;
natural plant extracts and nutriceuticals from the extensive global pharmacopeia including ginkgo biloba, huperzine, acai, curcumin, turmeric and others;
recent gene expression inhibition and activation strategies using small non-coding RNA, miRNA and anti-miRNA (antagomirs) to interfere with expression of disease causing genes;
combinatorial approaches to the above mentioned treatments
In conclusion, the need for more efficacious treatment approaches for the clinical management of AD is long past due. Research work and clinical trials up to mid-2008 have been previously reviewed by this author, and much of the earlier research findings have been updated, wherever appropriate, in the present communication [64]. Currently, clinicaltrials.gov, a publically accessible website maintained by the US National Institutes of Health, describes 1013 AD studies and human clinical trials at the planned, recruiting and completed stage to more effectively address this most serious and expanding healthcare concern [64,114,126,127].
10. Expert opinion
Despite 106 years of research effort, and the many hundreds of millions of dollars spent on AD research since Alois Alzheimer first described this neurological disorder, there is not one pharmacological strategy, nor drug formulation, currently available to effectively contain this rapidly expanding health care concern. We are seriously ill-prepared to deal with this immense health and socioeconomic problem that is now reaching epidemic proportions [165]. While some benefit may be realized in drugs that may help in the early stages of AD, there is sincere skepticism on the ability of any drug treatment to slow the rate of decline in patients with moderate-to-advanced AD. Of totally questionable usefulness are the frequent press releases of an ‘Alzheimer breakthrough’ with cures ‘just around the corner’ - these claims provide false hope to the public, and incorrectly allude to the dynamic progress being made in elucidating a cure for AD, which is, in reality, virtually nil. Perhaps it is well time to actively pursue other hypotheses concerning the AD mechanism that do not purely focus on the ‘amyloid cascade hypothesis’ so that real research progress can be made, and an efficacious treatment for AD can be found.
Acknowledgements
Thanks are extended to Drs P. Alexandrov, B. Bhattacharjee, C. Eicken, P.E. Fraser, C. Hebel, J.M. Hill, T.P.A. Kruck, M.E. Percy, A.I. Pogue, W. Poon, E. Rogaev, T. Saing and Y. Zhao for helpful discussions and ongoing collaboration and to Darlene Guillot for expert technical assistance.
Declaration of interest:
This work was supported in part by grants from the Translational Research Initiative (LSUHSC, New Orleans LA, USA), the Louisiana Biotechnology Research Network (LBRN; Baton Rouge LA, USA), an Alzheimer Association Investigator-Initiated Research Grant IIRG-09-131729 (Chicago IL, USA), and from National Institutes of Health NIA AG18031 and NIH NIA AG038834 (Bethesda MD, USA).
Bibliography
Research papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1. Alzheimer Association. 2011 Alzheimer’s Disease Facts and Figures statistical resource. http://wwwalzorg/downloads/Facts_Figures_2011pdf. • Compilation of AD-relevant facts and figures
- 2.Vellas B, Andrieu S, Sampaio C, Wilcock G. Disease-modifying trials in Alzheimer’s disease: a European Task Force group. Lancet Neurol. 2007;6:56–62. doi: 10.1016/S1474-4422(06)70677-9. [DOI] [PubMed] [Google Scholar]
- 3. Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. An English translation of Alzheimer’s 1907 paper “Uber eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 1995;8:429–431. doi: 10.1002/ca.980080612. •• For neuroscience graduate student, neurologists and AD researchers a must read
- 4.Lukiw WJ. One hundred years of Alzheimer’s disease research: are we any closer to a cure? Aging Health. 2007;3:279–282. [Google Scholar]
- 5. Hardy J. Alzheimer's disease: the amyloid cascade hypothesis: an update and reappraisal. J Alzheimers Dis. 2006;9:151–153. doi: 10.3233/jad-2006-9s317. •• What is wrong and what is right with the amyloid cascade hypothesis of AD.
- 6.Nilsson S, Koehler KF, Gustafsson JÅ. Development of subtype-selective oestrogen receptor-based therapeutics. Nat Rev Drug Discov. 2011;10:778–792. doi: 10.1038/nrd3551. [DOI] [PubMed] [Google Scholar]
- 7.Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10:698–712. doi: 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- 8. Zhao Y, Cui JG, Lukiw WJ. Natural secretory products of human neural and microvessel endothelial cells:Implications in pathogenic "spreading" and Alzheimer's disease. Molecular Neurobiology. 2006;34:181–192. doi: 10.1385/MN:34:3:181. •• Novel hypothesis on the pathological spreading of AD throughout the human brain.
- 9.Zhao Y, Zhao B. Natural antioxidants in prevention and management of Alzheimer's disease. Front Biosci (Elite Ed) 2012;4:794–808. doi: 10.2741/419. [DOI] [PubMed] [Google Scholar]
- 10.Armstrong RA. Plaques and tangles and the pathogenesis of Alzheimer's disease. Folia Neuropathol. 2006;44:1–11. [PubMed] [Google Scholar]
- 11. Hardy J. Alzheimer's disease: the amyloid cascade hypothesis: an update and reappraisal. J. Alzheimers Dis. 2006;9:151–153. doi: 10.3233/jad-2006-9s317. •• Comprehensive and critical review on the contribution of amyloid signaling to AD.
- 12. Selkoe DJ. Resolving controversies on the path to Alzheimer's therapeutics. Nat Med. 2011;17:1060–1065. doi: 10.1038/nm.2460. •• Comprehensive evaluation of current approaches to AD therapeutics
- 13.Esiri MM, Chance SA. Vulnerability to Alzheimer's pathology in neocortex: the roles of plasticity and columnar organization. J Alzheimers Dis. 2006;9:79–89. doi: 10.3233/jad-2006-9s310. [DOI] [PubMed] [Google Scholar]
- 14. Cui JG, Hill JM, Zhao Y, Lukiw WJ. Expression of inflammatory genes in the primary visual cortex of late-stage Alzheimer's disease. Neuroreport. 2007;18:115–119. doi: 10.1097/WNR.0b013e32801198bc. •• Pathological factors in AD brain – spreading from association to primary sensory areas.
- 15.van de Pol LA, Korf ES, van der Flier WM, Brashear HR, Fox NC, Barkhof F, Scheltens P. Magnetic resonance imaging predictors of cognition in mild cognitive impairment. Arch Neurol. 2007;64:1023–1028. doi: 10.1001/archneur.64.7.1023. [DOI] [PubMed] [Google Scholar]
- 16.Marshall GA, Monserratt L, Harwood D, Mandelkern M, Cummings JL, Sultzer DL, Laforce R, Jr, Rabinovici GD. Amyloid imaging in the differential diagnosis of dementia: review and potential clinical applications. Alzheimers Res Ther. 2011;3:31–34. doi: 10.1186/alzrt93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Velliquette RA, O'Connor T, Vassar R. Energy inhibition elevates beta-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: possible early events in Alzheimer's disease pathogenesis. J Neurosci. 2005;25:10874–10883. doi: 10.1523/JNEUROSCI.2350-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ewers M, Sperling RA, Klunk WE, Weiner MW, Hampel H. Neuroimaging markers for the prediction and early diagnosis of Alzheimer's disease dementia. Trends Neurosci. 2011;34:430–442. doi: 10.1016/j.tins.2011.05.005. •• An excellent and current review of the electronic imaging of AD brain neuropathology
- 19.Siemers ER, Quinn JF, Kaye J, Farlow MR, Porsteinsson A, Tariot P, Zoulnouni P, Galvin JE, Holtzman DM, Knopman DS, Satterwhite J, Gonzales C, Dean RA, May PC. Effects of a gamma-secretase inhibitor in a randomized study of patients with Alzheimer disease. Neurology. 2006;66:602–604. doi: 10.1212/01.WNL.0000198762.41312.E1. [DOI] [PubMed] [Google Scholar]
- 20. Tomita T. At the frontline of Alzheimer's disease treatment: gamma-secretase inhibitor/modulator mechanism. Naunyn Schmiedebergs Arch Pharmacol. 2008;377:295–300. doi: 10.1007/s00210-007-0206-2. •• A thoughtful review of Aβ42-lowering strategies for AD treatment
- 21.Siemers ER, Dean RA, Friedrich S, Ferguson-Sells L, Gonzales C, Farlow MR, May PC. Safety, tolerability, and effects on plasma and cerebrospinal fluid amyloid-beta after inhibition of gamma-secretase. Clin Neuropharmacol. 2007;30:317–325. doi: 10.1097/WNF.0b013e31805b7660. [DOI] [PubMed] [Google Scholar]
- 22.Epis R, Marcello E, Gardoni F, Luca MD. Alpha, beta-and gamma-secretases in Alzheimer's disease. Front Biosci (Schol Ed) 2012;4:1126–1150. doi: 10.2741/s322. [DOI] [PubMed] [Google Scholar]
- 23.Van Broeck B, Van Broeckhoven C, Kumar-Singh S. Current insights into molecular mechanisms of Alzheimer disease and their implications for therapeutic approaches. Neurodegener Dis. 2007;4:349–365. doi: 10.1159/000105156. [DOI] [PubMed] [Google Scholar]
- 24.Dong J, Lu K, Lakdawala A, Mehta AK, Lynn DG. Controlling amyloid growth in multiple dimensions. Amyloid. 2006;13:206–215. doi: 10.1080/13506120600960809. [DOI] [PubMed] [Google Scholar]
- 25.Li H, Wolfe MS, Selkoe DJ. Toward structural elucidation of the gamma-secretase complex. Structure. 2009;17:326–334. doi: 10.1016/j.str.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lukiw WJ. Docosahexaenoic acid and amyloid-beta peptide signaling in Alzheimer's disease. World Rev Nutr Diet. 2009;99:55–70. doi: 10.1159/000192996. [DOI] [PubMed] [Google Scholar]
- 27. Lukiw WJ, Bazan NG. Survival signaling in Alzheimer's disease. Biochem Soc Trans. 2006;34:1277–1282. doi: 10.1042/BST0341277. •• A comprehnsive review of DHA and NPD1 strategies for the treatment of AD
- 28.Thornton E, Vink R, Blumbergs PC, Van Den Heuvel C. Soluble amyloid precursor protein alpha reduces neuronal injury and improves functional outcome following diffuse traumatic brain injury in rats. Brain Res. 2006;1094:38–46. doi: 10.1016/j.brainres.2006.03.107. [DOI] [PubMed] [Google Scholar]
- 29.Dubé JB, Johansen CT, Hegele RA. Sortilin: an unusual suspect in cholesterol metabolism: from GWAS identification to in vivo biochemical analyses, sortilin has been identified as a novel mediator of human lipoprotein metabolism. Bioessays. 2011;33:430–437. doi: 10.1002/bies.201100003. [DOI] [PubMed] [Google Scholar]
- 30.Offe K, Dodson SE, Shoemaker JT, Fritz JJ, Gearing M, Levey AI, Lah JJ. The lipoprotein receptor LR11 regulates amyloid beta production and amyloid precursor protein traffic in endosomal compartments. J Neurosci. 2006;26:1596–1603. doi: 10.1523/JNEUROSCI.4946-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. http://wwwhealthscoutcom/animation/68/7/mainhtml#transcript; www.knol.google.com/k/bapineuzumab-pfizer-j-j-elan-review#.
- 32.Zhang H, Ma Q, Zhang YW, Xu H. Proteolytic processing of Alzheimer's β-amyloid precursor protein. J Neurochem. 2012;120:9–21. doi: 10.1111/j.1471-4159.2011.07519.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou ZD, Chan CH, Ma QH, Xu XH, Xiao ZC, Tan EK. The roles of amyloid precursor protein (APP) in neurogenesis: Implications to pathogenesis and therapy of Alzheimer disease. Cell Adh Migr. 2011;5:280–292. doi: 10.4161/cam.5.4.16986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, Katayama T, Baldwin CT, Cheng R, Hasegawa H, Chen F, Shibata N, Lunetta KL, Pardossi-Piquard R, Bohm C, Wakutani Y, Cupples LA, Cuenco KT, Green RC, Pinessi L, Rainero I, Sorbi S, Bruni A, Duara R, Friedland RP, Inzelberg R, Hampe W, Bujo H, Song YQ, Andersen OM, Willnow TE, Graff-Radford N, Petersen RC, Dickson D, Der SD, Fraser PE, Schmitt-Ulms G, Younkin S, Mayeux R, Farrer LA, St George-Hyslop P. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39:168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao Y, Cui JG, Lukiw WJ. Reduction of sortilin-1 in Alzheimer hippocampus and in cytokine-stressed human brain cells. Neuroreport. 2007;18:1187–1191. doi: 10.1097/WNR.0b013e32821c56c4. [DOI] [PubMed] [Google Scholar]
- 36.Lukiw WJ, Pappolla M, Pelaez RP, Bazan NG. Alzheimer's disease-a dysfunction in cholesterol and lipid metabolism. Cell Mol Neurobiol. 2005;25:475–483. doi: 10.1007/s10571-005-4010-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lukiw WJ. 100 years of Alzheimer’s disease research: are we any closer to a cure? Aging Health. 2007;3:279–282. ** Comprehensive overview review on progress in AD over the last 101 years.
- 38.Thal LJ. Prevention of Alzheimer disease. Alzheimer Dis Assoc Disord. 2006;20:S97–S99. doi: 10.1097/00002093-200607001-00015. [DOI] [PubMed] [Google Scholar]
- 39. Hardy J. A hundred years of Alzheimer's disease research. Neuron. 2006;52:3–13. doi: 10.1016/j.neuron.2006.09.016. ** A candid overview review on progress in AD over the last 100 years.
- 40.Tanzi RE, Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 41.Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, Hall K, Hasegawa K, Hendrie H, Huang Y, Jorm A, Mathers C, Menezes PR, Rimmer E, Scazufca M. Alzheimer's Disease International. Global prevalence of dementia: a Delphi consensus study. Lancet. 2005;366:2112–2117. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Delrieu J, Piau A, Caillaud C, Voisin T, Vellas B. Managing cognitivedysfunction through the continuum of Alzheimer's disease: role of pharmacotherapy. CNS Drugs. 2011;25:213–226. doi: 10.2165/11539810-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 43.Lyketsos CG, Colenda CC, Beck C, et al. Task Force of American Association for Geriatric Psychiatry. Position statement of the American Association for Geriatric Psychiatry regarding principles of care for patients with dementia resulting from Alzheimer disease. Am J Geriatr Psychiatry. 2006;14:561–572. doi: 10.1097/01.JGP.0000221334.65330.55. [DOI] [PubMed] [Google Scholar]
- 44.Reitz C. Dyslipidemia and Dementia: Current Epidemiology, Genetic Evidence, and Mechanisms Behind the Associations. J Alzheimers Dis. 2011 Sep 30; doi: 10.3233/JAD-2011-110599. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Marum RJ. Update on the use of memantine in Alzheimer's disease. Neuropsychiatr Dis Treat. 2009;5:237–247. doi: 10.2147/ndt.s4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Birks J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst Rev. 2006;1 doi: 10.1002/14651858.CD005593. CD00593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salomone S, Caraci F, Leggio GM, Fedotova J, Drago F. New pharmacological strategies for treatment of Alzheimer's disease: focus on disease-modifying drugs. Br J Clin Pharmacol. 2011 doi: 10.1111/j.1365-2125.2011.04134.x. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holzgrabe U, Kapkova P, Alptuzun V, Scheiber J, Kugelmann E. Targeting acetylcholin-esterase to treat neurodegeneration. Expert Opin Ther Targets. 2007;11:161–179. doi: 10.1517/14728222.11.2.161. [DOI] [PubMed] [Google Scholar]
- 49.Yanagisawa K. Newly approved drugs for Alzheimer disease: effectiveness and limitation. Brain Nerve. 2011;63:863–868. [PubMed] [Google Scholar]
- 50.Herrmann N, Chau SA, Kircanski I, Lanctôt KL. Current and emerging drug treatment options for Alzheimer's disease: a systematic review. Drugs. 2011;71:2031–2065. doi: 10.2165/11595870-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 51.Tanovice A, Alfaro V. Glutamate-related excitotoxicity neuroprotection with memantine, an uncompetitive antagonist of NMDA-glutamate receptor, in Alzheimer’s disease and vascular dementia. Rev Neurol. 2006;42:607–616. [PubMed] [Google Scholar]
- 52.Bordji K, Becerril-Ortega J, Buisson A. Synapses, NMDA receptor activity and neuronal Aβ production in Alzheimer's disease. Rev Neurosci. 2011;22:285–294. doi: 10.1515/RNS.2011.029. [DOI] [PubMed] [Google Scholar]
- 53.van Dyck CH, Tariot PN, Meyers B, Malca Resnick E and the Memantine MEM-MD-01 Study Group. A 24-week randomized, controlled trial of memantine in patients with moderate-to-severe Alzheimer disease. Alzheimer Dis Assoc Disord. 2007;21:136–143. doi: 10.1097/WAD.0b013e318065c495. [DOI] [PubMed] [Google Scholar]
- 54.Creeley CE, Wozniak DF, Nardi A, Farber NB, Olney JW. Donepezil markedly potentiates memantine neurotoxicity in the adult rat brain. Neurobiol Aging. 2008;29:153–167. doi: 10.1016/j.neurobiolaging.2006.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mount C, Downton C. Alzheimer's disease: progress or profit? Nature Medicine. 2006;3:1280–1284. doi: 10.1038/nm0706-780. •• A critical review on advancement, complexity and profiteering in AD treatment
- 56.Iliffe S. The National Institute for Health and Clinical Excellence (NICE) and drug treatment for Alzheimer's disease. CNS Drugs. 2007;21:177–184. doi: 10.2165/00023210-200721030-00001. [DOI] [PubMed] [Google Scholar]
- 57.McShane R, Areosa Sastre A, Minakaran N. Memantine for dementia. Cochrane Database Syst Rev. 2006 Apr;19(2) doi: 10.1002/14651858.CD003154.pub5. CD003154. [DOI] [PubMed] [Google Scholar]
- 58.Khairallah MI, Kassem LA. Alzheimer's disease: current status of etiopathogenesis and therapeutic strategies. Pak J Biol Sci. 2011;14:257–272. doi: 10.3923/pjbs.2011.257.272. [DOI] [PubMed] [Google Scholar]
- 59.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400 doi: 10.1038/22124. 173-7.60 54. [DOI] [PubMed] [Google Scholar]; McLaurin J, Cecal R, Kierstead ME, Tian X, Phinney AL, Manea M, French JE, Lambermon MH, Darabie AA, Brown ME, Janus C, Chishti MA, Horne P, Westaway D, Fraser PE, Mount HT, Przybylski M, St George-Hyslop P. Therapeutically effective antibodies against amyloid-beta peptide target amyloid-beta residues 4–10 and inhibit cytotoxicity and fibrillogenesis. Nat Med. 2002;8:1263–1269. doi: 10.1038/nm790. [DOI] [PubMed] [Google Scholar]
- 60.Olkhanud PB, Mughal M, Ayukawa K, Malchinkhuu E, Bodogai M, Feldman N, Rothman S, Lee J, Chigurupati S, Okun E, Nagashima K, Mattson MP, Biragyn A. DNA immunization with HBsAg-based particles expressing a B cell epitope of amyloid β-peptide attenuates disease progression and prolongs survival in a mouse model of Alzheimer's disease. Vaccine. 2012 Jan 13; doi: 10.1016/j.vaccine.2011.12.136. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grimm MO, Rothhaar TL, Hartmann T. The role of APP proteolytic processing in lipid metabolism. Exp Brain Res. 2011 Dec 17; doi: 10.1007/s00221-011-2975-6. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 62.Panza F, Frisardi V, Solfrizzi V, Imbimbo BP, Logroscino G, Santamato A, Greco A, Seripa D, Pilotto A. Immunotherapy for Alzheimer's disease: from anti-β-amyloid to tau-based immunization strategies. Immunotherapy. 2012;4:213–238. doi: 10.2217/imt.11.170. [DOI] [PubMed] [Google Scholar]
- 63.Imbimbo BP, Ottonello S, Frisardi V, Solfrizzi V, Greco A, Seripa D, Pilotto A, Panza F. Solanezumab for the treatment of mild-to-moderate Alzheimer's disease. Expert Rev Clin Immunol. 2012;8:135–149. doi: 10.1586/eci.11.93. [DOI] [PubMed] [Google Scholar]
- 64.Lukiw WJ. Emerging amyloid beta (Aβ) peptide modulators for the treatment of Alzheimer's disease (AD) Expert Opin Emerg Drugs. 2008;13:255–271. doi: 10.1517/14728214.13.2.255. [DOI] [PubMed] [Google Scholar]
- 65.Carlsson CM, Xu G, Wen Z, Barnet JH, Blazel HM, Chappell RJ, Stein JH, Asthana S, Sager MA, Alsop DC, Rowley HA, Fain SB, Johnson SC. Effects of atorvastatin on cerebral blood flow in middle-aged adults at risk for Alzheimer's disease: a pilot study. Curr Alzheimer Res. 2011 Nov 28; doi: 10.2174/156720512803251075. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Santos DB, Peres KC, Ribeiro RP, Colle D, Santos AA, Moreira EL, Souza DO, Figueiredo CP, Farina M. Probucol, a lipid-lowering drug, prevents cognitive and hippocampal synaptic impairments induced by amyloid β peptide in mice. Exp Neurol. 2011 Dec 8; doi: 10.1016/j.expneurol.2011.11.036. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 67.Gelosa P, Cimino M, Pignieri A, Tremoli E, Guerrini U, Sironi L. The role of HMG-CoA reductase inhibition in endothelial dysfunction and inflammation. Vasc Health Risk Manag. 2007;3:567–577. [PMC free article] [PubMed] [Google Scholar]
- 68. Jasińska M, Owczarek J, Orszulak-Michalak D. Statins: a new insight into their mechanisms of action and consequent pleiotropic effects. Pharmacol Rep. 2007;59:483–499. •• Critical review on the pleiotropic effects of statin apart from cholesterol down-regulation
- 69.Parsons RB, Farrant JK, Price GC, Subramaniam D, Austen BM. Regulation of the lipidation of beta-secretase by statins. Biochem Soc Trans. 2007;35:577–582. doi: 10.1042/BST0350577. [DOI] [PubMed] [Google Scholar]
- 70.Butterfield DA, Barone E, Di Domenico F, Cenini G, Sultana R, Murphy MP, Mancuso C, Head E. Atorvastatin treatment in a dog preclinical model of Alzheimer's disease leads to up-regulation of haem oxygenase-1 and is associated with reduced oxidative stress in brain. Int J Neuropsychopharmacol. 2011;18:1–7. doi: 10.1017/S1461145711001118. [DOI] [PubMed] [Google Scholar]
- 71. Cordy JM, Hooper NM, Turner AJ. The involvement of lipid rafts in Alzheimer's disease. Mol Membr Biol. 2006;23:111–122. doi: 10.1080/09687860500496417. •• the contribution of cholesterol to Aβ generation and signaling in AD
- 72.Warren MW, Hynan LS, Weiner MF. Lipids and adipokines as risk factors for Alzheimer's disease. J Alzheimers Dis. 2012 Jan 9; doi: 10.3233/JAD-2012-111385. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ledesma MD, Dotti CG. Peripheral cholesterol, metabolic disorders and Alzheimer's disease. Front Biosci (Elite Ed) 2012;4:181–194. doi: 10.2741/e368. [DOI] [PubMed] [Google Scholar]
- 74.Wolozin B, Manger J, Bryant R, Cordy J, Green RC, McKee A. Re-assessing the relationship between cholesterol, statins and Alzheimer's disease. Acta Neurol Scand Suppl. 2006;185:63–70. doi: 10.1111/j.1600-0404.2006.00687.x. [DOI] [PubMed] [Google Scholar]
- 75.Pac-Soo C, Lloyd DG, Vizcaychipi MP, Ma D. Statins: the role in the treatment and prevention of Alzheimer's neurodegeneration. J Alzheimers Dis. 2011;27:1–10. doi: 10.3233/JAD-2011-110524. [DOI] [PubMed] [Google Scholar]
- 76.Parri HR, Hernandez CM, Dineley KT. Research update: Alpha7 nicotinic acetylcholine receptor mechanisms in Alzheimer's disease. Biochem Pharmacol. 2011;82:931–942. doi: 10.1016/j.bcp.2011.06.039. [DOI] [PubMed] [Google Scholar]
- 77.Burnett JR, Telford DE, Barrett PH, Huff MW. The ACAT inhibitor avasimibe increases the fractional clearance rate of postprandial triglyceride-rich lipoproteins in miniature pigs. Biochim. Biophys. Acta. 2005;1738:10–18. doi: 10.1016/j.bbalip.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 78.Yatskar L, Fisher EA, Schwartzbard A. Ezetimibe: rationale and role in the management of hypercholesterolemia. Clin. Cardiol. 2006;29:52–55. doi: 10.1002/clc.4960290203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Davignon J, Leiter LA. Ongoing clinical trials of the pleiotropic effects of statins. Vasc Health Risk Manag. 2005;1:29–40. doi: 10.2147/vhrm.1.1.29.58937. •• Early reports on the benefits of statins to AD treatment
- 80. Arvanitakis Z, Schneider JA, Wilson RS, Bienias JL, Kelly JF, Evans DA, Bennett DA. Statins, incident Alzheimer disease, change in cognitive function, and neuropathology. Neurology. 2008;70:1795–1802. doi: 10.1212/01.wnl.0000288181.00826.63. •• review of large clinical trials examining effects of statins on AD markers
- 81.Sparks DL. Alzheimer disease: Statins in the treatment of Alzheimer disease. Nat Rev Neurol. 2011;7:662–663. doi: 10.1038/nrneurol.2011.165. [DOI] [PubMed] [Google Scholar]
- 82. Sabbagh MN, Sparks DL. Statins to treat Alzheimer's disease: an incomplete story. Expert Rev Neurother. 2012;12:27–30. doi: 10.1586/ern.11.171. • The ongoing story of statins as AD-relevant medications
- 83.Shepardson NE, Shankar GM, Selkoe DJ. Cholesterol level and statin use in Alzheimer disease: I. Review of epidemiological and preclinical studies. Arch Neurol. 2011;68:1239–1244. doi: 10.1001/archneurol.2011.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Garzone P, Koller M, Pastrak A, et al. Oral amyloid anti-aggregating agent ELND005 is measurable in CSF and brain of healthy adult men. Alzheimers Dementia. 2009:54323. [Google Scholar]
- 85.Huttunen HJ, Greco C, Kovacs DM. Knockdown of ACAT-1 reduces amyloidogenic processing of APP. FEBS Lett. 2007;581:1688–1692. doi: 10.1016/j.febslet.2007.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Aisen PS, Gauthier S, Vellas B, Briand R, Saumier D, Laurin J, Garceau D. Alzhemed: a potential treatment for Alzheimer's disease. Curr Alzheimer Res. 2007;4:473–478. doi: 10.2174/156720507781788882. [DOI] [PubMed] [Google Scholar]
- 87.Sabbagh MN. Drug development for Alzheimer's disease: where are we now and where are we headed? Am J Geriatr Pharmacother. 2009;7:167–185. doi: 10.1016/j.amjopharm.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. http://wwwmedicalnewstodaycom/articles/75040php; wwwmyriadcom/
- 89.Imbimbo BP, Giardina GA. γ-secretase inhibitors and modulators for the treatment of Alzheimer's disease: disappointments and hopes. Curr Top Med Chem. 2011;11:1555–1570. doi: 10.2174/156802611795860942. [DOI] [PubMed] [Google Scholar]
- 90.Myriad website. http://wwwmyriadcom/alzheimers/flurizanphp.
- 91.Mazza M, Capuano A, Bria P, Mazza S. Ginkgo biloba and donepezil: a comparison in the treatment of Alzheimer’s dementia in a randomized placebo-controlled double-blind study. Eur J Neurol. 2006;13:981–985. doi: 10.1111/j.1468-1331.2006.01409.x. [DOI] [PubMed] [Google Scholar]
- 92.Itil TM, Eralp E, Ahmed I, Kunitz A, Itil KZ. The pharmacological effects of ginkgo biloba, a plant extract, on the brain of dementia patients in comparison with tacrine. Psychopharmacol Bull. 1998;34:391–397. [PubMed] [Google Scholar]
- 93.Kim HG, Oh MS. Nutraceuticals and Prevention of Neurodegeneration Herbal Medicines for the Prevention and Treatment of Alzheimer's Disease. Curr Pharm Des. 2012 Jan 1; [Epub ahead of print] [PubMed] [Google Scholar]
- 94.Howes MJ, Perry E. The role of phytochemicals in the treatment and prevention of dementia. Drugs Aging. 2011;28:439–468. doi: 10.2165/11591310-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 95.Fujiwara H, Iwasaki K, Furukawa K, et al. Uncaria rhynchophylla, a Chinese medicinal herb, has potent anti-aggregation effects on Alzheimer’s beta-amyloid proteins. J Neurosci Res. 2006;84:427–433. doi: 10.1002/jnr.20891. [DOI] [PubMed] [Google Scholar]
- 96.Eubanks LM, Rogers CJ, Beuscher AE, Koob GF, Olson AJ, Dickerson TJ, Janda KD. A molecular link between the active component of marijuana and Alzheimer’s disease pathology. Mol Pharm. 2006;3:773–777. doi: 10.1021/mp060066m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. da Rocha MD, Viegas FP, Campos HC, Nicastro PC, Fossaluzza PC, Fraga CA, Barreiro EJ, Viegas C., Jr The role of natural products in the discovery of new drug candidates for the treatment of neurodegenerative disorders II: Alzheimer's disease. CNS Neurol Disord Drug Targets. 2011;10:251–270. doi: 10.2174/187152711794480429. •• Current update on natural products and ethnobotanical approaches to successful AD treatment.
- 98.Roberts BR, Ryan TM, Bush AI, Masters CL, Duce JA. The role of metallobiology and amyloid-β peptides in Alzheimer's disease. J Neurochem. 2012;120:149–166. doi: 10.1111/j.1471-4159.2011.07500.x. [DOI] [PubMed] [Google Scholar]
- 99.Squitti R. Metals in Alzheimer's disease: a systemic perspective. Front Biosci. 2012;17:451–472. doi: 10.2741/3938. [DOI] [PubMed] [Google Scholar]
- 100.Percy ME, Kruck TP, Pogue AI, Lukiw WJ. Towards the prevention of potential aluminum toxic effects and an effective treatment for Alzheimer's disease. J Inorg Biochem. 2011;105:1505–1512. doi: 10.1016/j.jinorgbio.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kruck TP, Cui JG, Percy ME, Lukiw WJ. Molecular shuttle chelation: the use of ascorbate, desferrioxamine and Feralex-G in combination to remove nuclear bound aluminum. Cell Mol Neurobiol. 2004;24:443–459. doi: 10.1023/B:CEMN.0000022773.70722.b2. •• Combinatorial use of antioxidants and chelators to neutralize neurotoxic metal effects on Aβ peptide aggregation in AD and other neurodegenerative diseases.
- 102. Kawahara M, Kato-Negishi M. Link between Aluminum and the pathogenesis of Alzheimer's disease: the integration of the aluminum and amyloid cascade hypotheses. Int J Alzheimers Dis. 2011;2011 doi: 10.4061/2011/276393. 276393. •• Multifactorial interactive events in AD
- 103. Kruck TP, Percy ME, Lukiw WJ. Metal sulfate-mediated induction of pathogenic genes and repression by phenyl butyl nitrone and Feralex-G. Neuroreport. 2008;19:245–249. doi: 10.1097/WNR.0b013e3282f4cb7e. •• Systematic analysis and the use of antioxidants and chelators in combination to neutralize neurotoxic metal effects in neurodegenerative disease.
- 104.Zhao Y, Calon F, Julien C, Winkler JW, Petasis NA, Lukiw WJ, Bazan NG. Docosahexaenoic acid-derived neuroprotectin D1 induces neuronal survival via secretase- and PPARγ-mediated mechanisms in Alzheimer's disease models. PLoS One. 2011;6:e15816. doi: 10.1371/journal.pone.0015816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cole GM, Frautschy SA. DHA may prevent age-related dementia. J Nutr. 2010;140:869–874. doi: 10.3945/jn.109.113910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Lukiw WJ, Cui JG, Marcheselli VL, Bodker M, Botkjaer A, Gotlinger K, Serhan CN, Bazan NG. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J Clin Invest. 2005;115:2774–2783. doi: 10.1172/JCI25420. •• Potential benefits of DHA and DHA-derived NPD1 in the treatment of AD.
- 107. Lukiw WJ. Cholesterol and 24S-hydroxycholesterol trafficking in Alzheimer's disease. Expert Rev Neurother. 2006;6:683–693. doi: 10.1586/14737175.6.5.683. •• A comprehensive review on cholesterol signaling in AD brain and potential treatment using inhibitors of cholesterol synthesis.
- 108. Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Pro. Natl. Acad. Sci. USA. 1998;95:6460–6464. doi: 10.1073/pnas.95.11.6460. •• A classical paper evaluating the cholesterol-Aβ peptide connection.
- 109.Epis R, Marcello E, Gardoni F, Luca MD. Alpha, beta-and gamma-secretases in Alzheimer's disease. Front Biosci (Schol Ed) 2012;4:1126–1150. doi: 10.2741/s322. [DOI] [PubMed] [Google Scholar]
- 110. Ballard C, Khan Z, Clack H, Corbett A. Nonpharmacological treatment of Alzheimer disease. Can J Psychiatry. 2011;56:589–595. doi: 10.1177/070674371105601004. • Recent overview of potential non-druh AD-relevant formulations
- 111. Burgener SC, Buettner L, Coen Buckwalter K, Beattie E, Bossen AL, Fick DM, Fitzsimmons S, Kolanowski A, Richeson NE, Rose K, Schreiner A, Pringle Specht JK, Testad I, Yu F, McKenzie S. Evidence supporting nutritional interventions for persons in early stage Alzheimer's disease (AD) J Nutr Health Aging. 2008;12:18–21. doi: 10.1007/BF02982159. •• Non-pharmaceutical and nutritional approaches to AD management.
- 112.Burns A, O'Brien J, Auriacombe S, Ballard C, Broich K, Bullock R, Feldman H, Ford G, Knapp M, McCaddon A, Iliffe S, Jacova C, Jones R, Lennon S, McKeith I, Orgogozo JM, Purandare N, Richardson M, Ritchie C, Thomas A, Warner J, Wilcock G, Wilkinson D BAP Dementia Consensus group. British Association for Psychopharmacology. Clinical practice with anti-dementia drugs: a consensus statement from British Association for Psychopharmacology. J Psychopharmacol. 2006;20:732–755. doi: 10.1177/0269881106068299. [DOI] [PubMed] [Google Scholar]
- 113.Sparks DL, Connor DJ, Sabbagh MN, Petersen RB, Lopez J, Browne P. Circulating cholesterol levels, apolipoprotein E genotype and dementia severity influence the benefit of atorvastatin treatment in Alzheimer's disease: results of the Alzheimer's Disease Cholesterol-Lowering Treatment (ADCLT) trial. Acta Neurol Scand Suppl. 2006;185:3–7. doi: 10.1111/j.1600-0404.2006.00690.x. [DOI] [PubMed] [Google Scholar]
- 114. wwwclinicaltrialsgov; www.alzforum.org/drg/drc/default.asp.
- 115.Li L, Cao D, Kim H, Lester R, Fukuchi K. Simvastatin enhances learning and memory independent of amyloid load in mice. Ann Neurol. 2006;60:729–739. doi: 10.1002/ana.21053. [DOI] [PubMed] [Google Scholar]
- 116.Hoglund K, Wiklund O, Vanderstichel H, Eikenberg O, Vanmechelen E, Blennow K. Plasma levels of beta-amyloid(1–40), beta-amyloid(1–42), and total beta-amyloid remain unaffected in adult patients with hypercholesterolemia after treatment with statins. Arch Neurol. 2004;61:333–337. doi: 10.1001/archneur.61.3.333. [DOI] [PubMed] [Google Scholar]
- 117.Mancuso C, Siciliano R, Barone E, Butterfield DA, Preziosi P. Pharmacologists and Alzheimer disease therapy: to boldly go where no scientist has gone before. Expert Opin Investig Drugs. 2011;20:1243–1261. doi: 10.1517/13543784.2011.601740. [DOI] [PubMed] [Google Scholar]
- 118. Wu TY, Chen CP, Jinn TR. Traditional Chinese medicines and Alzheimer's disease. Taiwan J Obstet Gynecol. 2011;50:131–135. doi: 10.1016/j.tjog.2011.04.004. •• Potential benefits of Chinese traditional and ethnobotanical medicines to treat AD
- 119.Chakrabortee S, Liu Y, Zhang L, Matthews HR, Zhang H, Pan N, Cheng CR, Guan SH, Guo DA, Huang Z, Zheng Y, Tunnacliffe A. Macromolecular and small molecule modulation of intracellular Aβ42 aggregation and associated toxicity. Biochem J. 2011 Dec 9; doi: 10.1042/BJ20111661. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 120. Kumar V. Potential medicinal plants for CNS disorders: an overview. Phytother Res. 2006;20:1023–1035. doi: 10.1002/ptr.1970. • Overview of potential AD-relevant natural medications
- 121. Steele M, Stuchbury G, Munch G. The molecular basis of the prevention of Alzheimer's disease through healthy nutrition. Exp Gerontol. 2007;42:28–36. doi: 10.1016/j.exger.2006.06.002. •• Non-pharmaceutical and dietary/nutritional approaches to AD treatment.
- 122. Adams M, Gmünder F, Hamburger M. Plants traditionally used in age related brain disorders-a survey of the ethnobotanical literature. J Ethnopharmacol. 2007;113:363–381. doi: 10.1016/j.jep.2007.07.016. •• Potential benefits of ethnobotanical medicines to AD treatment.
- 123. Kim HG, Oh MS. Nutraceuticals and Prevention of Neurodegeneration Herbal Medicines for the Prevention and Treatment of Alzheimer's Disease. Curr Pharm Des. 2012 Jan 1; [Epub ahead of print] •• Potential benefits of ethnobotanical medicine for the treatment of AD.
- 124.Clare L, Linden DE, Woods RT, et al. Goal-oriented cognitive rehabilitation for people with early-stage Alzheimer disease: a single-blind randomized controlled trial of clinical efficacy. American Journal of Geriatric Psych. 2010;18:928–939. doi: 10.1097/JGP.0b013e3181d5792a. [DOI] [PubMed] [Google Scholar]
- 125.Erickson KI, Voss MW, Prakash RS, et al. Exercise training increases size of hippocampus and improves memory. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:3017–3022. doi: 10.1073/pnas.1015950108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fisher Center for Alzheimer’s Research Foundation. 2011 wwwALZinfoorg; ClinicalTrials.gov; http://www.clinicaltrials.gov/ct2/results?term=alzheimer%27s+disease.
- 127.Mancuso C, Siciliano R, Barone E, Butterfield DA, Preziosi P. Pharmacologists and Alzheimer disease therapy: to boldly go where no scientist has gone before. Expert Opin Investig Drugs. 2011;20:1243–1261. doi: 10.1517/13543784.2011.601740. [DOI] [PubMed] [Google Scholar]
- 128. Lukiw WJ. Micro-RNA speciation in fetal, adult and Alzheimer's disease hippocampus. Neuroreport. 2007;18:297–300. doi: 10.1097/WNR.0b013e3280148e8b. •• First paper to report significant changes in micro RNA (miRNA) speciation in AD.
- 129. Lukiw WJ, Zhao Y, Cui JG. An NF-kB-sensitive micro RNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human brain cells. J Biol Chem. 2008;283:31315–31322. doi: 10.1074/jbc.M805371200. •• First paper to report a specific miRNA-mediated pathogenic mechanism in AD.
- 130.Sethi P, Lukiw WJ. Micro-RNA abundance and stability in human brain: specific alterations in Alzheimer's disease temporal lobe neocortex. Neurosci Lett. 2009;459:100–104. doi: 10.1016/j.neulet.2009.04.052. [DOI] [PubMed] [Google Scholar]
- 131. Cui JG, Li YY, Zhao Y, Bhattacharjee S, Lukiw WJ. Differential regulation of interleukin-1 receptor-associated kinase-1 (IRAK-1) and IRAK-2 by miRNA-146a and NF-kB in stressed human astroglial cells and in Alzheimer disease. J Biol Chem. 2010;285:38951–38960. doi: 10.1074/jbc.M110.178848. • Involvment of aberrant NF-kB signaling and the innate immune system in AD.
- 132.Disease-modifying drug fails in Alzheimer's study. Harv Ment Health Lett. 2010;26:7–11. [PubMed] [Google Scholar]
- 133.Wan HI, Jacobsen JS, Rutkowski JL, Feuerstein GZ. Translational medicine lessons from flurizan's failure in Alzheimer's disease (AD) trial: Implication for future drug discovery and development for AD. Clin Transl Sci. 2009;2:242–247. doi: 10.1111/j.1752-8062.2009.00121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Green RC, Schneider LS, Amato DA, Beelen AP, Wilcock G, Swabb EA, Zavitz KH Tarenflurbil Phase 3 Study Group. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA. 2009;302:2557–2564. doi: 10.1001/jama.2009.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Aisen PS. Development of a disease-modifying treatment for Alzheimer's disease: Alzhemed. Alzheimers Dement. 2006;2:153–154. doi: 10.1016/j.jalz.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 136.Aisen PS, Gauthier S, Ferris SH, Saumier D, Haine D, Garceau D, Duong A, Suhy J, Oh J, Lau WC, Sampalis J. Tramiprosate in mild-to-moderate Alzheimer's disease - a randomized, double-blind, placebo-controlled, multi-centre study (the Alphase Study) Arch Med Sci. 2011;7:102–111. doi: 10.5114/aoms.2011.20612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Saumier D, Aisen PS, Gauthier S, Vellas B, Ferris SH, Duong A, Suhy J, Oh J, Lau W, Garceau D, Haine D, Sampalis J. Lessons learned in the use of volumetric MRI in therapeutic trials in Alzheimer's disease: the ALZHEMED (Tramiprosate) experience. J Nutr Health Aging. 2009;13:370–372. doi: 10.1007/s12603-009-0047-4. [DOI] [PubMed] [Google Scholar]
- 138.Sato N, Shinohara M, Rakugi H, Morishita R. Dual effects of statins on Aβ metabolism: upregulation of the degradation of APP-CTF and Aβ clearance. Neurodegener Dis. 2012 Feb 1; doi: 10.1159/000334534. [Epub ahead of print] PubMed PMID: 22301944. [DOI] [PubMed] [Google Scholar]
- 139.Carlsson CM, Xu G, Wen Z, Barnet JH, Blazel HM, Chappell RJ, Stein JH, Asthana S, Sager MA, Alsop DC, Rowley HA, Fain SB, Johnson SC. Effects of atorvastatin on cerebral blood flow in middle-aged adults at risk for Alzheimer's disease: a pilot study. Curr Alzheimer Res. 2011 Nov 28; doi: 10.2174/156720512803251075. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Shepardson NE, Shankar GM, Selkoe DJ. Cholesterol level and statin use in Alzheimer disease: II. Review of human trials and recommendations. Arch Neurol. 2011;68:1385–1392. doi: 10.1001/archneurol.2011.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Butterfield DA, Barone E, Mancuso C. Cholesterol-independent neuroprotective and neurotoxic activities of statins: perspectives for statin use in Alzheimer disease and other age-related neurodegenerative disorders. Pharmacol Res. 2011;64:180–186. doi: 10.1016/j.phrs.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Serini S, Bizzarro A, Piccioni E, Fasano E, Rossi C, Lauria A, Cittadini AR, Masullo C, Calviello G. EPA and DHA differentially affect in vitro inflammatory cytokine release by peripheral blood mononuclear cells from Alzheimer's patients. Curr Alzheimer Res. 2012 Jan 30; doi: 10.2174/156720512803251147. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 143.Amtul Z, Keet M, Wang L, Merrifield P, Westaway D, Rozmahel RF. DHA supplemented in peptamen diet offers no advantage in pathways to amyloidosis: is it time to evaluate composite lipid diet? PLoS One. 2011;6:e24094. doi: 10.1371/journal.pone.0024094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mancuso C, Siciliano R, Barone E, Preziosi P. Natural substances and Alzheimer's disease: From preclinical studies to evidence based medicine. Biochim Biophys Acta. 2011 Sep 14; doi: 10.1016/j.bbadis.2011.09.004. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 145.Hashimoto M, Hossain S. Neuroprotective and ameliorative actions of polyunsaturated fatty acids against neuronal diseases: beneficial effect of docosahexaenoic acid on cognitive decline in Alzheimer's disease. J Pharmacol Sci. 2011;116:150–162. doi: 10.1254/jphs.10r33fm. [DOI] [PubMed] [Google Scholar]
- 146.Swanson D, Block R, Mousa SA. Omega-3 Fatty Acids EPA and DHA: Health Benefits Throughout Life. Adv Nutr. 2012;3:1–7. doi: 10.3945/an.111.000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lebbadi M, Julien C, Phivilay A, Tremblay C, Emond V, Kang JX, Calon F. Endogenous conversion of omega-6 into omega-3 fatty acids improves neuropathology in an animal model of Alzheimer's disease. J Alzheimers Dis. 2011;27:853–869. doi: 10.3233/JAD-2011-111010. [DOI] [PubMed] [Google Scholar]
- 148.Saravanan P, Davidson NC. Fish oil and arrhythmias. Pro-arrhythmic effects of fish oils. BMJ. 2009;338:b393. doi: 10.1136/bmj.b393. [DOI] [PubMed] [Google Scholar]
- 149.León H, Shibata MC, Sivakumaran S, Dorgan M, Chatterley T, Tsuyuki RT. Effect of fish oil on arrhythmias and mortality: systematic review. BMJ. 2008;337:a2931. doi: 10.1136/bmj.a2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bachurin S, Bukatina E, Lermontova N, Tkachenko S, Afanasiev A, Grigoriev V, Grigorieva I, Ivanov Y, Sablin S, Zefirov N. Antihistamine agent Dimebon as a novel neuroprotector and a cognition enhancer. Ann N Y Acad Sci. 2001;939:425–435. doi: 10.1111/j.1749-6632.2001.tb03654.x. [DOI] [PubMed] [Google Scholar]
- 151.Bezprozvanny I. The rise and fall of Dimebon. Drug News Perspect. 2010;23:518–523. doi: 10.1358/dnp.2010.23.8.1500435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wang J, Ferruzzi MG, Varghese M, Qian X, Cheng A, Xie M, Zhao W, Ho L, Pasinetti GM. Preclinical study of dimebon on β-amyloid-mediated neuropathology in Alzheimer's disease. Mol Neurodegener. 2011;6:7–11. doi: 10.1186/1750-1326-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, Brunden KR, Wilson DA, Landreth GE. ApoE-Directed Therapeutics Rapidly Clear β-Amyloid and Reverse Deficits in AD Mouse Models. Science. 2012 Feb 9; doi: 10.1126/science.1217697. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Houacine J, Bolmont T, Aeschbach L, Oulad-Abdelghani M, Fraering PC. Selective neutralization of APP-C99 with monoclonal antibodies reduces the production of Alzheimer's Aβ peptides. Neurobiol Aging. 2012 Feb 6; doi: 10.1016/j.neurobiolaging.2011.12.033. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 155.Carter C. Alzheimer's Disease: APP, Gamma Secretase, APOE, CLU, CR1, PICALM, ABCA7, BIN1, CD2AP, CD33, EPHA1, and MS4A2, and Their Relationships with Herpes Simplex, C. Pneumoniae, Other Suspect Pathogens, and the Immune System. Int J Alzheimers Dis. 2011;2011 doi: 10.4061/2011/501862. 501862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Carter CJ. Alzheimer's disease: a pathogenetic autoimmune disorder caused by herpes simplex in a gene-dependent manner. Int J Alzheimers Dis. 2010;2010 doi: 10.4061/2010/140539. 140539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Castellani RJ, Smith MA. Compounding artefacts with uncertainty, and an amyloid cascade hypothesis that is 'too big to fail'. J Pathol. 2011;224:147–152. doi: 10.1002/path.2885. [DOI] [PubMed] [Google Scholar]
- 158.Lee HG, Zhu X, Castellani RJ, Nunomura A, Perry G, Smith MA. Amyloid-beta in Alzheimer disease: the null versus the alternate hypotheses. J Pharmacol Exp Ther. 2007;321:823–829. doi: 10.1124/jpet.106.114009. [DOI] [PubMed] [Google Scholar]
- 159.Lee HG, Zhu X, Nunomura A, Perry G, Smith MA. Amyloid beta: the alternate hypothesis. Curr Alzheimer Res. 2006;3:75–80. doi: 10.2174/156720506775697124. [DOI] [PubMed] [Google Scholar]
- 160.Lee HG, Casadesus G, Zhu X, Takeda A, Perry G, Smith MA. Challenging the amyloid cascade hypothesis: senile plaques and amyloid-beta as protective adaptations to Alzheimer disease. Ann N Y Acad Sci. 2004;1019:1–4. doi: 10.1196/annals.1297.001. [DOI] [PubMed] [Google Scholar]
- 161.Lukiw WJ, Cui JG, Yuan LY, Bhattacharjee PS, Corkern M, Clement C, Kammerman EM, Ball MJ, Zhao Y, Sullivan PM, Hill JM. Acyclovir or Aβ42 peptides attenuate HSV-1-induced miRNA-146a levels in human primary brain cells. Neuroreport. 2010;21:922–927. doi: 10.1097/WNR.0b013e32833da51a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hill JM, Zhao Y, Clement C, Neumann DM, Lukiw WJ. HSV-1 infection of human brain cells induces miRNA-146a and Alzheimer-type inflammatory signaling. Neuroreport. 2009;20:1500–1505. doi: 10.1097/WNR.0b013e3283329c05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hill JM, Gebhardt BM, Azcuy AM, Matthews KE, Lukiw WJ, Steiner I, Thompson HW, Ball MJ. Can a herpes simplex virus type 1 neuroinvasive score be correlated to other risk factors in Alzheimer's disease? Med Hypotheses. 2005;64:320–327. doi: 10.1016/j.mehy.2003.11.045. [DOI] [PubMed] [Google Scholar]
- 164.Wozniak MA, Frost AL, Preston CM, Itzhaki RF. Antivirals reduce the formation of key Alzheimer's disease molecules in cell cultures acutely infected with herpes simplex virus type 1. PLoS One. 2011;6(10):e25152. doi: 10.1371/journal.pone.0025152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Holtzman JL. Are we prepared to deal with the Alzheimer's disease pandemic? Clin Pharmacol Ther. 2010;88:563–565. doi: 10.1038/clpt.2010.84. [DOI] [PubMed] [Google Scholar]