

Abstract
From the molecular mechanism of antagonist unbinding in the β1 and β2 adrenoceptors investigated by steered molecular dynamics, we attempt to provide further possibilities of ligand subtype and subspecies selectivity. We have simulated unbinding of β1-selective Esmolol and β2-selective ICI-118551 from both receptors to the extracellular environment and found distinct molecular features of unbinding. By calculating work profiles, we show different preference in antagonist unbinding pathways between the receptors, in particular, perpendicular to the membrane pathway is favourable in the β1 adrenoceptor, whereas the lateral pathway involving helices 5, 6 and 7 is preferable in the β2 adrenoceptor. The estimated free energy change of unbinding based on the preferable pathway correlates with the experimental ligand selectivity. We then show that the non-conserved K347 (6.58) appears to facilitate in guiding Esmolol to the extracellular surface via hydrogen bonds in the β1 adrenoceptor. In contrast, hydrophobic and aromatic interactions dominate in driving ICI-118551 through the easiest pathway in the β2 adrenoceptor. We show how our study can stimulate design of selective antagonists and discuss other possible molecular reasons of ligand selectivity, involving sequential binding of agonists and glycosylation of the receptor extracellular surface.
Keywords: adrenergic receptors, drug design, G protein-coupled receptors, molecular dynamics, selectivity
A major challenge in drug design is to find a small molecule that selectively binds to its target receptor and does not cause unintended side-effects by binding to other similar receptors. When a high-resolution structure of a receptor is available, a structure-based drug design paradigm is applicable to identify not only a small molecule ligand with high binding affinity, but also with good selectivity. However, the binding site architecture of closely related receptor subtypes and subspecies are often highly homologous, making the search for highly selective drugs, which relies on docking of small molecules into the crystal structures of receptors, impractical. This is particularly evident in the design of selective orthosteric agonists and antagonists in such a large and pharmaceutically important class of drug targets as the G protein-coupled receptors.
One strategy for improving selectivity is to account for differences in the ligand binding and unbinding pathways of closely related receptors caused by non-conserved residues outside the drug-binding site. In this work, we aim to explore the dynamic and kinetic causes of antagonist subtype selectivity in the β1 and β2 adrenergic receptors (AR). β1AR and β2AR represent one of the most extensively characterized subfamilies of the G protein-coupled receptors, which are expressed in many cell types and play a pivotal role in regulation of the cardiovascular, pulmonary, endocrine and central nervous systems (1). Antagonists of the adrenergic receptors (β-blockers) are hallmark drugs for treatment of ischaemic heart disease, hypertension and congestive heart failure (1,2). Although the primary cardiovascular use of β-blockers is antagonism of β1AR responses in the heart, their use may also result in antagonism of β2AR in airways, resulting in bronchospasm (1,2). To avoid this side-effect, β1AR-selective antagonists are required.
To identify the dynamic and kinetic bases of antagonist selectivity, we have studied the unbinding process of two selective antagonists, Esmolol, which is 76-fold selective to β1AR (3–5), and ICI-118551, which is 550-fold β2AR selective (6,7) (Figure 1), from human β1AR and β2AR, using a molecular dynamics approach. Given that a computer-aided drug design campaign requires fast evaluation of potential binders, and monitoring of ligand binding and unbinding requires a microsecond time scale that is not affordable in a high-throughput level, we accelerated unbinding by applying an external force to pull the antagonist from the binding site in several directions using steered molecular dynamics (sMD). Through the use of multiple sMD simulations of ligand unbinding events, the statistical importance of unbinding pathways, as well as specific residue interactions important to them, can be analysed and key receptor conformations possessing characteristic interactions can be exploited in future drug design efforts.
Figure 1.

Structures of β1AR-selective Esmolol and β2AR-selective ICI-118551. Biological activities are taken from references 5 and 7.
Recent simulations of the unbinding pathways of the non-selective inverse agonist Carazolol from β2AR using the random acceleration molecular dynamics method have shown that unbinding occurs primarily through the extracellular region of β2AR and only rarely through transmembrane helices, suggesting that pathways through the extracellular surface provide a specific route to ligand entry (8). To further investigate this phenomenon, we have performed simulations in which the selective antagonists are pulled from the binding site to the extracellular surface along three directions: one perpendicular to the membrane and two lateral paths. We use the estimated free energy change of ligand unbinding based on the preferable pathway to correlate the experimentally observed ligand selectivity. From monitoring the impact of different interactions on the process of ligand unbinding, we delineate common and uncommon features of the unbinding process between two receptors and two ligands, along with their implications for structure-based drugs design studies. We also compare our results with the recent publication of non-selective ligand binding to β2AR using conventional MD (9) and non-selective ligand unbinding from β1AR and β2AR using steered MD (10). Finally, we discuss other potential reasons for different ligand binding and unbinding pathways between the receptors, which can account for structural constraints in the design of selective small molecule modulators.
Methods
Preparation of ligand-protein complexes for simulations in the hydrated phospholipid bilayer
To be consistent with ligand selectivity analysis in the human adrenergic receptors, we chose the available high-resolution crystal structure of the human β2AR (2.4 Å) (PDB ID: 2RH1) and the 3D model of human β1AR was constructed based on this structure using the Prime module 2.2a of the Schrodinger suite 9.0b. The 3D models of receptors were prepared for ligand docking studies using the Schrodinger protein preparation utility. The docking of ICI-118551 and Esmolol into the structures of β1AR and β2AR was performed with Glide 5.6c with the Extra Precision algorithm. Ligands were processed using LigPrep 2.4 with the OPLS 2005 force filedb (11,12). The investigated antagonists were close structural analogues of the ligands in the crystal structures, thus the docking poses similar to the crystal structures were quickly identified in the docking studies. Concurrent to this study, the crystal structure of β2AR in complex with ICI-118551 was published, which exhibit a nearly identical binding pose of ICI-118551 to our docking pose (13). The superposition of the crystallographic and computational coordinates of the binding site with ICI-118551 is shown in Figure S1 of the Supporting Information. The System Builder of Maestro gui 9.0 was used to embed four ligand-receptor complexes and the unoccupied receptors into a bilayer composed of 1-palmitoyl-2-oleoylphosphatidylcholine, solvate the extracellular and intracellular sides of the bilayer, neutralize the biomolecular system and preserve physiological pH using 0.15 m sodium chloride concentration. The final biomolecular systems contained around 75 000 atoms.
Biomolecular dynamics
Conventional and sMD were performed using the Desmond package 2.2 (14). The temperature and pressure were controlled at 300 K and 1 atm, respectively, using the Berendsen algorithm. Long-range electrostatics was calculated by means of the Particle Mesh Ewald Method (15). The OPLS2005 force field was used for all simulationsb (11,12). The biomolecular systems were equilibrated using the following steps: (i) energy minimization (2000 cycles) of the solvent and lipid bilayer, while the ligand-receptor complex was frozen, using the conjugate gradient algorithm up to a convergence threshold of 0.5 kcal/mol/Å; (ii) heating of the system to 10 K in the NVT ensemble over 12 pseconds; (iii) heating of the system from 10 to 300 K in the NPT ensemble over 12 pseconds; (iv) equilibration for 7–9 nseconds at 300 K in which the complexes were still frozen; (v) equilibration for 2 nseconds in which the protein backbone was restrained; (vi) equilibration for 2 nseconds with no restraints. Following equilibration, the 30 nseconds production runs were carried out in the NPT ensemble. The steered MD simulations were tested with a pulling speed of 0.005, 0.01, 0.015, 0.02 and 0.03 Å/psecond and a spring constant of 4.5, 4, 3.5, 2.5 and 1.5 kca/mol/Å2 (16,17). The pulling speed of 0.02 Å/psecond and spring constant of 4.0 kcal/mol/Å2 were selected for the detailed studies presented below. Ten pulling simulations in each chosen direction were performed with different initial velocities. Figure 2 was prepared in PyMol 0.99d and movies were made in Maestrob.
Figure 2.

The extracellular side of the adrenergic receptors. (A) the β1 adrenergic receptor in the complex with Esmolol, (B) the β2 adrenergic receptor in the complex with ICI-118551, (C) the schematic representation of the binding site interactions between Esmolol and the β1 adrenergic receptor, and (D) the schematic representation of the binding site interactions between ICI-118551 and the β2 adrenergic receptor. The snapshots for the images were taken from the molecular dynamics trajectories. The binding site residues and residues involved in ligand unbinding are visualized in the stick-like representation in A and B. Pulling directions are shown in arrows.
Analysis of MD trajectories
Hydrogen bond occupancy along simulations trajectories was computed in VMD 1.8.9 (18). Hydrogen bonds with a defined threshold for distance of 3 Å and angle of 35° were calculated in VMD 1.8.9 (18). Carbon atoms at distance 1.4–5 Å were selected for hydrophobic contacts in Maestro of the schrodinger suite 9.0b. Water-mediated contacts at a distance of 3 Å with a minimum donor angle of 120° and a minimum acceptor angle of 90° were computed in Maestro. The radius of gyration of the lateral channel involving helices 5, 6 and 7 was calculated by selecting Cα atoms of F193, T195, H296 and N293 with VMD (18). The work required pulling antagonists from the adrenergic receptors and ΔPMF were calculated using the VMD scripts.
Grid generation
The hydrogen bond acceptor and hydrophobic probes for the selected snapshots from sMD trajectories were calculated in Site-Map V2.5 of Schrodingere. The probes were placed in the receptors at a radius of 6 Å from the ligand in the selected snapshot of the MD trajectory.
Results and Discussion
Dynamics of the extracellular side of the β1 and β2 adrenergic receptors with the selective ligands
We started our study with the performance of conventional molecular dynamics (cMD) simulations of the human β1 and β2 adrenergic receptors (AR) with docked Esmolol and ICI-118551 to initiate comparison of dynamics of the receptor extracellular cavity and ligand binding modes in each receptor, to define the ligand pulling directions from the receptor and to produce the starting coordinates for ligand unbinding simulations. The protein conformations stabilized following 2 nseconds of equilibration, at which point the root-mean-square deviations of protein backbone and ligand heavy atoms converged around 2 and 0.9 Å for the Esmolol-β1AR complex, 2.5 and 0.6 Å for the ICI-118551-β1AR complex, 2.1 and 0.6 Å for the Esmolol-β2AR complex and 1.9 and 0.7 Å for the ICI-118551-β2AR. The 30 nseconds cMD simulations of the complexes revealed higher fluctuation of the extracellular cavity, involving the extracellular regions of the helices and loops, in β1AR than in β2AR. As expected, the complex of the receptors with the selective antagonist was more rigid than the complex with the non-selective antagonist. The root-mean-square fluctuations of the Cα atoms of the extracellular cavity for two receptors are shown in Figure S2 in the Supporting Information. In Figure 2, we show one equilibrated snapshot of the extracellular cavity for each receptor, displaying amino acid residues of the extracellular side, together with the schematic representation of the antagonist binding site depicting the key residues involved in the interactions with the ligands.
In β1AR, a salt bridge between E205EL2 and R351EL3 was observed, with occupancy of more than 20% in each simulation, that dynamically connects the second extracellular loop (EL2) and the third extracellular loop (EL3) from the lateral side of the extracellular cavity in β1AR (Figure 2A). This salt bridge has also been observed in recent simulations of human β1AR by Gonzalez and co-workers (10) using the CHARMM force field. It is likely unique to the human β1AR as the residue corresponding to E205EL2 in turkey β1AR (Q188) is located 3.5–10 Å from the arginine side chain in the available crystal structures (19–21). In β2AR, these residues are substituted to E180EL2 and D300EL3.
Residues D192EL2 and K3057.46 (The Ballesteros and Weinstein nomenclature in superscript (22)), in β2AR form a salt bridge that splits the β2AR extracellular cavity into two sub-cavities (Figure 2B). This salt bridge, which is present in five of seven crystal structures of inactive β2AR bound to ligands (13,23,24), has an occupancy near 80% in simulations of the receptor complexes with Esmolol and ICI-118551. This is twice more than in our simulations of the ligand-free receptor, suggesting that antagonists stabilize this interaction, which likely holds the receptor in the inactive state. Although site-directed mutagenesis of D192EL2 and K3057.46 with emonstration of the role of these residues in ligand binding has not been documented in the literature, the different influence of ligands on the stability of the D192EL2-K3057.46 salt bridge has been recently described by an NMR study via monitoring different resonances of radiolabeled K3057.46 in the presence of agonists and antagonists (25). This mechanism of receptor modulation by extracellular salt bridges through their breakage and appearance was observed in the free fatty acid receptor 1 using mutagenesis and molecular modelling (26). β1AR misses this salt bridge as K3057.46 is substituted to D3567.46.
Hydrogen bonds between the secondary amine and β-hydroxyl group of both ligands with D138/1133.32, N363/3127.39 and Y367/3167.43 (Figure 2C,D) were preserved during simulations with β1AR and β2AR, while the aromatic ring of ICI-118551 was buried deeper in the pocket than Esmolol in both receptors. The ester group of Esmolol was engaged in hydrogen bonding with N344/2936.55 and S229/2045.43 in 40% and 8% of the simulations time in β1AR, while in β2AR this was reduced to 13% and 3% of the simulation time. The varying interactions of N344/2936.55 are owing to different accessibilities of N344/2936.55 in the receptors; N344/2936.55 is exposed to the extracellular cavity with χ1 mainly in the g− rotameric state, (171 ± 8°) in β1AR, whereas it is buried into the receptor with χ1 in the t rotameric state, (−87 ± 25°) in β2AR. These different states are owing to a hydrogen bond between N2936.55 and Y3087.35 in β2AR, shown in the available crystal structures (13,21,22,25,26), that is disrupted by the substitution of Y3087.35 to phenylalanine in β1AR (Figure 2C,D) (13,23,24,27,28). Our simulations show that this hydrogen bond is formed more than 50% of the time in the complexes of β2AR with Esmolol and ICI-118551, reinforcing the non-direct role of the N2936.55-Y3087.35 interaction in ligand selectivity. The impact of Y308A7.35 and N2936.55Q/L/A mutations in the reduction of β2AR ligand selectivity and, interestingly, the increase ligand selectivity for N2936.55F has been shown by mutagenesis studies (29–31). This hydrogen bond was constantly monitored in the MD simulations of β2AR in the complex with carazolol by Vanni and co-workers (32). In the unoccupied receptor, the N2936.55-Y3087.35 hydrogen bond is present in only 20% of the simulations time, suggesting that ligand binding facilitates this interaction.
The final snapshots of the 30 nseconds cMD simulations were used as starting points for sMD calculations. To investigate the impact of structural and dynamics differences of the extracellular cavity in ligand recognition, we pulled antagonists along three distinct directions, one perpendicular to the membrane (path B) and two lateral – one to the side of helices 1, 2 and 7 (path A) and the other to the side of helices 5, 6 and 7 (path C) (Figure 2).
Pulling of selective antagonists from the β1 and β2 adrenergic receptors
We used a steered MD algorithm to pull the antagonists from the binding pocket over 2 nseconds of simulation time. Pulling was induced along a particular pathway by the introduction of an external biasing potential along a predefined vector (as shown in Figure 2). The choice of the receptor and the antagonist pulling atoms (Figure 1) determined three ligand unbinding pathways, where the pulling directions were defined by the centre of mass of the receptor atoms are shown in Table 1. We tested simulations at varying pulling velocities and spring constants, which all exhibited unbinding in a similar fashion, and chose sMD parameters to balance computational cost with simulation accuracy (see Methods section). To investigate the pulling mechanics of selective antagonists, Esmolol and ICI-118551 from β1AR and β2AR in chosen directions, we calculated the rupture force profile along the pathways. Figure 3 shows the average force profile for the perpendicular and two lateral pathways of the receptors releasing either Esmolol or ICI-118551 projected onto the unbinding simulation time and the ligand separation distance from the original position. For each pathway, the average values were taken from ten trajectories simulated with a pulling velocity of 0.02 Å/psecond and a spring constant of 4.0 kcal/mol/Å2. Generally, complete unbinding of the ligand occurred within 1–1.5 nseconds. Although the receptors have a similar pattern of the force profile graphs between the pathways, there are notable differences in the size and dynamics of the force peaks, suggesting distinct properties of the unbinding process. In β1AR, the average force peak in path B had lower values than in paths A and C for both Esmolol and ICI-118551, while in β2AR the force peaks along path C are lower than in paths A and B. To estimate the work performed by the antagonist during unbinding along chosen pathways we integrated the force curve over the ligand separation distance from the ligand original position (Figure 3). The computed work highlights the different preference in unbinding pathways and pulling of selective and non-selective antagonists between the receptors. In particular, less work is required to pull antagonists from path B in β1AR and, among antagonists, more work is required to pull the β1AR-selective antagonist in chosen pathways; less work is needed to pull the antagonists from path C and similarly, the β2AR-selective antagonist is required more work to apply in β2AR. Jarzynski's equality states that the average work applied on the replicated ensembles can represent, in the first approximation, the overall change in the free energy between bound (a) and unbound (b) state or the potential of mean force (ΔPMF) along a reaction coordinate:
where β is 1/(kbT), kb is Boltzmann constant and T is the temperature in Kelvin. Because in path B of β1AR and path C of β2AR, the ligands are needed the smallest work to perform, we use these pathways to compare ΔPMF with the experimental ligand selectivity. Although the calculation of the free energy change accurately requires numerous replicates (100–1000), we nevertheless hope that our multiple but still few simulations will provide a relative estimate of the free energy of ligand unbinding and binding. We repeated unbinding simulations for the preferable pathways (path B in β1AR and path C in β2AR) another ten times and calculated ΔPMF using the Jarzynski's equality formula. The ΔPMF of Esmolol and ICI-118551 were −7 and −6 kcal/mol in path B of β1AR and −6.3 and −7.9 kcal/mol in path C of β2AR, respectively. The ΔPMF of ligands confirms antagonist preferences between the receptors. The calculated ΔPMF values from the available experimental constants of inhibition (Ki) (Figure 1) for Esmolol, were −7.3 and −6.8 kcal/mol and, for ICI-118551 were −10 and −12.6 kcal/mol, in β1AR and β2AR, respectively. The ΔPMF of unbinding from β2AR is slightly lower from experimental calculated values, suggesting the importance of additional replicates to increase accuracy. Below, we compare unbinding of each selective antagonist from both receptors with characterization of the specific interactions that contribute to ΔPMF.
Table 1.
The atoms of the receptors and ligands to which an external force has been applied to pull Esmolol and ICI-118551 in three extracellular directions from the β1 and β2 adrenergic receptors
| Path | Cα atom of a residue in β1/β2 adrenoceptors | Ligand atom (Esmolol/ICI-118551) |
|---|---|---|
| Path A (TM7, TM1 & TM2) | F5.47(233/208) | C10/C9 |
| F6.52 (341/290) | ||
| S5.46 (232/207) | ||
| T3.36 (143/118) | ||
| Path B (Straight) | F5.47 (233/208) | C10/C9 |
| M2.53 (107/82) | ||
| T3.36 (143/118) | ||
| W6.48 (337/286) | ||
| Path C (TM5, TM6 & TM7) | I3.40 (146/121) | C10/C9 |
| M2.53 (107/82) | ||
| T3.36 (143/118) | ||
| Y7.43 (367/316) |
Figure 3.

The average, rupture force and work profiles of antagonist unbinding from the adrenergic receptors in three extracellular pathways. The graphs were plotted against ligand separation from the initial coordinates in the binding site. The unbinding of β1AR-selective Esmolol and β2AR-selective ICI-118551 are shown in black and red, respectively.
Pulling from the β1 adrenergic receptor
Because path B required the smallest work to apply, we hypothesized that specific molecular features along this path might result in the experimentally observed ligand specificity of selective and non-selective antagonists. To examine this, we compared the unbinding of antagonists along path B and decomposed the rupture force into specific interactions, involving hydrogen bonds, hydrophobic, and water-mediated contacts, the results of which are shown in Figure 4 for one characteristic trajectory. From pharmacological studies, Esmolol binds in β1AR more tightly than ICI-118551, thus more work is required to pull it out from the receptor, as indicated in the work profiles. After some jiggling resulting from the bias potential, involving the iterative breakage and appearance of the hydrogen bond between the ester group and N3446.55, Esmolol ultimately lost its first hydrogen bond with the receptor via water-bridged interactions at early as 350 pseconds, corresponding to the first peak in the force profile. This facilitated the movement of the aromatic ring of Esmolol towards the extra-cellular surface. Around 650 pseconds, the secondary amine and β-hydroxyl group of the ligand broke hydrogen bonds with N3637.39 and Y3677.43, and the ligand approached the extracellular surface while still retaining the salt bridge with D1383.32 in the binding site. The centre peak in the force profiles corresponds to the breakage of the salt bridge at the binding site, which happens after a minimum of 800 pseconds. After breakage of the interaction with D1383.32, the amine of the ligand established interactions with D217EL2 in multiple trajectories and occasionally with D356EL3, followed by diffusion into the water within 250 pseconds. In the ligand binding process, these two negatively charged residues likely serve as beacons that recognize the charged amino group of a ligand within the solvent environment. In the case of ICI-118551, the applied force caused the aromatic moiety to move towards the receptor surface, which was followed by the breakage of interactions with the original binding site residues. The unbinding process occurred quicker for ICI-118551 then for Esmolol.
Figure 4.

Esmolol and ICI-118551 unbinding from the β1 adrenergic receptor on the example of one representative trajectory shown in the form of the rupture force, work, hydrogen bonds, hydrophobic and water-mediated contacts projected onto unbinding time and antagonist separation time from the initial position.
The disruption of ligand-receptor interactions followed a similar pattern in other pulling directions. The ester group, released from its original interactions with binding site residues in β1AR, formed hydrogen bonds with T220EL2 at 400–550 pseconds, followed by forming interactions with K3476.58 at 600–700 pseconds that lasted for 200 pseconds in seven of the simulation trajectories. After breakage of water-bridged interactions with K3476.58, the ester group formed its last interaction with the backbone of F218EL2. This profile of continuous interchange of hydrogen bond interactions between the ester and the receptor along the unbinding pathway is shown in Figure 5 and Movie S1 (where the role of K3476.58 is highlighted). We also observed direct and water-bridged interactions between the ester group and K3476.58 in five trajectories along path A. In contrast, ICI-118551 did not form any hydrogen bonds with K3476.58. In both cMD and sMD simulations, K3476.58 moved freely in the cavity for a majority of the time and formed a salt bridge with E352EL3 in only 8% of the simulation time. Therefore, we propose that K3476.58 might act as a `hydrogen bond translator' that interacts with a specific group of the ligand and facilitates the movement of the selective antagonist between the receptor extracellular surface and the binding site, along with preventing the exit of the molecule from the binding cavity. Potentially, during the ligand binding process, this residue might also facilitate Esmolol to enter the binding cavity with an orientation that would result in having optimal interactions with the binding site residues.
Figure 5.
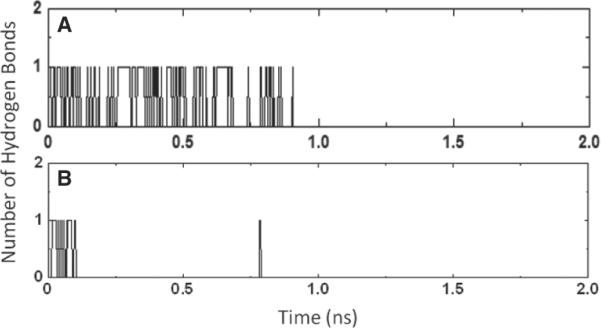
The hydrogen bond profile of the ester group during the Esmolol unbinding from the β1 and β2 adrenergic receptor.
Antagonists formed interactions with aromatic and aliphatic residues for 53% and 47% of the simulation time, correspondently. Helix 2 has been proposed to play a role in β1-selectivity of (−)-RO363 (33), and it is observed that the non-conserved I1182.64 held interactions with the ligand in 10% of the unbinding time, suggesting the potential role of I1182.64 in ligand selectivity, which was not found for (−)-RO363. During unbinding, the side chains of F218EL2 and F3597.35, which form the gate to the extracellular cavity, moved away from each other, increasing the centre of mass distance between the aromatic rings from 8 to 12 Å(Figure 6A,B). Once the ligand exited the receptor, their separation distance decreased to a minimum of 5 Å, similar to the value observed in the unoccupied receptor (Figure 6C). This suggests that these two phenylalanines form gates that open and close during ligand binding, discriminate whether the ligand can enter into the binding cavity, and if so, guide the ligand into the receptor.
Figure 6.
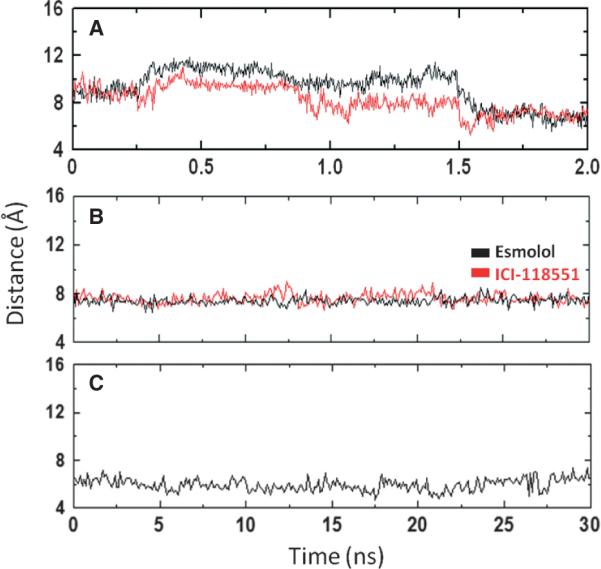
Evolution of the distance between the centre of the aromatic ring of F218EL2 and F3597.35 along the unbinding pathway B (A), 30 nseconds of conventional molecular dynamics of the complexes with Esmolol or ICI-118551 (B) and 30 ns of the unoccupied β1 adrenergic receptor (C).
As the antagonists was pulled from the binding cavity, water-mediated interactions formed to replace protein-ligand interactions (Figure 4). For example, the breakage of the salt bridge between the amine group and D1383.32 occurred when a water molecule reached the salt bridge, and was often observed to initially form interactions with D1383.32, followed by interacting with the secondary amine, resulting in the breakage of the direct protein-ligand interaction. To appreciate the impact of water molecules on unbinding, we simulated the ligand escape in vacuum and found the maximum value of the rupture force increased to 32 kcal/mol/Å (twice that observed in the solvated systems), suggesting that water-mediated interactions facilitate the ligand's movement along the extracellular cavity (Figure S3). Movement of antagonists along path B led to the flooding of 15 water molecules into the binding cavity (Figure S4) through the region surrounding the secondary amine of the antagonists.
Path C in β1AR required the highest force for both ligands. When pulled along this trajectory, both ligands exited through the EL2 in 6–7 simulations with the rupture forces reaching up to 20 kcal/mol/Å. In the case of b2AR, the ligand escaped along path C without going through EL2 in all trajectories. EL2 has high mobility and therefore tends to cover the extracellular mouth in β1AR, whereas it has a rigid position and localizes close to the side of helices in b2AR owing to restriction caused by the extracellular salt bridge. The average root-mean-square deviation values of the β1AR backbone along three pathways, provided in Figure S5, show higher fluctuation of the receptor in path C, indicating that large structural rearrangements are required to allow the ligand to exit the receptor along this pathway. This is due not only to a steric clash with the hydrophobic tail of K3476.58, but was also a result of the required breakage of the salt bridge between E205EL2 and R351EL3 in several trajectories, which formed and remained stable during equilibration in our cMD simulations.
Pulling from the β2 adrenergic receptor
Simulations of ligand unbinding from b2AR showed that, contrary to what was observed for β1AR, path C required the application of the smallest force to bring the antagonists to the surface. This is consistent with recently published work on binding of the β-adrenoblockers to b2AR using cMD, where the authors showed that ligands bind preferably to the receptor from the extracellular side of helices 5, 6 and 7 (9). In our simulations, we monitored the hydrogen bond network between the side chains of N2936.55, H2966.58 and Y3087.35 (Figure 7A,B), which drives them close to helix 6, enlarges the lateral channel, and thus facilitates the ligand's movement along path C. To monitor the changes in the size of the lateral channel, we calculated the radius of gyration along path C, as shown in Figure 7C. The hydrogen bond network that exists only during the unbinding process increases the radius of gyration compared to its value in the cMD of the antagonist-receptor complexes (Figure 7D). The dynamics of this network of interactions in the unbinding of ICI-118551 from b2AR through path C are shown in Movie S2.
Figure 7.
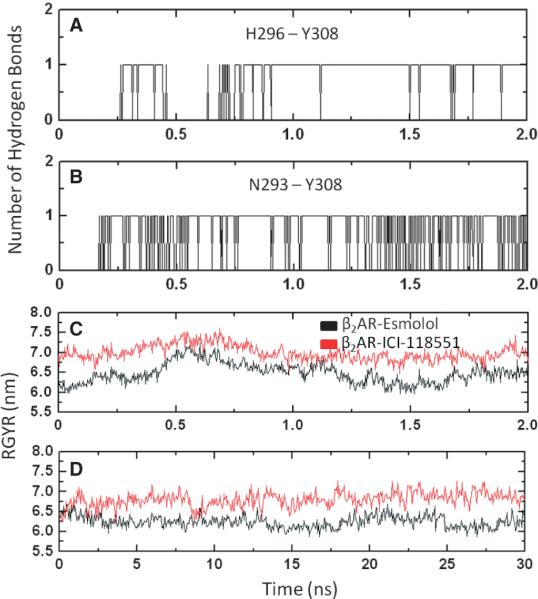
The lateral channel to the extracellular surface composing helices 5, 6 and 7. The H296-Y308-N293 hydrogen bond network of interactions in the β2 adrenergic receptor (A, B) along the ligand unbinding trajectories, radius of gyration of the lateral channel, along unbinding pathway C (C) and radius of gyration of the lateral channel in 30 nseconds classical MD of the ligand-receptor complexes (D).
In paths A and B, the higher peaks in the force profile appear to be a result of the antagonists breaking the D192EL2-K3057.46 salt bridge at the extracellular part, in addition to the disruption of the salt bridge in the binding site. Along these paths, the antagonists escaped without the breakage of the salt bridge in only seven of 40 trajectories, while the unbinding along pathway C did not require the breakage of the salt bridge in all MD trajectories. The average root-mean-square deviation values of the b2AR backbone along the three pathways, provided in the Figure S5, shows larger deviation of the receptor in paths A and B at the time of the salt bridge breakage, supporting a somewhat obstacle role of the salt bridge.
The sequence of breakage of the original interactions during the unbinding of Esmolol and ICI-118551 was similar to that in β1AR. Figure 8 shows the time dependence of different ligand-protein interactions along unbinding pathway C. The ester group of Esmolol formed and broke interactions with S2045.43 and occasionally with N2936.55 at the beginning of the unbinding process, while final breakage of this interaction resulted in the first peak in the force profile. Overall, the ester group of Esmolol formed fewer hydrogen bonds along the unbinding pathway in b2AR than in β1AR, as shown in Figure 5, while both ligands left the receptor surface by breaking interactions with D300EL3. Our data, as well as the recently published cMD simulations of ligand binding to b2AR, suggest that D300EL3 likely plays the recognition role of the charged amine of the ligand in a cellular environment, similar to D217EL2 and D356EL3 in β1AR.
Figure 8.

Esmolol and ICI-118551 unbinding from the β2 adrenergic receptor on the example of one representative trajectory shown in the form of the rupture force, work, hydrogen bonds, hydrophobic and water-mediated contacts projected onto unbinding time and antagonist separation time from the initial position.
It was also observed that Esmolol escaped quicker than ICI-118551 from b2AR. This is a result of ICI-118551 forming extra aromatic and hydrophobic interactions with Y3087.35, H2966.58, F193EL2 and F194EL2 through its large hydrophobic moiety along the pathway, as shown in Figure 8. F194EL2 formed hydrophobic interactions with ICI-118551 and followed the ligand to the solvent environment in several trajectories through movement of its backbone and side chain. The distance between the centre of the aromatic ring of H2966.58 and antagonists remained 5Å during 250ps in several trajectories, suggesting formation of aromatic interactions. We propose that the side chain of F194EL2 and H2966.58 steer the ligand into the binding site during a binding event. Unlike their counterparts F218EL2 and F3597.35 in β1AR, the distance between the centre of the aromatic ring of F193EL2 and Y3087.35 showed only small fluctu ations around 8–9 Å throughout the simulations, indicating that the narrow cleft is enough to progress the antagonist into the binding pocket in β2AR (Figure S6). This distance is preserved when the antagonists were pulled to the direction of path B. Because the mobility of F193EL2 is restricted by the D192EL2-K3057.46 salt bridge and Y3087.35 is constrained by the hydrogen bond with N2936.55, F193EL2 and Y3087.35 thus function as open gates.
Conclusion and Future Directions
Side-effects caused by many β-antagonists are owing to their promiscuous binding to closely related subtypes of the adrenoceptors (β1AR and β2AR). Therefore, understanding the molecular basis of β-antagonist selectivity is likely to provide a novel rationale for the discovery of selective ligands. In this work, we have compared the unbinding process of β1- and β2-selective antagonists from β1AR and β2AR by applying steered MD simulations and have shown the potential for a kinetic basis of antagonist selectivity, in addition to the dynamic binding site selectivity caused by different geometry of the conserved N3446.55. Remarkably, the calculated average rupture force and work profiles from multiple unbinding trajectories suggest that the perpendicular to the membrane direction (path B) is likely the preferable pathway for both ligands to dissociate from β1AR, whereas the lateral direction, composing helices 5, 6 and 7 (path C) is always favourable in β2AR. The calculated ΔPMF of the antagonists for these pathways correlates with the observed ligand selectivity between the receptors. We expect that accuracy of ΔPMF calculations involving hundreds replicates will increase and provide more precise estimation of the ligand binding affinities. However, this approach could be challenging for examining binding activities in the high-throughput scale for many potential ligands.
When pulled in each of the three directions to the extracellular surface, the non-conserved K3476.58, the E205EL2 -R351EL3 salt bridge, which is localized in the lateral side in β1AR, and the D193 EL2-K305 EL3 salt bridge, which faces the binding cavity in β2AR, hinder the antagonists from unbinding in the other directions. The relatively equal force profile of path A and B are similar to two favourable unbinding channels in β1AR identified recently by Gonzalez and co-workers (10), while the preference of path C in our short simulations of unbinding from β2AR is in agreement with the recently published 1–19 μseconds conventional MD simulations of ligand binding to β2AR (9).
Within the preferable pathways for ligand unbinding from β1AR and β2AR, we found that the distinct moieties of antagonists have a different pattern of interactions in the receptors. The unbinding of Esmolol is governed through the interchange of hydrogen bonds of the ester group, involving K3476.58 in β1AR, whereas ICI-118551 does not form interactions with this residue. In contrast, the unbinding from β2AR is likely driven by hydrophobic and aromatic interactions with Y3087.35, H2966.58, F193EL2 and F194EL2 and, thus, the hydrophobic moiety of ICI-118551 spends more time interacting with these residues than Esmolol. The ester group of Esmolol forms few hydrogen bonds in β2AR during unbinding. By simulating unbinding from the adrenoceptors, we assume that the binding process takes place in an opposite but a similar fashion; thus, the proposed residues underling the unbinding likely play important roles during binding as well. The hydrophobic surfaces between alprenolol and Y3087.35, H2966.58, F193EL2 have been recently observed by Dror and co-workers in long simulations of alprenolol binding (9).
In our simulations, K3476.58 coordinates the antagonist unbinding by restricting the escape from one side of the extracellular surface and utilizing a hydrogen bond to translate the antagonist to the extracellular surface through another route, providing selective features of antagonist unbinding in β1AR. Interestingly, K3476.58 in human β1AR is replaced by asparagine in turkey β1AR, which can play a different role in ligand binding and unbinding, which explains recent simulations of dihydroalprenolol binding that showed the binding pathway was similar to β2AR (9). In addition, in a recent pharmacological study, it was observed that the β1AR-selective antagonist, CGP20712A, in human is not selective in turkey (34). With both species β1AR receptors having identical binding sites, we hypothesize that the residue difference at the extracellular surface is responsible for the different binding profiles of antagonists between these two proteins.
Our study suggests a kinetics basis of antagonist selectivity in the adrenoceptors in which the non-conserved residues at the extracellular surface form selectivity filters that recognize the molecule from the soluble environment by its shape and physicochemical properties, and bring it into the binding site through a cascade of specific intermediate interactions. The mapping of structures taken from the unbinding trajectories by hydrophobic and electrostatic probes highlights differences in the physicochemical properties of the extracellular surface (Figure 9). For example, there is preference for a hydrogen bond acceptor region in β1AR and a hydrophobic region in β2AR, especially in the snapshots where the selective antagonist forms specific interactions during unbinding (Figure 9B–C, E–D). The kinetics-based preferable regions are poorly seen in the ligand-receptor complexes, as shown by the initial structures in Figure 9A,D. Receptor conformations from these unbinding studies can play an important role within a drug design application, for example by designing a ligand with unique binding kinetics by docking molecules or fragments within the binding and unbinding channel, in addition to classical docking to the binding site, or by growing a ligand in such way that it can interact with the extracellular residues forming the preferable maps. Although, there is no experimental proof of kinetic basis of antagonist selectivity in the adrenergic receptors at this moment, our computational study suggests amino acid residues of the receptor extracellular surface for site-directed mutagenesis studies or medicinal chemistry optimization to explore this hypothesis experimentally.
Figure 9.

Hydrophobic and hydrogen bond acceptor grids in the structural snapshots of the β1 and β2 adrenergic receptors taken from the antagonist unbinding trajectories.
In this work, we have simulated the unbinding event of two antagonists that do not cause conformational changes leading to activation and signalling. The process of agonist binding and unbinding is thought to be more complicated as it involves a series of conformational changes in the receptor structure, resulting in formation of the active state. Fluorescence spectroscopy studies have shown a sequential binding model of agonists in which the partial agonist, dopamine, has rapid binding, whereas a full agonist, noradrenaline, preserves the biphasic binding kinetics in β2-AR (35). We anticipate that there are different interactions in the binding and unbinding pathways of agonists and antagonists. Simulations of the agonist binding process, in which step-by-step conformational changes of the receptor undergoing a transition from the inactive conformation towards the active one, may provide novel molecular features in ligand kinetics, which in turn, could help to interpret ligand efficacy and facilitate design of selective ligands with desired efficacy. The recently published crystal structure of β2AR in complex with the Gs protein, representing the active state, provides the opportunity for a such study (27). Thus, transition from inactive to active states of β2AR in the presence of ligands with different efficacy using adaptive biasing simulations recently performed by Provasi and co-workers revealed energetics of the receptor conformational space during activation (36). Transition from active to inactive states in the presence of agonists and atomic details of activation have been recently suggested by Dror and co-workers (37).
Our simulations in a hydrated lipid bilayer at physiological pH were aimed to approximate the realistic in vivo environment. However, GPCRs are often subjected to N- or O-glycosylation within their extracellular regions, which the current computational works on GPCR simulations and our study have neglected. Pharmacological studies have identified that the asparagine residues at positions 6N(N - the N-terminus of the receptor), 15N and 187EL2 in β2AR and at position 15N in β1AR are N-glycosylated (38,39). The non-conserved N187EL2 is located in the second extracellular loop faces the extracellular cavity; thus, it may be directly involved in ligand binding through paths C and B in β2AR. N187EL2 is replaced by D212EL2 in β1AR, without the capability to be glycosylated. It has been shown for several GPCRs that the blockage of glycosylation results in non-functional receptors that are often incapable of ligand binding (40–42). Glycosylation regulates surface expression and dimerization of β1AR (43). It is not clear how many oligosaccharide chains are present for each glycosylation site in GPCRs; however, it is expected that it could vary from a minimum pentasaccharide structure to a complex oligosaccharide structure containing several oligosaccharide chains (44). Such a 'sweet cloud' confronts the extracellular cavity and likely influences ligand binding and unbinding, providing further structural differences between the receptors. The oligosaccharide cloud can form specific intermediate binding sites, where not only an orthosteric ligand can stick on the way to its binding site, but also an allosteric ligand could interact with this site to modulate receptor activation, enlarging the spectrum of the recently proposed putative allosteric binding sites of β1AR and β2AR (45). Importantly, glycosylation produces a stable hydration shell, which causes different entropic cost during ligand binding and unbinding compared to a less ordered hydration shell in the absence of oligosaccharides. Taken together, the next challenge in understanding the molecular basis of ligand binding and unbinding is involvement of receptor active states and glycosylation in GPCR simulations.
Supplementary Material
Acknowledgments
We thank Dr. John King for a generous donation to establish a medicinal chemistry laboratory at the School of Pharmacy of Queen's University Belfast and for providing a Ph.D. studentship for B.S. This study used the high-performance computational facilities of Queen's University Belfast. I.G.T. is supported by the Royal Society. We thank Prof. J. Andrew McCammon, Prof. Daniel Fourmy, Prof. Riccardo Baron, Dr. Anthony Ivetac and Dr. Phineus R. L. Markwick for helpful discussion and Dr. Richard Rankin, Dr. Vaughan Pur-nell and Derek McPhee for technical support. J. W. was supported by Award Number F32GM093581 from the United States National Institute of General Medical Sciences.
Footnotes
Prime 2.0. (2008) New York, NY, USA: Schrödinger, LLC.
(2010) New York, NY, USA: Schrodinger, LLC.
Glide 5.6. (2009) New York, NY, USA: Schrödinger, LLC.
The PyMOL Molecular Graphics System 0.99. (2002) San Carlos, CA, USA: DeLano Scientific, LLC.
SiteMap 2.4. (2010) New York, NY, USA: Schrödinger, LLC.
Supporting Information Additional Supporting Information may be found in the online version of this article:
Please note: Wiley-Blackwell is not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Zaugg M, Schaub MC, Pasch T, Spahn DR. Modulation of beta-adrenergic receptor subtype activities in perioperative medicine: mechanisms and sites of action. Br J Anaesth. 2002;88:101–123. doi: 10.1093/bja/88.1.101. [DOI] [PubMed] [Google Scholar]
- 2.Rosskopf D, Michel MC. Pharmacogenomics of G protein-coupled receptor ligands in cardiovascular medicine. Pharmacol Rev. 2008;60:513–535. doi: 10.1124/pr.108.000612. [DOI] [PubMed] [Google Scholar]
- 3.Jahn P, Eckrich B, Schneidrowski B, Volz-Zang C, Schulte B, Mutschler E, Palm D. Beta 1-adrenoceptor subtype selective antagonism of esmolol and its major metabolite in vitro and in man. Investigations using tricresylphosphate as red blood cell carboxylesterase inhibitor. Arzneimittelforschung. 1995;45:536–541. [PubMed] [Google Scholar]
- 4.Jackman GP, Iakovidis D, Nero TL, Anavekar NS, Rezmann-Vitti LA, Louis SN, Mori M, Drummer OH, Louis WJ. Synthesis, beta-adrenoceptor pharmacology and toxicology of S-(−)-1-(4-(2-ethoxyethoxy)phenoxy)-2-hydroxy-3-(2-(3,4-dimethoxyphenyl)et hylamino)propane hydrochloride, a short acting beta(1)-specific antagonist. Eur J Med Chem. 2002;37:731–741. doi: 10.1016/s0223-5234(02)01399-5. [DOI] [PubMed] [Google Scholar]
- 5.Atsushi N, Fumiyuki T, Masakazu Y, Yoichiro K, Koji T, Takashi T. Pharmacological properties of esmolol hydrochloride. Jpn Pharmacol Ther. 2003;31:21–30. [Google Scholar]
- 6.Baker JG. The selectivity of beta-adrenoceptor antagonists at the human beta1, beta2 and beta3 adrenoceptors. Br J Pharmacol. 2005;144:317–322. doi: 10.1038/sj.bjp.0706048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoffmann C, Leitz MR, Oberdorf-Maass S, Lohse MJ, Klotz KN. Comparative pharmacology of human beta-adrenergic receptor subtypes–characterization of stably transfected receptors in CHO cells. Naunyn Schmiedebergs Arch Pharmacol. 2004;369:151–159. doi: 10.1007/s00210-003-0860-y. [DOI] [PubMed] [Google Scholar]
- 8.Wang T, Duan Y. Ligand entry and exit pathways in the beta2-adrenergic receptor. J Mol Biol. 2009;392:1102–1115. doi: 10.1016/j.jmb.2009.07.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan Y, Xu H, Shaw DE. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc Natl Acad Sci USA. 2011;108:13118–13123. doi: 10.1073/pnas.1104614108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonzalez A, Perez-Acle T, Pardo L, Deupi X. Molecular Basis of Ligand Dissociation in beta-Adrenergic Receptors. PLoS One. 2011;6:e23815. doi: 10.1371/journal.pone.0023815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaminski G, Friesner R, Tirado-Rives J, Jorgensen W. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 12.Jorgensen W, Maxwell D, TiradoRives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 13.Wacker D, Fenalti G, Brown MA, Katritch V, Abagyan R, Cherezov V, Stevens RC. Conserved binding mode of human beta(2) adrenergic receptor inverse agonists and antagonist revealed by X-ray crystallography. J Am Chem Soc. 2010;132:11443–11445. doi: 10.1021/ja105108q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossváry I, Moraes MA, Sacerdoti FD, Salmon JK, Shan Y, Shaw DE. Scalable algorithms for molecular dynamics simulations on commodity clusters. Proceedings of the ACM/ IEEE Conference on Supercomputing; Tampa, Florida. 2006. [Google Scholar]
- 15.Darden T, York D, Pedersen L. Particle mesh Ewald – an N.Log(n) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 16.Grubmuller H, Heymann B, Tavan P. Ligand binding: molecular mechanics calculation of the streptavidin biotin rupture force. Science. 1996;271:997–999. doi: 10.1126/science.271.5251.997. [DOI] [PubMed] [Google Scholar]
- 17.Isralewitz B, Gao M, Schulten K. Steered molecular dynamics and mechanical functions of proteins RID D-5561-2009. Curr Opin Struct Biol. 2001;11:224–230. doi: 10.1016/s0959-440x(00)00194-9. [DOI] [PubMed] [Google Scholar]
- 18.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 19.Warne T, Moukhametzianov R, Baker JG, Nehme R, Edwards PC, Leslie AG, Schertler GF, Tate CG. The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature. 2011;469:241–244. doi: 10.1038/nature09746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moukhametzianov R, Warne T, Edwards PC, Serrano-Vega MJ, Leslie AG, Tate CG, Schertler GF. Two distinct conformations of helix 6 observed in antagonist-bound structures of a beta1-adrenergic receptor. Proc Natl Acad Sci USA. 2011;108:8228–8232. doi: 10.1073/pnas.1100185108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 23.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EY, Velasquez J, Kuhn P, Stevens RC. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure. 2008;16:897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bokoch MP, Zou Y, Rasmussen SG, Liu CW, Nygaard R, Rosenbaum DM, Fung JJ, et al. Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature. 2010;463:108–112. doi: 10.1038/nature08650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sum CS, Tikhonova IG, Costanzi S, Gershengorn MC. Two arginine-glutamate ionic locks near the extracellular surface of FFAR1 gate receptor activation. J Biol Chem. 2009;284:3529–3536. doi: 10.1074/jbc.M806987200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rasmussen SG, Devree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, et al. Crystal structure of the beta(2) adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 29.Isogaya M, Sugimoto Y, Tanimura R, Tanaka R, Kikkawa H, Nagao T, Kurose H. Binding pockets of the beta(1)- and beta(2)-adrenergic receptors for subtype-selective agonists. Mol Pharmacol. 1999;56:875–885. doi: 10.1124/mol.56.5.875. [DOI] [PubMed] [Google Scholar]
- 30.Kikkawa H, Kurose H, Isogaya M, Sato Y, Nagao T. Differential contribution of two serine residues of wild type and constitutively active beta2-adrenoceptors to the interaction with beta2-selective agonists. Br J Pharmacol. 1997;121:1059–1064. doi: 10.1038/sj.bjp.0701229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pooput C, Rosemond E, Karpiak J, Deflorian F, Vilar S, Costanzi S, Wess J, Kirk KL. Structural basis of the selectivity of the beta(2)-adrenergic receptor for fluorinated catecholamines. Bioorg Med Chem. 2009;17:7987–7992. doi: 10.1016/j.bmc.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vanni S, Neri M, Tavernelli I, Rothlisberger U. Observation of “ionic lock” formation in molecular dynamics simulations of wild-type beta 1 and beta 2 adrenergic receptors. Biochemistry. 2009;48:4789–4797. doi: 10.1021/bi900299f. [DOI] [PubMed] [Google Scholar]
- 33.Sugimoto Y, Fujisawa R, Tanimura R, Lattion AL, Cotecchia S, Tsujimoto G, Nagao T, Kurose H. Beta(1)-selective agonist (−)-1-(3,4-dimethoxyphenetylamino)-3-(3,4-dihydroxy)-2-propanol [(−)-RO363] differentially interacts with key amino acids responsible for beta(1)-selective binding in resting and active states. J Pharmacol Exp Ther. 2002;301:51–58. doi: 10.1124/jpet.301.1.51. [DOI] [PubMed] [Google Scholar]
- 34.Baker JG. A full pharmacological analysis of the three turkey beta-adrenoceptors and comparison with the human beta-adrenoceptors. PLoS One. 2010;5:e15487. doi: 10.1371/journal.pone.0015487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Swaminath G, Xiang Y, Lee TW, Steenhuis J, Parnot C, Kobilka BK. Sequential binding of agonists to the beta2 adrenoceptor. Kinetic evidence for intermediate conformational states. J Biol Chem. 2004;279:686–691. doi: 10.1074/jbc.M310888200. [DOI] [PubMed] [Google Scholar]
- 36.Provasi D, Artacho MC, Negri A, Mobarec JC, Filizola M. Ligand-induced modulation of the free-energy landscape of g protein-coupled receptors explored by adaptive biasing techniques. PLoS Comput Biol. 2011;7:e1002193. doi: 10.1371/journal.pcbi.1002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dror RO, Arlow DH, Maragakis P, Mildorf TJ, Pan AC, Xu H, Borhani DW, Shaw DE. Activation mechanism of the beta2-adrenergic receptor. Proc Natl Acad Sci USA. 2011;108:18684–18689. doi: 10.1073/pnas.1110499108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mialet-Perez J, Green SA, Miller WE, Liggett SB. A primate-dominant third glycosylation site of the beta2-adrenergic receptor routes receptors to degradation during agonist regulation. J Biol Chem. 2004;279:38603–38607. doi: 10.1074/jbc.M403708200. [DOI] [PubMed] [Google Scholar]
- 39.Rands E, Candelore MR, Cheung AH, Hill WS, Strader CD, Dixon RA. Mutational analysis of beta-adrenergic receptor glycosylation. J Biol Chem. 1990;265:10759–10764. [PubMed] [Google Scholar]
- 40.Russo D, Chazenbalk GD, Nagayama Y, Wadsworth HL, Rapoport B. Site-directed mutagenesis of the human thyrotropin receptor: role of asparagine-linked oligosaccharides in the expression of a functional receptor. Mol Endocrinol. 1991;5:29–33. doi: 10.1210/mend-5-1-29. [DOI] [PubMed] [Google Scholar]
- 41.Nehring RB, Richter D, Meyerhof W. Glycosylation affects agonist binding and signal transduction of the rat somatostatin receptor subtype 3. J Physiol Paris. 2000;94:185–192. doi: 10.1016/s0928-4257(00)00203-5. [DOI] [PubMed] [Google Scholar]
- 42.Kaushal S, Ridge KD, Khorana HG. Structure and function in rhodopsin: the role of asparagine-linked glycosylation. Proc Natl Acad Sci USA. 1994;91:4024–4028. doi: 10.1073/pnas.91.9.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He J, Xu J, Castleberry AM, Lau AG, Hall RA. Glycosylation of beta(1)-adrenergic receptors regulates receptor surface expression and dimerization. Biochem Biophys Res Commun. 2002;297:565–572. doi: 10.1016/s0006-291x(02)02259-3. [DOI] [PubMed] [Google Scholar]
- 44.Wheatley M, Hawtin SR. Glycosylation of G-protein-coupled receptors for hormones central to normal reproductive functioning: its occurrence and role. Hum Reprod Update. 1999;5:356–364. doi: 10.1093/humupd/5.4.356. [DOI] [PubMed] [Google Scholar]
- 45.Ivetac A, McCammon JA. Mapping the druggable allosteric space of G-protein coupled receptors: a fragment-based molecular dynamics approach. Chem Biol Drug Des. 2010;76:201–217. doi: 10.1111/j.1747-0285.2010.01012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.