

Abstract
The molecular mechanisms by which receptors regulate the Ras Binding Domains of the PIP3-generating, class I PI3Ks remain poorly understood, despite their importance in a range of biological settings, including tumorigenesis, activation of neutrophils by pro-inflammatory mediators, chemotaxis of Dictyostelium and cell growth in Drosophila. We provide evidence that G protein-coupled receptors (GPCRs) can stimulate PLCβ2/β3 and diacylglycerol-dependent activation of the RasGEF, RasGRP4 in neutrophils. The genetic loss of RasGRP4 phenocopies knock-in of a Ras-insensitive version of PI3Kγ in its effects on PI3Kγ-dependent PIP3 accumulation, PKB activation, chemokinesis and reactive oxygen species (ROS) formation. These results establish a new mechanism by which GPCRs can stimulate Ras, and the broadly important principle that PLCs can control activation of class I PI3Ks.
Keywords: neutrophil, PI3K, PLCβ, Ras, RasGRP4
Introduction
G protein-coupled receptors (GPCRs) control a variety of neutrophil responses, including chemotaxis and reactive oxygen species (ROS) formation, which are important in health and disease (Simon and Green, 2005; Ley et al, 2007). The intracellular signals coordinating these responses are complex, operate over time scales spanning seconds to hours, are GPCR and context dependent and far from understood. There are, however, a relatively limited number of primary intracellular signals that encode the spatiotemporal characteristics of GPCR and G-protein activation that are of known physiological importance. These include class I PI3Ks (phosphoinositide 3 kinases, particularly PI3Kγ), PLCβs (phospholipase C) and small GTPases such as Rac1 and 2, cdc42, RhoA and Rap1 and 2. In this work, we have focused on mechanisms controlling activation of PI3Kγ.
PI3Kγ is a key effector in a number of myeloid-derived cells (Hirsch et al, 2000; Li et al, 2000; Sasaki et al, 2000) that can be activated directly by Gβγ subunits (Stoyanov et al, 1995; Stephens et al, 1997). PI3Kγ synthesizes the signalling lipid PIP3 and hence can drive activation of PIP3-binding proteins, such as PKB and several specific regulators of small GTPases. It comprising a regulatory (p84 or p101) and a catalytic (p110γ) subunits (Stoyanov et al, 1995; Stephens et al, 1997; Suire et al, 2005; Voigt et al, 2006). Full activation by Gβγs depends on p101 (Suire et al, 2006). The p110γ subunit contains a Ras Binding Domain (RBD) capable of binding and conveying activation by Ras-family GTPases (Rodriguez-Viciana et al, 1994; Pacold et al, 2000; Suire et al, 2002). The RBD is clearly essential for activation of PI3Kγ by GPCRs (Suire et al, 2006). It is unclear, however, whether Ras proteins regulate the RBD of PI3Kγ in vivo in the context of studies indicating that other small GTPases can control class I PI3Ks (Shin et al, 2005).
Current thinking about small GTPases, such as Ras, suggests that they are molecular switches that exist in either a GTP-bound, signalling-competent state or a GDP-bound, basal state. GTP-, but not GDP-, bound GTPases can activate specific proteins with domains, for example, CRIB or RBD, evolved to bind them. Guanine nucleotide Exchange Factors (GEFs) act to stimulate GTP loading. GTPase Activating Proteins (GAPs) act to return the GTPases to their GDP-bound state. Hence, Ras can in principle be activated by activation or inhibition of relevant GEFs and/or GAPs.
Ras-family GTPases, in neutrophils K- and N-Ras, are activated rapidly and substantially following engagement of GPCRs (Zheng et al, 1997). This signal is important for activation of the protein kinase, Raf (Marshall, 1996), and hence the canonical p42/p44-MAPK pathway and also has the potential to activate PI3Kγ (see above and below) and hence a range of PIP3-regulated responses.
Despite the central role of Ras proteins in neutrophils the mechanism by which they are activated is entirely unknown. Further, there is no clear picture of how Ras can be activated by GPCRs in other cell types that could be assumed to apply in neutrophils (Downward, 2003).
PLCβ2 and PLCβ3 are expressed in neutrophils and can be activated by a range of GPCRs, via both Gβγ and Gαq subunits (Smrcka et al, 1991; Taylor et al, 1991; Camps et al, 1992), to hydrolyse PI(4,5)P2 and drive accumulation of its products, diacylglycerol (DAG) and Ins(1,4,5)P3. DAG can activate DAG/phorbol ester-binding, C1 domain-containing effectors, such as the conventional or novel PKCs, and Ins(1,4,5)P3 activates intracellular Ca2+ mobilization, both of which are important regulators of ROS formation and cell migration (Li et al, 2000; Shi et al, 2007). Although PI3Kγ and PLCβ2/β3 use a common substrate lipid it is thought they represent independent, master regulators of ‘parallel’, GPCR-sensitive, signalling cassettes in neutrophils (Tang et al, 2011).
In summary, it is clear that the RBD of PI3Kγ is crucial for GPCR-stimulated accumulation of PIP3 (Suire et al 2006), despite this and the demonstrated importance of the RBDs of other class I PI3Ks in many other broadly important biological settings such as chemotaxis in Dictyostelium (Sasaki et al, 2004), control of cell size in Drosophila (Orme et al, 2006) and tumorigenesis (Gupta et al, 2007), the signalling networks controlling these domains and whether Ras-, or another family of, GTPases, regulate the RBDs in vivo are unclear. Furthermore, the molecular mechanisms by which GPCRs activate Ras, in both neutrophils and more broadly, are poorly defined.
We have addressed these issues by attempting to define how GPCRs control Ras and the RBD of PI3Kγ in neutrophils.
Results
A targeted shRNAi screen to identify a RasGEF in a human neutrophil-like cell line required for fMLP-stimulated activation of Ras
We addressed the nature of the GPCR-sensitive mechanism driving activation of Ras in neutrophils. On the basis of the rapid response, the very low basal levels of Ras-GTP and precedent, we hypothesized that a relevant RasGEF would be receptor sensitive. We screened human neutrophils for expression of all of the potential RasGEFs in the human genome (cdc25 domain-containing proteins that were either known to use Ras-family proteins as substrates or were of unknown specificity). Nine candidates could be detected by reverse transcriptase PCR (RT–PCR) in mRNA from either differentiated PLB-985 cells (human neutrophil-like) or human peripheral blood-derived neutrophils (RasGEF1c, RasGEF2, RasGRF1, Bcar3, Sos1, Sos2, RasGRP3, RasGRP4 and Sh2d3c). We positively selected PLB-985 cells stably expressing a single shRNAi directed against candidate RasGEFs or a control target (typically three different, stable, shRNAi-expressing cell lines for each target). fMLP-stimulated activation of Ras was then assessed by a Raf-RBD ‘pull-down assay’ in differentiated cells. This revealed that shRNAi suppression of RasGRP4 (about 85% reduction in protein levels, see Supplementary Figure 1) but not any of the other candidate RasGEFs reduced activation of Ras by fMLP (Figure 1). Further experiments showed that suppression of RasGRP4 also reduced fMLP-stimulated PIP3 accumulation, indicating that the GTP-Ras which we were assaying was relevant to activation of PI3Kγ (see Supplementary Figure 1).
Figure 1.

An shRNAi screen for suppressors of fMLP-stimulated Ras activation in PLB-985 cells. Nine potential RasGEFs were detected in human neutrophils or differentiated PLB-985 cells. A panel of PLB-985 cell lines was established expressing, shRNAi directed against these RasGEFs, control targets or vector alone (two, or mostly three, independent cell lines each expressing a different shRNAi for each target were derived by lentiviral transduction and use of bis-cistronically expressed eGFP as a selectable marker). Cell lines were differentiated, stimulated with fMLP (100 nM, 30 s) and Ras activation was assayed by pull-down assay with Raf-RBD. (A) Representative immunoblots showing fMLP (+), or its vehicle (−), stimulated Ras activation in parental PLB-985 cells or in lines expressing shRNAi against the indicated RasGEFs. Immunoblots of the total Ras in the lysates from which the pull downs were prepared are also shown. (B) Quantification of data, like that shown in (A), from at least three experiments for each shRNAi-expressing cell line presented as the relative amount of GTP-Ras corrected for total Ras, in the form of mean values±s.e. Figure source data can be found with the Supplementary data.
RasGRP4 message has a restricted distribution within the myeloid compartment and although expressed strongly in mast cell lineages, it is also detectable in monocytes and granulocytes (Reuther et al, 2002; Yang et al, 2002). RasGRP4 has RasGEF activity (Reuther et al, 2002; Yang et al, 2002), can regulate the MAPK pathway and transcriptional targets in mast cells (Katsoulotos et al, 2008). Aberrant expression of RasGRP4 may be associated with disease (Watanabe-Okochi et al, 2009). Recent reports have suggested that FcεRI receptor-dependent, and PMA-induced, responses in mouse mast cells and inflammatory reactions in, some but not all, mouse models of inflammation are reduced in the genetic absence of RasGRP4 (Adachi et al, 2012; Zhu et al, 2012). However, the position of RasGRP4 in the fabric of intracellular signalling and its physiological roles in neutrophils are unclear.
Disruption of the mouse RasGRP4 gene
To validate the results of the screen and address the physiological role of RasGRP4 and Ras activation in mouse neutrophils, we disrupted the mouse RasGRP4 gene using standard homologous targeting strategies (see Supplementary Methods and Supplementary Figure 2). Two independent, correctly targetted ES clones were used to derive mouse strains carrying the targeted RasGRP4 allele in their germline. RasGRP4+/+ and RasGRP4kof/kof mice were derived from each strain and were used in the experiments described below. We observed no significant differences between the two independent RasGRP4kof/kof strains. RasGRP4kof/kof mice were; born at expected Mendelian ratios, of normal weight and appearance, fertile and had normal blood cell counts (see Supplementary Figure 2). These results suggested that haematopoiesis was unperturbed by loss of RasGRP4 expression. Isolated, bone marrow-derived neutrophils from RasGRP4kof/kof mice appeared to be fully responsive in a number of assays (details below), suggesting that they were fully differentiated and broadly functional. We crossed RasGRP4kof/kof mice with FlpE- and Cre-expressing strains sequentially to derive a strain, RasGRP4−/−, containing minimal heterologous DNA and lacking the cDNA for exons 5 and 6, encoding the catalytic domain, of RasGRP4 (see Supplementary Figure 3 and Supplementary Materials and methods). RasGRP4−/− mice were viable and fertile and their neutrophils responded indistinguishably to those from RasGRP4kof/kof mice in specific experiments (see below).
RasGRP4 is required for fMLP-stimulated activation of Ras and PI3K γ pathways
fMLP failed to stimulate significant activation of Ras in neutrophils from RasGRP4kof/kof mice (Figure 2A). The simplest explanation of this result is that RasGRP4 has a unique role as an fMLP-sensitive RasGEF in both mouse and human neutrophils.
Figure 2.

fMLP-stimulated activation of Ras, p42/p44 MAPKs, PKB and PIP3 accumulation is reduced in RasGRP4kof/kof mouse neutrophils. Neutrophils were prepared from either RasGRP4kof/kof or RasGRP4+/+ mice and their responses to fMLP (10 μM) in a range of assays was compared. (A) The lower part shows representative immunoblots measuring fMLP-, or vehicle- (plotted at 0 s in the graphs), stimulated Ras activation (by RafRBD pull down) and p42/p44 MAPK activation, with anti-phospho-T202/Y204 antibodies, and the amount of β-actin, as a loading control, in aliquots of the lysates from which the pull downs were performed. The upper part presents quantification of data from three independent experiments, like those shown in the lower part, showing activation of Ras. Data are presented as means±s.e., n=3 and are expressed as % of the samples stimulated for 1 min (100%), normalized to their respective loading control. (B) Quantification of p42/p44 MAPK phosphorylation from three separate experiments, like those shown in the lower part of (A) is shown. Data are presented as means±s.e., n=3 and are expressed as % of the samples stimulated for 1 min (100%), normalized to their respective loading controls. (C) Quantification of stearoyl/arachidonoyl-PIP3, as a ratio with the recovered PIP3-ISD, in fMLP- (10 μM) or vehicle (plotted at 0 s) -stimulated neutrophils, from three independent experiments is shown. Data are presented as means±s.e., n=6 (D) The upper part shows a representative immunoblot measuring fMLP-, or vehicle- (plotted at 0 s in the graph), stimulated activation of PKB, with anti-phospho-S473-PKB antibodies, aliquots of lysates were from the experiment shown in (A). The lower part shows quantification of phosphorylation of S473-PKB from three independent experiments for RasGRP4+/+ and two independent experiments for RasGRP4kof/kof, like that shown in the upper part, as means±s.e., n=3 for RasGRP4+/+ or as means±s.d., n=2 for RasGRP4kof/kof and are expressed as a % of the samples stimulated for 1 min (100%), normalized via their respective loading controls. Figure source data can be found with the Supplementary data.
Next, we addressed whether the reduction in Ras activation in RasGRP4kof/kof neutrophils also reduced activation of predicted Ras effector pathways. We found fMLP-stimulated phosphorylation of p42/p44 MAPKs (T202/Y204), PIP3 accumulation and phosphorylation of PKB (S473) were severely reduced in RasGRP4kof/kof neutrophils (Figure 2). This indicates that the reduction in fMLP-stimulated Ras activation in neutrophils lacking RasGRP4 is sufficient to suppress stimulation of the p42/p44-MAPK and PI3Kγ/PKB pathways.
Role of RasGRP4 in regulation of Rac and Rap GTPases in neutrophils
Rac GTPases are central regulators of neutrophil responsiveness and can be rapidly activated by GPCRs via the RacGEFs DOCK2 (Kunisaki et al, 2006) and PRex-1 (Welch et al, 2005). PIP3 is thought to regulate the intracellular distribution of both RacGEFs but the activity of only PRex-1. We measured activation of Rac1 and 2 by fMLP using a PAK-CRIB pull-down assay and found that in RasGRP4kof/kof neutrophils fMLP-induced activation of Rac1 and 2 was not significantly changed after brief stimulation, but slightly reduced after longer times of stimulation (Figure 3B). The timescale over which the absence of RasGRP4 has an effect on Rac activation is in-keeping with the relatively slow appearance of a phenotype in the activation of another PIP3 effector, PKB (Figure 2D) and the multifactorial regulation of Rac activity in these cells. This suggests that rapid activation of Rac by fMLP is largely unchanged by the absence of RasGRP4 and hence that this phenotype is not a result of a broad reduction in G-protein signalling and that the weak reduction in Rac activation at later times probably results from reduced PIP3-dependent activation of RacGEFs.
Figure 3.

fMLP-stimulated activation of Rap1 and Rac1 and 2 in RasGRP4kof/kof mouse neutrophils. Bone marrow-derived neutrophils were prepared from either RasGRPkof/kof or RasGRP+/+ mice and assayed for their ability to support fMLP-stimulated activation of Rap1 and Rac1 and 2. (A) The lower part shows a representative immunoblot of fMLP (+, 10 μM), or vehicle (−), induced activation of Rap1, measured by pull down with RalGDS bait, in RasGRP4+/+ (+/+) or RasGRP4kof/kof (k/k) neutrophils. The upper part shows quantification of data, like that shown in the lower part, from five independent experiments, presented as the relative amount of Rap1-GTP/total Rap1, normalized between experiments by correcting the overall mean ratio for each experiment to the same value, in the form of means±s.e. (n=4 or 5). The reduction in Rap1 activation was judged significant by comparing the ratio of the area under the curve for RasGRP4kof/kof/RasGRP4+/+ by a one sample of a t-test, P=0.0357. (B) fMLP-induced activation of Rac1 and Rac2 was quantified by pull down with PAK-CRIB bait and immunoblotting with anti-Rac1 or Rac2-specific antibodies. Results were normalized via the amount of GTP-Rac in unstimulated RasGRP4+/+ neutrophils in each experiment. The data are presented as means±s.e., n=4 (the reduction in activation, in RasGRP4kof/kof compared with RasGRP4+/+ neutrophils, of Rac2 was significant at P=0.03 while the one of Rac1 was not significant at P=0.11). Figure source data can be found with the Supplementary data.
In the context of the similarities between Ras and Rap GTPases and the observation that RasGRP2 is a physiological RapGEF (Crittenden et al, 2004), we assayed fMLP-stimulated activation of Rap1 using RalGDS-RBD and found it to be reduced in RasGRP4kof/kof compared with RasGRP4+/+ neutrophils (Figure 3A). This result was unexpected in the context of work that indicated RasGRP4 is a RasGEF but not a RapGEF when transfected into cells (Reuther et al, 2002) and genetic evidence that suggested RasGRP2 is required for GPCR-stimulated Rap1 activation in mouse neutrophils (Bergmeier et al, 2007). The simplest explanation of this result is that RasGRP4 can be both a Ras and Rap1GEF although it remains possible that the reduction in GTP-bound Rap1 is an indirect result of inhibition of Ras activation.
The role of RasGRP4 in regulation of ROS formation and neutrophil migration
We sought to establish the consequences of loss of RasGRP4 on the functional responses of neutrophils. We measured ROS formation in response to the GPCR ligands C5a, LTB4, fMLP and the phorbol ester, PMA, that bypasses cell surface receptors and activates a subset of C1 domain-containing proteins, such as PKCs and RasGRPs, directly. The amount of ROS produced in response to all three GPCR ligands was reduced substantially in neutrophils from both RasGRP4kof/kof and RasGRP4−/−, compared with RasGRP4+/+, mice, across all times of stimulation and doses of agonist (Figure 4A and B). In contrast, PMA-stimulated ROS formation was increased slightly in RasGRP4kof/kof and RasGRP4−/− compared with RasGRP4+/+ mouse neutrophils (Figure 4A and B).
Figure 4.

ROS responses displayed by neutrophils from RasGRP4kof/kof and RasGRP4−/− mice. (A) Neutrophils were prepared from either RasGRP4kof/kok (right side) or RasGRP4+/+ (left side) mice and stimulated with different concentrations of fMLP, C5a, LTB4 or PMA (from top to bottom panels) and the rate of ROS formation was measured. The data presented are from a single experiment typical of three or four experiments. (B) In the upper four panels, data from experiments shown in (A) were integrated over time to generate an estimate of the total ROS generated in response to each stimulus (RasGRP4+/+ in solid black, RasGRP4kof/kof in white). The two lower panels show integrated ROS in response to either fMLP (left) or PMA (right) for neutrophils derived from RasGRP4+/+ (solid black) or RasGRP4−/− (chequered) mice. The data are means±s.e., from four independent experiments (fMLP and PMA for RasGRP4kof/kof) or three experiments (C5a, LTB4 and RasGRP4−/− data).
These results are entirely consistent with past work showing that either loss of PI3Kγ or knock-in of a Ras-insensitive mutant PI3Kγ substantially reduced fMLP-stimulated ROS formation (Li et al, 2000; Suire et al, 2006) and hence that RasGRP4 and Ras regulate ROS formation via the RBD of PI3Kγ. These results also suggest that PMA-stimulated ROS formation is not dependent on activation of Ras and its downstream targets and is presumably mediated entirely by conventional and/or novel PKC species. In addition, the results suggest that the reduction in GPCR-mediated ROS formation in both the RasGRP4kof/kof and RasGRP4−/− neutrophils is extremely likely a result of loss of RasGRP4 specifically.
We also assayed chemotaxis of isolated mouse neutrophils in gradients of fMLP in an EZTaxiscan chamber. These experiments revealed that RasGRP4kof/kof neutrophils moved in the fMLP gradients with the same migratory index, mean velocity when moving and mean total accumulated distance on both glass and fibrinogen-coated glass as RasGRP4+/+ neutrophils (Figure 5B). However, the proportion of RasGRP4kof/kof neutrophils that moved in response to fMLP on both glass and fibrinogen-coated glass was lower than for RasGRP4+/+ neutrophils (Figure 5C), which is remarkably similar to the phenotype of PI3Kγ-deficient cells (Ferguson et al, 2007). Surprisingly, however, when we tested the ability of neutrophils to migrate into an aseptically inflamed peritoneum, there was no difference between RasGRP4kof/kof and RasGRP4+/+ mice, despite the fact that a number of mouse models with loss of PI3Kγ function show reduced responses in these assays (Suire et al, 2006; Figure 5A). We consider this is in part a result of the fact that PI3Kγ has roles in endothelial cells, which do not express RasGRP4, that support extravasation of neutrophils (Puri et al, 2005) and in part due to an undefined, additional, inhibitory, PI3Kγ-independent role for RasGRP4 in migration of neutrophils into the peritoneum (see below, possibly involving PLCβ2/β3).
Figure 5.

Neutrophil migration in vitro and in vivo. (A) RasGRP4kof/kof and RasGRP4+/+ mice were injected intraperitoneally with thioglycollate. After 3.5 h, the neutrophils that had migrated into the peritoneum were quantified. The data are presented as means±s.e. from 4 independent experiments including a total of 14 RasGRP4+/+ and 15 RasGRP4kof/kof mice. (B) Neutrophils were isolated from RasGRP4kof/kof or RasGRP4+/+ mice and assayed for their ability to respond to, and migrate within, fMLP gradients on either untreated glass or fibrinogen-coated glass. Data are presented as centre-zeroed individual tracks (untreated glass only) for cells that moved a total accumulated distance of >25 μm. (C) Data were extracted from individual cell tracks like those in (B) using the ImageJ-plug-in Chemotaxis tool. The % of cells that moved (a total accumulated distance of >25 μM) are presented as means±s.e. A univariate analysis of variance showed 1P=0.014 and 2P=0.041.
The molecular mechanism regulating activation of RasGRP4
We sought to define the molecular mechanism by which fMLP activates RasGRP4. Other members of the RasGRP4 family are either argued to be activated by increased free cytosolic Ca2+ acting via the tandem EF-hand domain in the case of RasGRP2 (Stefanini et al, 2009) (the C1 domain of RasGRP2 does not appear to bind DAG (Johnson et al, 2007) or via coincident PKC-mediated phosphorylation of T133/T184 and binding of DAG to the C1 domain in the case of RasGRP1/3 (Zheng et al, 2005). RasGRP4 possesses a similar overall topology to its other family members but is very unlikely to be regulated by Ca2+ as its tandem EF hand-like domain lacks key residues known to be required for binding of Ca2+. Furthermore, RasGRP4 lacks the PKC sites equivalent to those required for activation of RasGRP1/3 (Stone, 2011). Interestingly, the mouse genome-sequencing project has predicted the existence of two splice variants of RasGRP4, differing on the basis of a 5aa insert into a loop forming part of the lipid-binding pocket of the C1 domain (Johnson et al, 2007) (the longer variant is not found in the human genome). The sequence of the shorter variant and human RasGRP4 fits well with a subfamily of C1 domains known to bind phorbol esters and DAGs (e.g., PKCδ (C1b), β2-chimaerin, RasGRP1 and 3) and have been demonstrated to bind these molecules both in vitro and in transfected cells (Reuther et al, 2002; Yang et al, 2002; Johnson et al, 2007); the longer variant disrupts the binding motif and has been shown unable to bind lipid species (Johnson et al, 2007). We investigated public mRNA-seq data sets from mouse neutrophils for raw sequence information spanning the key region of the potential 15 bp insert and found three independent sequence reads that spanned the splice boundary in a manner consistent with the shorter, and not the longer, variant. We concluded that the shorter, DAG-binding, variant of RasGRP4 is expressed in mouse neutrophils and that DAG could be a signal controlling RasGRP4.
DAG metabolism is complex, with a number of potential routes of synthesis and degradation spread across several membrane compartments. Both PLD activity, via hydrolysis of PtdCho and production and dephosphorylation of PtdOH, and PLC activity, via hydrolysis of PIP2, can mediate receptor-stimulated increases in DAG. It is possible to preferentially monitor the pool of DAG produced by PLCs and thought to regulate signalling effectors like PKCs, by specifically measuring the levels of the molecular species of DAG that are enriched in PIP2, particularly stearoyl/arachidonoyl-(C18:0/C20:4)-DAG (Pessin and Raben, 1989). Previously, this has been achieved by derivatization and HPLC (Pettitt and Wakelam, 1993) or by lipidomic/mass spectrometry-based methods (Gorden et al, 2011), but in both cases demands significant input material. We have developed a method to quantify stearoyl/arachidonoyl-DAG in small numbers of neutrophils (0.5 × 106) based on use of an internal spike (ISD) of deuterated, D6-stearoyl/arachidonoyl-DAG to trace recovery of the endogenous lipid through a neutral lipid extraction, in-line chromatography on a C4 column and analysis by electrospray mass spectrometry using Multiple Reaction Monitoring. This assay revealed that fMLP stimulated a two-fold increase in stearoyl/arachidonoyl-DAG in mouse neutrophils (Figure 6B).
Figure 6.

Genetic blockade of PLCβ2/β3 signalling inhibits fMLP activation of Ras, p42/p44 MAPK, PKB and PIP3 accumulation. Neutrophils were prepared from either PLCβ2−/− × PLCβ3−/− or PLCβ2+/+ × PLCβ3+/+ mice, stimulated with fMLP (10 μM for 1 min), or its vehicle, and a range of responses were quantified. (A) Lower part shows representative immunoblots of Ras activation by pull down, and phosphorylation of S473-PKB (P-PKB) and T202/Y204-p42/p44 MAPK (P-p42/p44-MAPK) and total Ras (as a loading control) in aliquots of the lysates from which the pull downs were performed. The upper part shows quantification of data, like that below, from three independent experiments and are expressed as means±s.e., n=8, as a % of the stimulated samples in that experiment, normalized via their respective loading controls. (B, C) Measurements of neutrophil stearoyl/arachidonoyl-DAG or stearoyl/arachidonoyl-PIP3 as response ratios with the ISD recovered in that sample are shown. The data presented are means±s.d., n=8, from three independent experiments. Figure source data can be found with the Supplementary data.
GPCR-stimulated increases in Ins(1,4,5)P3, cytosolic-free Ca2+ and PKC activation are abolished in PLCβ2−/− × PLCβ3−/− mouse neutrophils (Li et al, 2000). We found fMLP-stimulated accumulation of stearoyl/arachidonoyl-DAG was also abolished in PLCβ2−/− × PLCβ3−/− neutrophils (Figure 6B). This result indicates that PLCβ2/β3 are responsible for fMLP-stimulated DAG formation in mouse neutrophils.
To test the hypothesis that RasGRP4 is activated by DAG generated by PLCβ2/β3, we measured fMLP-elicited activation of Ras in PLCβ2−/− × PLCβ3−/− neutrophils and found it much reduced compared with PLCβ2+/+ × PLCβ3+/+ neutrophils (Figure 6A). Furthermore, acute treatment with the partially selective (Klein et al, 2011) PLC inhibitor U73122, but not the inactive analogue U73343, completely inhibited fMLP-stimulated activation of Ras in wild-type mouse neutrophils (see Supplementary Figure S4). A PLD1/2-selective inhibitor, VU0155056 (Scott et al, 2009), only inhibited the response weakly (see Supplementary Figure 4). Together, these results indicate that PLCβ2/β3-generated DAG can activate RasGRP4.
We tested that idea that PLCβ2/β3 are required for full activation of MAPK and PIP3 signalling in fMLP-stimulated neutrophils. We found that PIP3 accumulation and phosphorylation of both PKB and p42/p44 MAPKs were substantially reduced in fMLP-stimulated PLCβ2−/− × PLCβ3−/− mouse neutrophils compared with their wild-type controls (Figure 6A and C). Furthermore, PIP3 and PKB phosphorylation were also inhibited by U73122 (see Supplementary Figure 4). These results suggest that PLCβ2/β3 powerfully regulate fMLP-stimulated PI3Kγ and Raf/p42/p44 MAPK signalling.
We tested whether loss of RasGRP4 had an unexpected impact on other DAG effectors such as PKCs. We measured fMLP-stimulated, PKCδ-dependent phosphorylation of T154 in p40PHOX (Chessa et al, 2010). It was not significantly reduced in RasGRP4kof/kof compared with RasGRP4+/+ mouse neutrophils (Supplementary Figure 5) and hence concluded activation of other DAG effectors was normal in the absence of RasGRP4.
The simplest explanation for this body of results is that following GPCR activation of neutrophils PLCβ2/β3-generated DAG regulates RasGRP4 and hence activation of Ras and the PI3Kγ and Raf/MAPK pathways. There are some details in our results that suggest this is not the complete story. The extent of the inhibition of PI3Kγ by blockade of PLCβ appears greater than that inflicted by removing RasGRP4, or knocking-in a Ras-insensitive version of PI3Kγ, suggesting that although the major route by which PLCβs control this cassette is through RasGRP4 and Ras there may be an additional route by which PLCs modulate this pathway.
Discussion
The above results indicate that RasGRP4 is the major fMLP-sensitive RasGEF in mouse and human neutrophils and that it is regulated by PLCβ2/β3-derived DAG. They also indicate that Ras is the direct, dynamic regulator of the RBD domain of PI3Kγ in vivo and, therefore, that PLCβ signals shape class I PI3K responses in these cells (Figure 7). Many receptors are capable of simultaneously activating PLCs and class I PI3Ks and in some cells (e.g., B cells and mast cells) activation of PLCγs can depend on class I PI3K signalling (Huber et al, 1998; Scharenberg et al, 1998). This is the first clear demonstration, however, that PLCs can regulate PI3Ks.
Figure 7.

A schematic mapping RasGRP4 as an effector of PLCβ-derived DAG and a regulator of PI3Kγ upstream of ROS formation. There is strong biochemical and genetic evidence indicating that these connections are important in neutrophil function, with the possible exception of the idea that Rac can regulate PLCβ, where, although the underpinning biochemical data are very clear, the genetic evidence is contradictory.
Previous work has suggested that the RBDs of PI3Ks are an important regulatory input but that they are likely to work in synergy with other signals, for example, with Gβγ subunits in the case of PI3Kγ (Suire et al, 2006). This is consistent with the inability of phorbol esters to directly stimulate PIP3 accumulation or PKB activation in neutrophils or neutrophil-like cells, although they can drive p42/p44-MAPK activation (Stephens et al, 1993; Poon and Stone, 2009), which is presumably a function of the fact that Ras activation is sufficient for Raf, but not PI3Kγ, activation in vivo.
Given the restricted distribution of RasGRP4 message it is unlikely that RasGRP4 is a universal link between GPCRs and Ras activation, however, given the history of molecules that had been declared not to be expressed in tissues in which they were subsequently found to have important roles by genetic deletion, this is not yet completely clear. Superficially, it would seem that both RasGRP1 and 3 could be activated by GPCR receptors via PLC-generated DAG and associated PKC activity. However, it seems that although they are expressed in mouse neutrophils they do not fulfill this role. This apparent segregation in function could be a consequence of either the relatively weak expression of RasGRP3 in these cells or an unappreciated need for the enzymes to be associated with a phosphotyrosine-based signalling complex to organize efficient PKC-mediated phosphorylation. Similarly, there is strong evidence that RasGRPs can be activated by Ca2+ signals in brain; however, it is not clear whether they can respond to a pure GPCR stimulus.
Our data also suggest that RasGRP4 can act as a GPCR-sensitive, DAG-regulated Rap1GEF. As RasGRP2 is a GPCR-sensitive, Ca2+-regulated Rap1GEF, this indicates that Rap1 activity is independently controlled via different signals, with contrasting spatiotemporal properties, that both emerge from activation of PLCβs. Perhaps, this is related to the range of GPCR-driven cell responses, spanning from sub-second to many minutes, with which Rap GTPases are associated in myeloid cells.
Given the above results, and the phenotypes of both p110γ−/− and Ras-insensitive p110γ-knock-in mice, we had expected that migration of neutrophils into an inflamed peritoneum would be reduced in RasGRP4kof/kof compared with RasGRP4+/+ mice. Hence, we were surprised to find migration was unchanged. We note, however, that in the absence of PLCβ2/β3 neutrophil migration to sites of inflammation in vivo is, for unexplained reasons, increased (Li et al, 2000). We would argue that this presumably inhibitory role for endogenous PLCβ2/β3 in part involves RasGRP4 and this, combined with the fact neutrophil migration into the peritoneum depends on p110γ in endothelial cells that do not express RasGRP4, may underlie the unexpected contrast in the phenotypic consequences of loss of PI3Kγ versus RasGRP4 in this regard.
A recent study suggested, in contrast to this manuscript, that PLCβ2/β3 and PI3Kγ operate on ‘parallel’ and not ‘serial’ signalling pathways downstream of GPCRs in mouse neutrophils (Tang et al, 2011). The only result in that study which appears inconsistent with our work is that fMLP-stimulated activation of PKB was not reduced in the absence of PLCβ2/β3, where we find a 60% reduction (and larger reductions in fMLP-stimulated PIP3 accumulation and Ras activation). We have no explanation for this difference as we imported both the PLCβ2−/− × PLCβ3−/− mice and their controls from the animal facility that supported the study, to Babraham for these experiments.
Previous work has shown that GPCR-mediated activation of Ras in neutrophils is conveyed by pertussis toxin-sensitive G proteins (Zheng et al, 1997). This result is in-keeping with the idea that PLCβ2/β3, like PI3Kγ, are activated by relatively large amounts of Gβγ subunits released from these abundant G-protein species. PLCβ2/β3 can also be regulated directly by Rac GTPases and GTP-bound Gαq-family subunits. It is unclear to what extent these different mechanisms contribute to how, where and when PLCβ2/β3, Ca2+ and DAG signals are delivered in vivo and may read-through to downstream targets such as PI3Kγ. Previous work has indicated that the RBD of PI3Kγ, but not Gβγs/p101, are required for GPCR regulation of ROS formation and yet other PI3Kγ-dependent neutrophil responses were similarly dependent on both routes of activation. It was concluded that this was likely based upon the principle that Ras regulation directed the synthesis of a spatially discrete pool of PIP3 capable of controlling specific responses, for example, ROS formation (Suire et al, 2006). This work is consistent with that and supports the idea that the PLCβ/DAG/RasGRP4/Ras pathway provides an input to PI3Kγ that determines both the intensity and location, perhaps at Ras-centric nano-clusters, of its signals.
In conclusion, our results suggest that RasGRP4 is a central hub in GPCR-triggered pro-inflammatory pathways in both human and mouse neutrophils. They also reveal an unappreciated role for PLCs and Ras in the regulation of class I PI3Ks that could have wide spread implications, given that other RasGEFs in the RasGRP family can be activated by receptor tyrosine kinases and PLCγ in parallel with PI3Ks α, β and δ (Stone, 2011).
Materials and methods
Cell lines, antibodies and reagents
All materials used were of the lowest endotoxin level available and were purchased from Sigma unless stated otherwise. The polyclonal antibody against hRasGRP4 was raised in rabbit against 14-mer peptide (MNRKDSKRKSHQEC) found in the N-terminal end of the protein and affinity purified. The other antibodies used for western blots were commercially available: anti-pan-Ras (1:1000; Calbiochem); anti-P-PKB-S473 (1:2000; Cell Signalling); anti-Rap1 (1:750; Epitomics); anti-βactin (1:10 000; Sigma); anti-P-p42/44 MAPK (T202/Y204) (1:2000; Cell Signalling), anti-Rac1 (1:3000; Millipore), anti-Rac-2 (1:300; Millipore), anti-phospho-p40phox (Thr154) (1:1000; Cell Signalling) and anti-βcop (1:40, gift from Dr N Ktistakis). The PLD1/2 inhibitor VU0155056 (Scott et al, 2009) was synthesized at BI by JC and tested extensively in our laboratory (Norton et al, 2011). Deuterated and un-deuterated 18:0/20:4-DAG were made by JC/IN by modification of a published procedure (Chen et al, 1996). fMLP, PMA and C5a were from Sigma. Leukotriene B4 (LTB4) was from Enzo Life Sciences.
PLB-985 cells were cultured in RPMI supplemented with 10% fetal calf serum (FCS) and 1% penicillin/streptomycin at 37°C in 5% CO2 in a humidified chamber. Prior to any cell assay, the PLB-985 were differentiated during 6–8 days in medium containing 0.5% N, N-dimethylfomamide without antibiotics.
HEK 293FT (Invitrogen) were cultured in DMEM, complemented with 10% FBS, 1% penicillin/streptomycin at 37°C in 5% CO2 in a humidified chamber.
Reverse transcriptase PCR analysis
mRNA was extracted from PLB-985 cells using a QIAamp RNA Blood kit (Qiagen) according to manufacturer's instructions. The reverse transcription was performed with Omniscript Reverse Transcription and the amplification of the cDNA was performed with HotstartTaq DNA polymerase (Qiagen) using the following primers in order to check the expression of known RasGEFs in PLB-985 cells. The sequences of the relevant primers are shown in Supplementary data.
Production of lentivirus RasGRP4 shRNA
Using Dharmacon siDESIGN Center, we designed oligonucleotides (2–3 individual sets per RasGEF) of 19 mers of sense and antisense strands separated by a loop (flanked by a 5′ BglII and 3′ ClaI/HindIII restriction sites) for the following RasGEFs: RasGef1c, RapGef2, RasGrf1, Sos1, Sos2, RasGRP3, sh2d3c and Bcar3, RasGRP4.
Once annealed, the respective double-stranded oligonucleotides were introduced into the bis-cistronic pCMS3-H1p-EGFP plasmid (via the BglII and HindIII sticky ends) and then cloned into a lentivirus expression vector plasmid pLVTH via EcoRI/ClaI sites. Recombinant lentivirus were produced by co-transfecting 293FT cells with the above, pMD4-VSVG and pCMV-DR8.91 using standard procedures. Independent, stable, mixed, populations of cells expressing a single shRNAi were then selected by fluorescence activated cell sorting of GFP-expressing cells.
Generation of RasGRP4 knockout mice
We targeted mouse RasGRP4 using a vector created by the High Throughput Gene Targeting group at the Wellcome Trust Sanger Institute (RasGRP4tm2a(KOMP)Wtsi, we abbreviate this to a ‘knockout-first’ (kof) allele); see Supplementary Figure 2. The vector was linearized with AsiSI and transfected into C57/Bl6-derived ES cells (Bruce 4) and appropriately targeted clones (from a total of 172) were identified by Southern blotting (5′, 3′ and internal [32P]-oligonucleotide probes). Two correctly targeted, independent clones of ES cells (E09/AE1 and E09/AE12) were injected into C57/Bl6Tyr−/−-derived blastocysts by the Gene Targeting Facility at Babraham and male chimeras were mated with C57/Bl6Tyr−/− females. Southern blotting and PCR-based approaches were used to confirm germline transmission RasGRP4tm2a(KOMP)Wtsi/tm2a(KOMP)Wtsi (‘RasGRP4kof/kof’) and RasGRP4+/+ mice were derived from each ES cell line. Genotyping of the mice was routinely performed by PCR amplification of the area containing intron 6 and the adjacent lox-P site, generating a 315-bp fragment for Wt and 390-bp fragment from RasGRP4 targeted alleles. The results presented in this manuscript are derived from mice from both of the strains. We crossed RasGRP4kof/kof × FlpE expressing strain (Farley et al, 2000, C57/bl-6 background) then progeny of this cross was mated with a Cre-deleter strain (Schwenk et al, 1995, C57/bl-6 background) to remove the drug selection and reporter cassettes and derive a RasGRP4−/− strain with minimal heterologous DNA in the RasGRP4 locus (see Supplementary Figure 3 and result Figure 4B).
We obtained PLCβ2−/− × PLCβ3−/− mice (Li et al, 2000) and their colony-related wild-type controls in a C57/bl-6 background from Yale University. We included both imported PLCβ2+/+ × PLCβ3+/+ mice and wild-type C57/bl-6 mice from the Babraham Institute animal facility in experiments alongside imported PLCβ2−/− × PLCβ3−/− mice. We found neutrophils from these two types of ‘control’ mice responded indistinguishably in terms of fMLP-stimulated PIP3 accumulation and phosphorylation of S473-PKB and hence pooled results from these strains to give rise to the data set described as ‘PLCβ2+/+ × PLCβ3+/+’ in those assays in Figure 6.
All of the mice used in experiments were kept under specific pathogen-free conditions in Transgenics at the Babraham Institute. This work was performed under Home Office Project license PPL 80/2335.
Purification of mouse neutrophils
Murine neutrophils were isolated at room temperature from bone marrow using Percoll (62 and 55%; Anderson et al, 2008) (purity, as assessed by cytospin and staining with May-Grumwald-Giemsa, was typically between 65 and 85% neutrophils). For Rac activation and chemotaxis assays, the neutrophils were purified at 4°C (Ferguson et al, 2007) (typically between 55 and 75% neutrophils).
‘Pull-down’ assays
In experiments to measure Ras, Rac1 or 2 or Rap1 activation, neutrophils were stimulated while in suspension (4 × 106/ml in HBSS, at 37°C) rapidly diluted with ice-cold PBS, sedimented by brief centrifugation, aspirated and solubilized into ice-cold lysis buffer containing 1% Triton X-100, 0.12 M NaCl, 1 mM EGTA, 1 mM EDTA, 20 mM HEPES/NaOH pH 7.4 at 4°C. The lysates were sheared through a 25-gauge needle and centrifuged (12 000 r.p.m., 10 min, 4°C). Aliquots of the supernatants mixed with 4 × SDS–PAGE sample buffer and Ras, Rap1 and Rac 1/2 pull-down assays were performed using GST-Raf-RBD, GST-RalGDS and GST-PAK-CRIB proteins, respectively, freshly pre-bound to glutathione-sepharose beads (about 25 μl of packed beads with 50 μg of protein bait).
Western blot
After incubation with relevant primary and secondary, HRP-labelled, antibodies, membranes were incubated with ECL reagents (GE Healthcare) and exposed onto light-sensitive film. Protein levels were quantified by 2D densitometry using Aida Image Analyzer software v.3.27. Phosphorylation of T154-p40PHOX was measured by immunoblotting with a phospho-specific antibody as described (Chessa et al, 2010).
In-vivo migration
The migration of murine neutrophils into the peritoneum was assessed after 3.5 h after intra-peritoneal injection of thioglycollate (0.25 ml of 3% thioglycollate) by flushing the peritoneal cavity with 2 × 8 ml PBS/5 mM EDTA. After lysis of the red blood cells, leucocytes were double stained for Gr-1 and Mac-1 and analysed by FACS. Neutrophils (double positive) were counted. Parallel, cytospins were prepared and stained with May-Grumwald-Giemsa stain.
ROS assay
fMLP, C5a, LTB4 and PMA-stimulated ROS formation was measured with an HRP/luminol-based assay and a multiplate luminometer at 37°C (Anderson et al, 2008).
EZ-Taxiscan chamber assay
This assay was used to quantify migration of mouse bone marrow-derived neutrophils in gradients of fMLP (optimized to give a clear but submaximal response) as described (Ferguson et al, 2007). The cells were imaged every 20 s, with an exposure time of 50 ms, for 33.33 min (100 frames) using a × 10 objective (long-range, 0.3 NA) on an Olympus IX 81 microscope with a Hamamatsu Orca camera, Marzhauser SCAN-IM motorized stage and Till phototonics Polychrome V illuminator. The system light path was configured in a reflection mode using incident light at 488/10 nm and reflected light acquired through a 483/32 emission filter. The glass coverslips were washed with concentrated H2SO4 and, after rinsing with H2O, with 0.25 M NaOH before being washed with H2O and stored under ethanol. Cleaned coverslips were used either directly or coated with 2.5 mg/ml fibrinogen in HBSS at 37°C for 1 h then washed with the buffer used in the chemotaxis experiments.
Analysis of migration movies
Movies taken of the EZ-Taxiscan chambers were converted into stacks of TIF files and analysed within the ImageJ plug-in ‘manual tracking’. Only cells that remained within both the field of view and the bridge were tracked. The tabbed text files that were created were analysed within the ImageJ plug-in ‘chemotaxis tool’. The proportion of cells that moved more than a total accumulated distance of 25 μm was determined (‘moved in response to fMLP’) and was corrected for the purity of neutrophils in the cell preparation in each experiment. The population of motile cells was then used to calculate the velocity of the cells (using a velocity threshold of 0.006 μm/s to make this an estimate of the velocity of moving cells and not a mixture of moving and stationary cells), the migratory index (a measure of how straight the paths taken by the cells are; distance from origin/total distance travelled) and mean total accumulated distance. The data in Figure 5B and C are based on the following numbers of observations: on glass surfaces the number of cells that were tracked and moved >25 μm were 141 for RasGRP4+/+ and 120 cells for RasGRP4kof/kof, in 3 experiments; on fibrinogen-coated glass the numbers were 10 and 21 cells, respectively, in 2 experiments.
Quantification of phosphoinositides and DAG
Phosphoinositides in PLB-985 cells were quantified by [32P]-Pi labelling of the cells followed by extraction, deacylation and anion-exchange HPLC (Condliffe et al, 2005). PIP2 and PIP3 in mouse neutrophils were quantified by mass spectrometry (Clark et al, 2011), but with adaptions to the procedure to allow both DAG and phosphoinositides to be measured in the same aliquots of cells. Aliquots of neutrophil suspensions (135 μl, 0.5 × 106) were stimulated with fMLP (15 μl, 100 μM, 10 μM final concentration) or vehicle alone. After an appropriate time, incubations were quenched with 750 μl of a solvent mixture containing MeOH/CHCl3 (2:1) that created a single homogenous phase. Two internal standards were then added to correct recoveries; D6-1-stearoyl-2-arachidonyl-DAG (10 pg) and C16:0/C17:0-PIP3 (1 ng). In all, 725 μl CHCl3 and 193 μl H2O were added, the samples mixed, and the resultant two phases (Folch et al, 1957) separated by centrifugation (5 min at 2000 g). Approximately 1 ml of the lower phase containing the neutral lipids, and none of the PIP2 or PIP3, was removed, dried under N2, and resuspended in 100 μl of methanol: water (4:1). These samples were further processed to quantify stearoyl/arachidonoyl-DAG as described below. The upper phase was acidified and resolved into two clear phases by the addition of 500 μl MeOH/CHCl3 (2:1), 500 μl CHCl3 and 170 μl 2 M HCl, thorough mixing and centrifugation (5 min at 2000 g). The lower phase, now containing the PIP2 and PIP3, was derivatized using TMS-diazomethane, resolved by in-line HPLC and analysed by mass spectrometry, as described previously (Clark et al, 2011); values for endogenous C18/C20:4 PIP2 and PIP3 were corrected for recovery of the C16:0/C17:0-PIP3 internal standard.
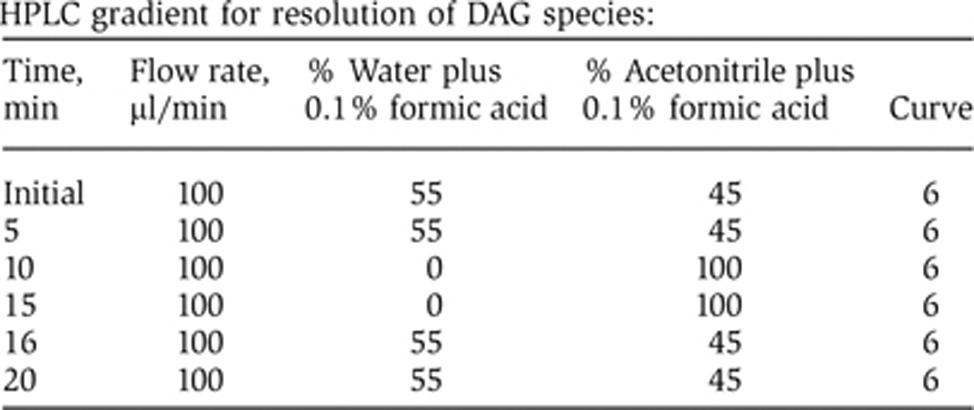
To measure 1-stearoyl-2-arachidonoyl-sn-glycerol (18:0/20:4 DAG) by mass spectroscopy, the samples in methanol/water (4:1) were injected (45 μl) onto a Waters Acquity UPLC BEH300 C4 1.0 × 100 mm column at 294 K and eluted with a 45–100% acetonitrile in water gradient with 0.1% formic acid added at a flow rate of 100 μl/min over 20 min (see the gradient structure, below). The eluent was then passed into an AB Sciex 4000 QTrap mass spectrometer and the MRM transitions 645.6>341.4 for the 18:0/20:4-DAG and 651.6>347.4 for the deuterated internal standard were monitored, corresponding to loss of an arachidonate group (as the most sensitive MRM transition we identified) (mass spec machine settings are defined below). Both the labelled and unlabelled 18:0–20:4-DAG eluted at 10.94 min. Values for endogenous 18:0/20:4-DAG were corrected for the recovery of the internal standard.
AB Sciex Instruments 4000 QTrap Mass spectrometer parameters are Scan Type: MRM; Polarity: Positive; Ion Source: Turbo Spray; Resolution Q1: Unit; Resolution Q2: Low; Dwell: 50 ms; CUR: 20; IS: 4500; TEM: 300; GS1: 18; GS2: 20; Ihe: ON; CAD: Medium; DP: 100; EP: 10; CE: 35; CXP: 10.
Statistics
Professional statistical support and advice was provided by the Bio-Informatics Facility at the Babraham Institute. Statistics tests are defined where applied.
Supplementary Material
Acknowledgments
We would like to acknowledge Dianqing Wu (Yale university) for access to PLCβ2−/− × PLCβ3−/− mice, Arnaud Deladérière (Babraham) for help with some experiments measuring neutrophil migration in vivo, the High Throughput Gene Targeting group at the Wellcome Trust Sanger Institute for help in obtaining the RasGRP4 gene targeting vector, Felix Kreuger for help with analysis of mRNA-seq data bases and Anne Segonds-Pichon for help with statistics. This work was supported by a grant from the BBSRC, BB/D013593/1 and the British Lung Foundation.
Author contributions: SS performed experiments, analysed data and wrote the paper; CE, KA, GD, IN, HG and DP performed experiments and analysed data; KD and DP performed experiments; JC analysed data, wrote the paper and developed the DAG-assay methodology at all levels; PTH and LS planned work, performed experiments, analysed the data and wrote the paper.
Footnotes
The authors declare that they have no conflict of interest.
References
- Adachi R, Krillis SA, Nigrovic PA, Hamilton MJ, Chung K, Thakurdas SM, Boyce JA, Anderson P, Stevens RL (2012) Ras guanine nucleotide releasing protein-4 (RasGRP4) involvement in experimental arthritis and colitis. J Biol Chem 287: 20047–20055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KE, Boyle KB, Davidson K, Chessa TA, Kulkarni S, Jarvis GE, Sindrilaru A, Scharffetter-Kochanek K, Rausch O, Stephens LR (2008) CD18-dependent activation of the neutrophil NADPH oxidase during phagocytosis of Escherichia coli or Staphylococcus aureus is regulated by class III but not class I or II PI3Ks. Blood 112: 5202–5211 [DOI] [PubMed] [Google Scholar]
- Bergmeier W, Goerge T, Wang HW, Crittenden JR, Baldwin AC, Cifuni SM, Housman DE, Graybiel AM, Wagner DD (2007) Mice lacking the signaling molecule CalDAG-GEFI represent a model for leukocyte adhesion deficiency type III. J Clin Invest 117: 1699–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camps M, Carozzi A, Schnabel P, Scheer A, Parker PJ, Gierschik P (1992) Isozyme-selective stimulation of phospholipase C-beta 2 by G protein beta gamma-subunits. Nature 360: 684–686 [DOI] [PubMed] [Google Scholar]
- Chen J, Profit AA, Prestwich GD (1996) Synthesis of photoactivatable 1,2-O-diacyl-sn-glycerol derivatives of 1-L-phosphatidyl-D-myo-inositol 4,5-bisphosphate (PtdInsP(2)) and 3,4,5-trisphosphate (PtdInsP(3)). J Org Chem 61: 6305–6312 [DOI] [PubMed] [Google Scholar]
- Chessa TA, Anderson KE, Hu Y, Xu Q, Rausch O, Stephens LR, Hawkins PT (2010) Phosphorylation of threonine 154 in p40phox is an important physiological signal for activation of the neutrophil NADPH oxidase. Blood 116: 6027–6037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark J, Anderson KE, Juvin V, Smith TS, Karpe F, Wakelam MJ, Stephens LR, Hawkins PT (2011) Quantification of PtdInsP3 molecular species in cells and tissues by mass spectrometry. Nat Methods 8: 267–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condliffe AM, Davidson K, Anderson KE, Ellson CD, Crabbe T, Okkenhaug K, Vanhaesebroeck B, Turner M, Webb L, Wymann MP, Hirsch E, Ruckle T, Camps M, Rommel C, Jackson SP, Chilvers ER, Stephens LR, Hawkins PT (2005) Sequential activation of class IB and class IA PI3K is important for the primed respiratory burst of human but not murine neutrophils. Blood 106: 1432–1440 [DOI] [PubMed] [Google Scholar]
- Crittenden JR, Bergmeier W, Zhang Y, Piffath CL, Liang Y, Wagner DD, Housman DE, Graybiel AM (2004) CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation. Nat Med 10: 982–986 [DOI] [PubMed] [Google Scholar]
- Downward J (2003) Role of receptor tyrosine kinases in G-protein-coupled receptor regulation of Ras: transactivation or parallel pathways? Biochem J 376: e9–e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley FW, Soriano P, Steffen LS, Dymecki SM (2000) Widespread recombinase expression using FLPeR (flipper) mice. Genesis 28: 106–110 [PubMed] [Google Scholar]
- Ferguson GJ, Milne L, Kulkarni S, Sasaki T, Walker S, Andrews S, Crabbe T, Finan P, Jones G, Jackson S (2007) PI(3)Kgamma has an important context-dependent role in neutrophil chemokinesis. Nat Cell Biol 9: 86–91 [DOI] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH (1957) A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 226: 497–509 [PubMed] [Google Scholar]
- Gorden DL, Ivanova PT, Myers DS, McIntyre JO, VanSaun MN, Wright JK, Matrisian LM, Brown HA (2011) Increased diacylglycerols characterize hepatic lipid changes in progression of human nonalcoholic fatty liver disease; comparison to a murine model. PLoS One 6: e22775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, Nye E, Stamp G, Alitalo K, Downward J (2007) Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell 129: 957–968 [DOI] [PubMed] [Google Scholar]
- Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann MP (2000) Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science 287: 1049–1053 [DOI] [PubMed] [Google Scholar]
- Huber M, Helgason CD, Damen JE, Liu L, Humphries RK, Krystal G (1998) The src homology 2-containing inositol phosphatase (SHIP) is the gatekeeper of mast cell degranulation. Proc Natl Acad Sci USA 95: 11330–11335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JE, Goulding RE, Ding Z, Partovi A, Anthony KV, Beaulieu N, Tazmini G, Cornell RB, Kay RJ (2007) Differential membrane binding and diacylglycerol recognition by C1 domains of RasGRPs. Biochem J 406: 223–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsoulotos GP, Qi M, Qi JC, Tanaka K, Hughes WE, Molloy TJ, Adachi R, Stevens RL, Krilis SA (2008) The diacylglycerol-dependent translocation of ras guanine nucleotide-releasing protein 4 inside a human mast cell line results in substantial phenotypic changes, including expression of interleukin 13 receptor alpha2. J Biol Chem 283: 1610–1621 [DOI] [PubMed] [Google Scholar]
- Klein RR, Bourdon DM, Costales CL, Wagner CD, White WL, Williams JD, Hicks SN, Sondek J, Thakker DR (2011) Direct activation of human phospholipase C by its well known inhibitor u73122. J Biol Chem 286: 12407–12416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisaki Y, Nishikimi A, Tanaka Y, Takii R, Noda M, Inayoshi A, Watanabe K, Sanematsu F, Sasazuki T, Sasaki T, Fukui Y (2006) DOCK2 is a Rac activator that regulates motility and polarity during neutrophil chemotaxis. J Cell Biol 174: 647–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley K, Laudanna C, Cybulsky MI, Nourshargh S (2007) Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 7: 678–689 [DOI] [PubMed] [Google Scholar]
- Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, Wu D (2000) Roles of PLC-beta2 and -beta3 and PI3Kgamma in chemoattractant-mediated signal transduction. Science 287: 1046–1049 [DOI] [PubMed] [Google Scholar]
- Marshall CJ (1996) Ras effectors. Curr Opin Cell Biol 8: 197–204 [DOI] [PubMed] [Google Scholar]
- Norton LJ, Zhang Q, Saqib KM, Schrewe H, Macura K, Anderson KE, Lindsley CW, Brown HA, Rudge SA, Wakelam MJ (2011) PLD1 rather than PLD2 regulates phorbol-ester-, adhesion-dependent and Fcγ-receptor-stimulated ROS production in neutrophilsp. J Cell Sci 124: 1973–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orme MH, Alrubaie S, Bradley GL, Walker CD, Leevers SJ (2006) Input from Ras is required for maximal PI(3)K signalling in Drosophila. Nat Cell Biol 8: 1298–1302 [DOI] [PubMed] [Google Scholar]
- Pacold ME, Suire S, Perisic O, Lara-Gonzalez S, Davis CT, Walker EH, Hawkins PT, Stephens L, Eccleston JF, Williams RL (2000) Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell 103: 931–943 [DOI] [PubMed] [Google Scholar]
- Pessin MS, Raben DM (1989) Molecular species analysis of 1,2-diglycerides stimulated by alpha-thrombin in cultured fibroblasts. J Biol Chem 264: 8729–8738 [PubMed] [Google Scholar]
- Pettitt TR, Wakelam MJ (1993) Bombesin stimulates distinct time-dependent changes in the sn-1,2-diradylglycerol molecular species profile from Swiss 3T3 fibroblasts as analysed by 3,5-dinitrobenzoyl derivatization and h.p.l.c. separation. Biochem J 289: Pt 2487–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon HY, Stone JC (2009) Functional links between diacylglycerol and phosphatidylinositol signaling systems in human leukocyte-derived cell lines. Biochem Biophys Res Commun 390: 1395–1401 [DOI] [PubMed] [Google Scholar]
- Puri KD, Doggett TA, Huang CY, Douangpanya J, Hayflick JS, Turner M, Penninger J, Diacovo TG (2005) The role of endothelial PI3Kgamma activity in neutrophil trafficking. Blood 106: 150–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuther GW, Lambert QT, Rebhun JF, Caligiuri MA, Quilliam LA, Der CJ (2002) RasGRP4 is a novel Ras activator isolated from acute myeloid leukemia. J Biol Chem 277: 30508–30514 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD, Downward J (1994) Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature 370: 527–532 [DOI] [PubMed] [Google Scholar]
- Sasaki AT, Chun C, Takeda K, Firtel RA (2004) Localized Ras signaling at the leading edge regulates PI3K, cell polarity, and directional cell movement. J Cell Biol 167: 505–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B, Wakeham A, Itie A, Bouchard D, Kozieradzki I, Penninger JM (2000) Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science 287: 1040–1046 [DOI] [PubMed] [Google Scholar]
- Scharenberg AM, El-Hillal O, Fruman DA, Beitz LO, Li Z, Lin S, Gout I, Cantley LC, Rawlings DJ, Kinet JP (1998) Phosphatidylinositol-3,4,5-trisphosphate (PtdIns-3,4,5-P3)/Tec kinase-dependent calcium signaling pathway: a target for SHIP-mediated inhibitory signals. EMBO J 17: 1961–1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk F, Baron U, Rajewsky K (1995) A Cre-transgenic strain for the ubquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res 23: 5080–5081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott SA, Selvy PE, Buck JR, Cho HP, Criswell TL, Thomas AL, Armstrong MD, Arteaga CL, Lindsley CW, Brown HA (2009) Design of isoform-selective phospholipase D inhibitors that modulate cancer cell invasiveness. Nat Chem Biol 5: 108–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G, Partida-Sanchez S, Misra RS, Tighe M, Borchers MT, Lee JJ, Simon MI, Lund FE (2007) Identification of an alternative G{alpha}q-dependent chemokine receptor signal transduction pathway in dendritic cells and granulocytes. J Exp Med 204: 2705–2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin HW, Hayashi M, Christoforidis S, Lacas-Gervais S, Hoepfner S, Wenk MR, Modregger J, Uttenweiler-Joseph S, Wilm M, Nystuen A, Frankel WN, Solimena M, De Camilli P, Zerial M (2005) An enzymatic cascade of Rab5 effectors regulates phosphoinositide turnover in the endocytic pathway. J Cell Biol 170: 607–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon SI, Green CE (2005) Molecular mechanics and dynamics of leukocyte recruitment during inflammation. Annu Rev Biomed Eng 7: 151–185 [DOI] [PubMed] [Google Scholar]
- Smrcka AV, Hepler JR, Brown KO, Sternweis PC (1991) Regulation of polyphosphoinositide-specific phospholipase C activity by purified Gq. Science 251: 804–807 [DOI] [PubMed] [Google Scholar]
- Stefanini L, Roden RC, Bergmeier W (2009) CalDAG-GEFI is at the nexus of calcium-dependent platelet activation. Blood 114: 2506–2514 [DOI] [PubMed] [Google Scholar]
- Stephens L, Eguinoa A, Corey S, Jackson T, Hawkins PT (1993) Receptor stimulated accumulation of phosphatidylinositol (3,4,5)-trisphosphate by G-protein mediated pathways in human myeloid derived cells. EMBO J 12: 2265–2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens LR, Eguinoa A, Erdjument-Bromage H, Lui M, Cooke F, Coadwell J, Smrcka AS, Thelen M, Cadwallader K, Tempst P, Hawkins PT (1997) The G beta gamma sensitivity of a PI3K is dependent upon a tightly associated adaptor, p101. Cell 89: 105–114 [DOI] [PubMed] [Google Scholar]
- Stone JC (2011) Regulation and Function of the RasGRP Family of Ras Activators in Blood Cells. Genes Cancer 2: 320–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoyanov B, Volinia S, Hanck T, Rubio I, Loubtchenkov M, Malek D, Stoyanova S, Vanhaesebroeck B, Dhand R, Nurnberg B (1995) Cloning and characterization of a G protein-activated human phosphoinositide-3 kinase. Science 269: 690–693 [DOI] [PubMed] [Google Scholar]
- Suire S, Hawkins P, Stephens L (2002) Activation of phosphoinositide 3-kinase gamma by Ras. Curr Biol 12: 1068–1075 [DOI] [PubMed] [Google Scholar]
- Suire S, Coadwell J, Ferguson GJ, Davidson K, Hawkins P, Stephens L (2005) p84, a new Gbetagamma-activated regulatory subunit of the type IB phosphoinositide 3-kinase p110gamma. Curr Biol 15: 566–570 [DOI] [PubMed] [Google Scholar]
- Suire S, Condliffe AM, Ferguson GJ, Ellson CD, Guillou H, Davidson K, Welch H, Coadwell J, Turner M, Chilvers ER, Hawkins PT, Stephens L (2006) Gbetagammas and the Ras binding domain of p110gamma are both important regulators of PI(3)Kgamma signalling in neutrophils. Nat Cell Biol 8: 1303–1309 [DOI] [PubMed] [Google Scholar]
- Tang W, Zhang Y, Xu W, Harden TK, Sondek J, Sun L, Li L, Wu D (2011) A PLCβ/PI3Kγ-GSK3 signalling pathway regulates cofilin phosphatase slingshot2 and neutrophil polarization and chemotaxis. Dev Cell 21: 1038–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SJ, Chae HZ, Rhee SG, Exton JH (1991) Activation of the beta 1 isozyme of phospholipase C by alpha subunits of the Gq class of G proteins. Nature 350: 516–518 [DOI] [PubMed] [Google Scholar]
- Voigt P, Dorner MB, Schaefer M (2006) Characterization of P87Pikap, a novel regulatory subunit of phosphoinositide 3-kinase gamma that is highly expressed in heart and interacts with PDE3B. J Biol Chem 281: 9977–9986 [DOI] [PubMed] [Google Scholar]
- Watanabe-Okochi N, Oki T, Komeno Y, Kato N, Yuji K, Ono R, Harada Y, Harada H, Hayashi Y, Nakajima H, Nosaka T, Kitaura J, Kitamura T (2009) Possible involvement of RasGRP4 in leukemogenesis. Int J Hematol 89: 470–481 [DOI] [PubMed] [Google Scholar]
- Welch HC, Condliffe AM, Milne LJ, Ferguson GJ, Hill K, Webb LM, Okkenhaug K, Coadwell WJ, Andrews SR, Thelen M, Jones GE, Hawkins PT, Stephens LR (2005) P-Rex1 regulates neutrophil function. Curr Biol 15: 1867–1873 [DOI] [PubMed] [Google Scholar]
- Yang Y, Li L, Wong GW, Krilis SA, Madhusudhan MS, Sali A, Stevens RL (2002) RasGRP4, a new mast cell-restricted Ras guanine nucleotide-releasing protein with calcium- and diacylglycerol-binding motifs. Identification of defective variants of this signaling protein in asthma, mastocytosis, and mast cell leukemia patients and demonstration of the importance of RasGRP4 in mast cell development and function. J Biol Chem 277: 25756–25774 [DOI] [PubMed] [Google Scholar]
- Zheng L, Eckerdal J, Dimitrijevic I, Andersson T (1997) Chemotactic peptide-induced activation of Ras in human neutrophils is associated with inhibition of p120-GAP activity. J Biol Chem 272: 23448–23454 [DOI] [PubMed] [Google Scholar]
- Zheng Y, Liu H, Coughlin J, Zheng J, Li L, Stone JC (2005) Phosphorylation of RasGRP3 on threonine 133 provides a mechanistic link between PKC and Ras signaling systems in B cells. Blood 105: 3648–3654 [DOI] [PubMed] [Google Scholar]
- Zhu M, Fuller DM, Zhang W (2012) The role of Ras Guanine nucleotide releasing protein 4 in FcεRI-mediated signalling, mast cell function and T cell development. J. Biol Chem 287: 8135–8143 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.