

Abstract
Purpose
Neonatal nonketotic hyperglycinemia is an autosomal recessive inborn disorder of glycine metabolism in which large quantities of glycine accumulate in all body tissues. It is characterized by a progressive lethargy, hypotonia, myoclonic jerks, and early death secondary to respiratory problems. As a result of early diagnosis and treatment protocols, more patients survive the critical neonatal period with profound mental retardation, delayed developmental milestones, seizures, and spasticity. There are no reports about the orthopaedic manifestations of neonatal nonketotic hyperglycinemia. The purpose of this study is to evaluate the musculoskeletal findings of neonatal nonketotic hyperglycinemia.
Methods
This is a retrospective IRB-approved study of all patients in our Orthopaedic and Genetics Clinics with the diagnosis of neonatal nonketotic hyperglycinemia during a 10-year period. Demographic, clinical, and imaging data were analyzed.
Results
Twelve patients with neonatal nonketotic hyperglycinemia were evaluated, with a mean age of 7 years and 2 months (range: 5 months to 21 years). Seven were male and five were female. Eleven patients (92 %) have evidence of progressive early-onset neuromuscular scoliosis with a mean Cobb angle of 55° (range: 30–95°). Five children (42 %) presented evidence of progressive hip dislocation secondary to spasticity. All the patients have severe multiple joint contractures.
Conclusion
Neonatal nonketotic hyperglycinemia is a rare metabolic disorder presented in the past as a lethal condition. Recent advances in early diagnosis and neonatal care improve overall outcome. As pediatric orthopaedic surgeons, we need to establish treatment based on update information of the disease and probability to improve quality of life.
Keywords: Metabolic disorder, Scoliosis, Joint contractures, Hip dislocation
Introduction
Neonatal nonketotic hyperglycinemia (NKH) (McKusick 23830, OMIM 605899) [1], also known as glycine encephalopathy, is an autosomal recessive disorder of glycine metabolism [2] that causes an excessive accumulation of glycine in all body fluids and tissues, including the brain and nerve tissues [1, 3–13]. Glycine functions as both excitatory and inhibitory neurotransmitters. It acts as a neuromodulator with an excitatory effect at the N-methyl-d-aspartate receptor channel complex located at the cortex and forebrain, causing intractable seizures, and acts as an inhibitory neuromodulator at the spinal cord and brainstem level, causing muscular hypotonia, neonatal apnea, and hiccupping [1–4, 8, 11, 12].
The incidence is 1 in 12,000 live births in Finland and 1 in 63,000 live births in British Columbia, Canada [3, 6, 8]. There is an elevated presentation among the Arab-Israeli population, New Zealand Maori, and southern part of the Netherlands [11, 14].
NKH has been classified based on the age of manifestation and clinical outcome as neonatal, infantile, late-onset, and transient types [8]. The majority of the cases presented in the neonatal form, also known as classical type, with progressive lethargy, hypotonia, and myoclonic jerks in the first hours to days of life, leading to apnea and death after an uneventful pregnancy and delivery [2–4, 9, 11, 13–15]. The patients, who rarely survived, have profound intellectual disability and intractable seizures. The initial hypotonia converts to a severe spasticity before 6 months of age [4]. The second most frequent presentation is infantile with hypotonia, progressive spasticity secondary to the intractable seizures with developmental delay, and severe psychomotor retardation after a normal pregnancy, labor, and neonatal period. The other presentations range from milder disease, with onset from late infancy to adulthood, to rapidly progressing and severe disease with late onset. The overall life expectancy from this metabolic entity varies by clinical form, with an evident poor life expectancy in the neonatal form compared with those patients with infantile or atypical presentation, who have a better prognosis [1, 4].
Additional clinical features of NKH anecdotically reported include cleft lip/cleft palate, dysplastic ears, congenital hernia, cryptorchidism, pulmonary hypertension [15, 16], and brain malformations, such as hypoplasia of the corpus callosum [10]. The occurrences of those manifestations are always associated with a poor clinical course [1]. The orthopaedic manifestations of NKH have not been previously described in the literature. The purpose of this study is to present the natural history and musculoskeletal manifestations of a neonatal NKH patient group.
Materials and methods
This is a retrospective study of all neonatal NKH patients diagnosed and followed in the Orthopaedic and Genetics Clinics of Mayaguez Medical Center, Puerto Rico, from 2000 to 2010. Approval was given by the Institutional Review Board of the Ponce School of Medicine, Puerto Rico. All the patients were diagnosed due to the presence of an increased cerebrospinal fluid (CSF) to plasma glycine ratio (>0.08, normal level <0.02), together with a CSF glycine concentration over 80 μmol/L (normal level <20 μmol/L) at birth, motivated by the clinical picture of neonatal hypotonia, seizures, and respiratory problems. The following clinical demographic data were collected: age, gender, birth history, weight, treatment, family history, symptoms, hospitalizations, and age at death. The physical examination included joint range of motion and deformities. The radiograph evaluation consisted of a serial pelvis and spine radiograph done every 6 months over the 10-year study period.
Results
Twelve neonatal NKH patients were evaluated during the 2000–2010 study period. Seven patients were male (58 %) and the remaining five patients were female (42 %). The mean age at the last evaluation was 7 years and 2 months (range: 5 months to 21 years). The mean follow-up time was 5 years and 4 months (range: 5 months to 10 years). None of the patients were lost to follow-up. Of the 12 patients studied, five patients are surviving at the time of writing the study, with a mean age of 5 years and 6 months (range: 2–12 years). All the patients presented severe spasticity associated to intractable seizures, developmental delay, deep psychomotor retardation, and respiratory problems that required multiple annual hospitalizations. Seven patients died during the follow-up period, at a mean age of 8 years and 5 months (range: 5 months to 21 years), secondary to respiratory problems, such as aspiration pneumonia, infections, or respiratory insufficiency.
The clinical findings and orthopaedic manifestations of the 12 patients are presented in Table 1. Seizures, neonatal hypotonia, late-onset spasticity with multiple joint contractures, failure to thrive, nonambulatory, and severe global mental retardation were present in all patients. Four of twelve of the patients required mechanical ventilation during the neonatal period. The mean birth weight percentile was within the normal range for gestational age and gender (mean: 43th, range: 10–97th). Progressively, these patients develop a decrease of their percentile secondary to failure to thrive (mean: 27th, range: 3–50th). Grade IV gastro-esophageal reflux was documented in all patients requiring gastrostomy, with fundoplication in 10 of 12 patients.
Table 1.
Patient demographics
| Patient number | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gender | M | M | F | F | F | M | M | M | M | F | M | F |
| Birth weight percentile | 10th | 50th | 50th | 50th | 75th | 97th | 25th | 10th | 50th | 25th | 50th | 25th |
| Last weight percentile | 10th | 3th | 10th | 50th | 3th | 25th | 25th | 50th | 50th | 50th | 3th | 50th |
| Last age | 5 months | 21 years | 6 years | 4 years | 14 years | 12 years | 5 years | 2 years | 6 years | 2 years | 12 years | 2 years |
| Last status | Expired | Expired | Expired | Expired | Expired | Alive | Alive | Alive | Alive | Expired | Expired | Alive |
| Follow-up | 5 months | 10 years | 6 years | 4 years | 10 years | 10 years | 5 years | 2 years | 6 years | 2 years | 10 years | 2 years |
| Scoliosis (Cobb) (°) | Neg. | 57 | 85 | 45 | 95 | 63 | 45 | 30 | 56 | 52 | 45 | 30 |
| Hip dislocation | Neg. | Both | Right | Neg. | Both | Neg. | Left | Neg. | Neg. | Neg. | Left | Neg. |
| Pathologic fracture | Neg. | Neg. | Pos. | Neg. | Neg. | Pos. | Pos. | Neg. | Neg. | Neg. | Pos. | Neg. |
| Joint contractures | Pos. | Pos. | Pos. | Pos. | Pos. | Pos. | Pos. | Pos. | Pos. | Pos. | Pos. | Pos. |
| G-tube | Neg. | Pos. | Pos. | Pos. | Pos. | Pos. | Pos. | Pos. | Pos. | Neg. | Pos. | Pos. |
All the patients had orthopaedic manifestations of diverse severity. Except for one patient that died at 5 months of age, the rest of the patients had early-onset progressive neuromuscular scoliosis. The curve involves the entire thoracic and lumbar spine with the apex being near the thoracolumbar junction, with an evident pelvic obliquity as the main presentation (see Fig. 1). The magnitude of the spine progressive deformity was defined using the Cobb measurement [17]. The mean Cobb angle was 55°, with a range of 30–95°. The mean Cobb angles in the surviving patients at study preparation was 45° (range: 30–63°) versus 63° (range: 45–95°) in those patients that expired before the preparation of the manuscript (p = 0.5208; no significant differences). The clinical features combined with the spine deformity confirmed that our population suffered a clear picture of thoracic insufficiency syndrome [18], secondary to the inability of the chest to maintain an adequate chest and lung growth and function. This limitation is secondary to the chest muscle spasticity and limited lung space available, due to the severe scoliosis. Once the patient progressed to more than 40°, they were treated with bracing without success to control the progression of deformity.
Fig. 1.
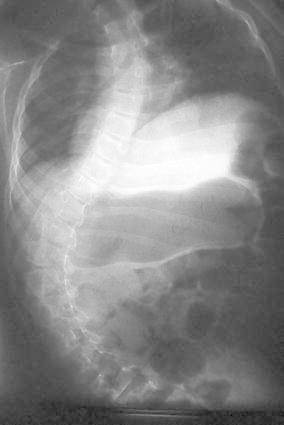
Spine AP radiograph—17-year-old male patient with progressive neuromuscular scoliosis
Besides joint contractures and scoliosis, other musculoskeletal manifestations included neuromuscular hip dislocation, pathologic fractures, and rigid pes equinovarus. Five of twelve patients presented painful hip dislocation secondary to the adductor contractures, with evidence of progressive hip dysplasia (see Fig. 2). The rest of the patients were found with limited abduction (less than 40°) with coxa valga without evident hip dislocation. All patients have bilateral knee extension contractures with bilateral ankle equinovarus deformity secondary to the joint contractures. The upper extremities showed bilateral shoulder adduction contractures, bilateral elbow extension contractures, and bilateral wrist flexion contractures with thumb in palm. They have an opisthotonus presentation. Due to the osteoporotic bone, four patients had pathologic fractures that were treated conservatively.
Fig. 2.
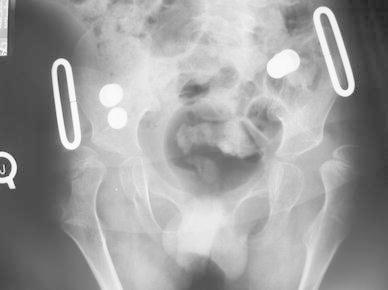
Pelvic AP radiograph—14-year-old male patient with bilateral hip dislocation
Discussion
NKH is an autosomal recessive inborn error of glycine degradation resulting in an excessive accumulation of glycine in all tissues, predominantly in the central nervous system [5]. The diagnosis of NKH is based on an increase of glycine in plasma and CSF, with an increase in the CSF to plasma glycine ratio [1, 3–5, 8, 14]. Enzymatic confirmation relies on measurement of the glycine cleavage enzyme activity in the liver obtained by open biopsy or by molecular genetic testing [1–6, 8, 11, 14].
The classification of NKH is based on the age at initial presentation and ultimate clinical outcome [4]. No effective treatment exists to counteract the impaired glycine metabolism in neonatal NKH [1–3, 8, 10, 12, 14]. The current treatment is targeted to control the cell damage through a reduction of the plasma concentration of glycine with sodium benzoate [1, 4, 5]. Clinical data on the CNS glycine level changes as a result of these medications is still limited [2, 4]. Blockers of glycine receptors such as ketamine, dextromethorphan, or felmate, together with an antiepileptic medication such as benzodiazepines, has been used with variable outcomes [1, 4, 7, 8].
NKH needs to be considered in the differential diagnosis of a newborn with unexplained apnea, lethargy, myoclonic jerks, and seizures [3, 4]. Although this is a rare recessive metabolic disorder, we have evaluated 12 cases of neonatal NKH that survived and which were followed in our institution for a 10-year period. Diagnosis was based upon clinical features, together with the increase in CSF and plasma elevated glycine levels, as well as elevated CSF to plasma ratio. Molecular diagnosis was not performed due to the high costs of sequencing and the lack of medical insurance coverage for this test. Enzymatic confirmation was not performed due to the surgical risk of an open liver biopsy. As an autosomal recessive condition with a 25 % of chance of recurrence on each pregnancy [4], two patients had a sibling with NKH that died during the neonatal period.
The clinical orthopaedic manifestations seen in these patients are secondary to the excitatory effect at the cortex and forebrain level, leading to an extreme spasticity, joint limit range of motion, and deformity such as painful hip dislocation, pes equinovarus, and pathologic fractures that impair quality of life. All the patients suffered from an early-onset neuromuscular scoliosis. During the 10-year period of this study, 58 % of patients died secondary to thoracic insufficiency syndrome [18].
The current literature presented this disorder as a neonatal lethal condition jeopardizing the success of any medical or surgical treatment. Hoover-Fong et al. [4] in 2004, reported 4 years and 2 months as the mean age of death in those patients that survived the newborn period; in our study, the mean age of death was 8 years. However, recent work has reported that patients with a similar clinical presentation, such as severe cerebral palsy, have a life span that reaches the age of 60 years [19]. The message of this work is that children with NKH should not be considered as a severely spastic cerebral palsy patients.
Although none of our patients had surgery, we recognize that, with recent advances in pediatric post-surgical care and in non-fusion spine instrumentation techniques, select patients with painful dislocated hips and/or progressive scoliosis with thoracic insufficiency syndrome may benefit from surgery, in spite of their limited life span. The treatment decision must involve the family and must be in the patient’s best interest.
Contributor Information
Norman Ramirez, Phone: +1-787-2642066, FAX: +1-787-2644483, Email: normanpipe@aol.com.
John M. Flynn, Email: jmflynnmd@aol.com
Francisco Casalduc, Email: francisco.casalduc@upr.edu.
Stephanie Rodriguez, Email: stephy_0.7@hotmail.com.
Alberto S. Cornier, Email: sancor@hotmail.con
Simón Carlo, Email: simoncarlo3@yahoo.com.
References
- 1.Hennermann J. Clinical variability in glycine encephalopathy. Future Neurol. 2006;5:621–630. doi: 10.2217/14796708.1.5.621. [DOI] [Google Scholar]
- 2.Lu FL, Wang PJ, Hwu WL, Tsou Yau KI, Wang TR. Neonatal type of nonketotic hyperglycinemia. Pediatr Neurol. 1999;20:295–300. doi: 10.1016/S0887-8994(98)00157-X. [DOI] [PubMed] [Google Scholar]
- 3.Hamosh A, Scharer G, Van Hove J (2009) Glycine encephalopathy. In: Pagon RA, Bird TD, Dolan CR, Stephens K (eds) GeneReviews [Internet]. University of Washington, Seattle, WA, 1993–2002
- 4.Hoover-Fong JE, Shah S, Van Hove JL, Applegarth D, Toone J, Hamosh A. Natural history of nonketotic hyperglycinemia in 65 patients. Neurology. 2004;63:1847–1853. doi: 10.1212/01.WNL.0000144270.83080.29. [DOI] [PubMed] [Google Scholar]
- 5.Applegarth DA, Toone JR. Nonketotic hyperglycinemia (glycine encephalopathy): laboratory diagnosis. Mol Genet Metab. 2001;74:139–146. doi: 10.1006/mgme.2001.3224. [DOI] [PubMed] [Google Scholar]
- 6.Applegarth DA, Toone JR. Glycine encephalopathy (nonketotic hyperglycinemia): comments and speculations. Am J Med Genet A. 2006;140:186–188. doi: 10.1002/ajmg.a.31030. [DOI] [PubMed] [Google Scholar]
- 7.Matalon R, Michals K, Naidu S, Hughes J. Treatment of non-ketotic hyperglycinaemia with diazepam, choline and folic acid. J Inher Metab Dis. 1982;5:3–5. doi: 10.1007/BF01799799. [DOI] [Google Scholar]
- 8.Bhamkar RP, Colaco P. Neonatal nonketotic hyperglycinemia. Indian J Pediatr. 2007;74:1124–1126. doi: 10.1007/s12098-007-0212-x. [DOI] [PubMed] [Google Scholar]
- 9.Atay E, Bozaykut A, Sezer G. Four cases of neonatal non-ketotic hyperglycinaemia. Ann Trop Paediatr. 2004;24:345–347. doi: 10.1179/027249304225019172. [DOI] [PubMed] [Google Scholar]
- 10.Paupe A, Bidat L, Sonigo P, Lenclen R, Molho M, Ville Y. Prenatal diagnosis of hypoplasia of the corpus callosum in association with non-ketotic hyperglycinemia. Ultrasound Obstet Gynecol. 2002;20:616–619. doi: 10.1046/j.1469-0705.2002.00869.x. [DOI] [PubMed] [Google Scholar]
- 11.Korman SH, Gutman A. Pitfalls in the diagnosis of glycine encephalopathy (non-ketotic hyperglycinemia) Dev Med Child Neurol. 2002;44:712–720. doi: 10.1111/j.1469-8749.2002.tb00275.x. [DOI] [PubMed] [Google Scholar]
- 12.Suzuki Y, Kure S, Oota M, Hino H, Fukuda M. Nonketotic hyperglycinemia: proposal of a diagnostic and treatment strategy. Pediatr Neurol. 2010;43:221–224. doi: 10.1016/j.pediatrneurol.2010.04.018. [DOI] [PubMed] [Google Scholar]
- 13.Boneh A, Allan S, Mendelson D, Spriggs M, Gillam LH, Korman SH. Clinical, ethical and legal considerations in the treatment of newborns with non-ketotic hyperglycinaemia. Mol Genet Metab. 2008;94:143–147. doi: 10.1016/j.ymgme.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 14.García-Pérez A, Martínez-Granero MA, Martín-Ancel AM, Bonet-Serra B, García-Muñoz MJ, García-Segura JM, Viaño J, Lama-More RA. Clinical progress of neonatal non-ketotic hyperglycinemia under treatment. Rev Neurol. 2004;39:727–730. [PubMed] [Google Scholar]
- 15.Cataltepe S, van Marter LJ, Kozakewich H, Wessel DL, Lee PJ, Levy HL. Pulmonary hypertension associated with nonketotic hyperglycinaemia. J Inherit Metab Dis. 2000;23:137–144. doi: 10.1023/A:1005613715351. [DOI] [PubMed] [Google Scholar]
- 16.Al-Shareef I, Arabi M, Dabbagh O. Cardiac involvement in nonketotic hyperglycinemia. J Child Neurol. 2011;26:970–973. doi: 10.1177/0883073811399150. [DOI] [PubMed] [Google Scholar]
- 17.Cobb JR (1948) Outline for the study of scoliosis. In: Edwards JW (ed) Instructional course lectures. The American Academy of Orthopaedic Surgeons, Ann Arbor, vol 5, pp 261–275
- 18.Campbell RM, Jr, Smith MD, Mayes TC, Mangos JA, Willey-Courand DB, Kose N, Pinero RF, Alder ME, Duong HL, Surber JL. The characteristics of thoracic insufficiency syndrome associated with fused ribs and congenital scoliosis. J Bone Joint Surg Am. 2003;85-A(3):399–408. doi: 10.2106/00004623-200303000-00001. [DOI] [PubMed] [Google Scholar]
- 19.Turk MA. Health, mortality, and wellness issues in adults with cerebral palsy. Dev Med Child Neurol. 2009;51:24–29. doi: 10.1111/j.1469-8749.2009.03429.x. [DOI] [PubMed] [Google Scholar]