

Abstract
Mannose-binding lectin (MBL) is an evolutionarily conserved protein that functions in human innate immunity by binding to microbial surfaces and promoting opsonophagocytosis. MBL has been shown to bind to Cryptosporidium sporozoites, and earlier work has suggested that the protective role of MBL may be most important in childhood. We evaluated the association between polymorphisms in the MBL gene (MBL2), serum MBL deficiency, and infection with Cryptosporidium, Entamoeba histolytica, and Giardia intestinalis in children. A large, prospective cohort of Bangladeshi preschool children was followed up for >3 years. Clinical outcomes, serum MBL levels, and MBL2 polymorphisms and haplotypes were determined. Statistically significant associations with E. histolytica and G. intestinalis were not found. Serum MBL deficiency, polymorphisms in the −221 promoter region, and the YO/XA MBL2 haplotype were strongly associated with Cryptosporidium infections, particularly recurrent infection. Children with multiple infections with Cryptosporidium were more likely to be MBL deficient (odds ratio [OR], 10.45), carry the −221 promoter variant (OR, 4.02), and have the YO/XA haplotype (OR, 4.91). We have identified a potentially important component of the human innate immune response to Cryptosporidum infection. Further work is needed to evaluate the mechanism of protection of MBL in Cryptosporidium infection.
The purpose of this work was to determine whether genetic polymorphisms in the human mannose-binding lectin (MBL) gene (MBL2) and serum levels of MBL are associated with susceptibility to childhood infections caused by the enteric protozoa responsible for a large burden of childhood diarrhea: Cryptosporidium species, Entamoeba histolytica, and Giardia intestinalis.
MBL is an evolutionarily conserved component of innate immunity [1, 2]. As a soluble protein of the collectin family, MBL contains a calcium-dependent (C-type) lectin domain and a collagenous tail. The human collectins include MBL and surfactant proteins A and D, and they serve as a group of pattern-recognition molecules that bind to sugar ligands found on microbial surfaces via their carbohydrate-recognition domain (CRD) [3]. Using the CRD or lectin domain to bind to microbial surfaces and the collagenous tail to interface with phagocytes, collectins promote opsonophagocytosis of pathogens and activate the lectin arm of the complement system.
Clinically significant defects in serum levels of MBL were first observed in 1968 and were later described for a cohort of 10 children with recurrent infectious diseases; 2 of the 10 children had severe diarrhea [4, 5]. MBL has been shown to adhere to a variety of intracellular and extracellular bacterial, viral, fungal, and protozoan microbes in vitro, including Cryptosporidium parvum [6, 7]. However, the clinical significance of MBL in the prevention and control of infectious diseases remains an area of interest and controversy [2, 8-12]. Low serum MBL levels have been associated with a variety of infectious, rheumatologic, and thrombotic diseases, and recent studies have attempted to standardize the cutoffs for low MBL levels associated with susceptibility to disease [1, 2, 13, 14]. Large clinical studies involving multiple ethnic and geographic groups have demonstrated that adults with either low serum MBL levels or low-producing MBL2 genotypes are at higher risk for respiratory tract infections, sepsis, tuberculosis, and severe bacterial infections, particularly death from pneumococcal infection [7, 14-16].
The genetic contribution to deficient or low serum levels of MBL results from polymorphisms in the MBL2 gene (MBL1 is a pseudogene), which create low MBL-producing MBL2 genotypes in ~5% of the world’s population. Multiple global population-based studies of MBL2 have demonstrated 6 possible genotypes on the basis of mutations in exon 1 (“structural” polymorphisms B, C, and D [together called O], which prevent multimerization of MBL2) and 6 promoter (“regulatory”) variants. Genotypes containing structural polymorphisms in exon 1 and/or 1 promoter variant (the X/Y polymorphism at position −221) in particular have been shown to be associated with statistically significantly decreased serum levels of MBL [17-21].
Additionally, MBL is proposed to serve a more clinically important role in young children than in adults, especially during the vulnerable months after weaning (age 6–18 months) and before the adaptive immune system (particularly immunoglobulins) has matured [1, 22]. Low or deficient serum MBL levels and low–MBL-producing MBL2 genotypes have been associated with increased risk for acute respiratory tract infections, meningococcal disease, malaria, and severe infections requiring hospitalization in children, as well as pulmonary morbidity in preterm infants [22-27].
Enteric protozoan infections causing diarrhea, particularly those with Cryptosporidium and E. histolytica, are more common and often more severe in very young children [28, 29]. Earlier work demonstrated that young Haitian children with diarrhea due to infection with Cryptosporidium species had lower levels of serum MBL than did control children [17]. MBL has been detected in duodenal aspirates of patients with AIDS and cryptosporidiosis, and Cryptosporidium sporozoites are bound by MBL in a concentration-dependent manner [8]. By observing a distinct population of Bangladeshi preschool children, we sought to determine the contribution of serum MBL levels, genetic polymorphisms, and haplotypes of MBL2 to symptomatic infection with Cryptosporidium species, E. histolytica, and Giardia intestinalis.
METHODS
Study population
The study was approved by the institutional review boards of the University of Vermont, the University of Virginia, and the International Centre for Diarrhoeal Disease Research, Bangladesh. The human experimentation guidelines of the participating institutions and the US Department of Health and Human Services were followed in conducting the research. Informed consent was obtained from the parents or guardians prior to enrollment. Briefly, from 1999 through 2002 a total of 289 preschool children (24–60 months old) from an urban slum of Dhaka (Mirpur) were enrolled, and they were followed up every other day for at least 3 years, as described elsewhere [30]. Children were categorized in 3 groups corresponding to their baseline age at enrollment: 24–35 months, 36–47 months, and 48–60 months. Parents and children were visited and interviewed every other day by health care workers to track symptoms of diarrhea. Treatment for Cryptosporidium infection in children has been recently described [31], but it was not available during the period of the study.
Definitions of diarrhea, recurrent infection, and asymptomatic infection in this cohort have been described elsewhere [32]. Briefly, diarrhea was defined as ≥3 loose or watery stools within 24 h. A case of cryptosporidiosis or Cryptosporidium diarrhea (ie, symptomatic Cryptosporidium infection) was defined as a child with diarrhea and a concurrent stool specimen that tested positive for Cryptosporidium oocysts by an antigen-detection test (Cryptosporidium I; Techlab). Similarly, cases of E. histolytica or Giardia infection were defined as described above, except that antigen-specific diagnostic tests (E. histolytica I and Giardia I; Techlab) were used. Asymptomatic infection with 1 of the 3 protozoa was defined as a stool specimen positive by an antigen-detection test for Cryptosporidium, E. histolytica, or Giardia but no accompanying diarrhea or enteric symptoms. When stool specimens were positive for any of these protozoa after ≥2 months of negative stool surveillance results, infection was counted as a distinct episode. Analysis was performed for single or multiple (≥2) episodes of the abovedescribed categories.
Stool specimens were collected every 4 months and also during acute episodes of diarrhea. Serum specimens were collected every 4 months and as soon as possible after an episode of diarrhea. Genomic DNA was extracted from 200 μL of peripheral blood (Qiagen). For genetic analysis, only 1 individual from a nuclear family was included.
Genotyping
Genotype determination of the MBL2 gene polymorphisms was performed by polymerase chain reaction (PCR) and direct sequencing. Analysis of polymorphisms was started with a 14-primer set, but the methods were modified as described below because of the inability to interpret indistinct bands [33]. Four single-nucleotide polymorphisms of interest were determined: 3 located in the structural region of exon 1, and 1 located in the promoter region (Table 1). The 3 structural polymorphisms occur at codon 54 (glycine [GGC] to aspartic acid [GAC] substitution), at codon 57 (lysine [GGA] to glutamic acid [GAA] substitution), and at codon 52 (arginine [CGT] to cysteine [TGT] substitution). The wild-type allele is termed A at all 3 positions, and polymorphisms are referred to as B, C, and D, respectively [4]. The structural polymorphisms B, C, and D are collectively referred to as O. The promoter polymorphism under study is located at position −221 (X/Y variant), where there is a G→C nucleotide substitution [18]. Three primers (F3, 5 ′-CATTTGTTCTCACTGCCACC-3 ′; F4, 5 ′-CATTTGTTCTCACTGCCACG-3 ′; and R2, 5 ′-GGGCTGGCAAGACAACTATTA-3 ′) were used to encompass all 4 sites of interest on the MBL2 gene.
Table 1. Alleles of the MBL2 Variants.
| SNP | Location | Region | Haplotype |
|---|---|---|---|
| rs1800450 | Exon 1, codon 54 | Structural | A/B |
| rs1800451 | Exon 1, codon 57 | Structural | A/C |
| rs5030737 | Exon 1, codon 52 | Structural | A/D |
| rs7906206 | −221 | Promoter | X/Y |
NOTE. The ancestral allele is A, and the 3 structural polymorphisms are B, C, and D (collectively referred to as O). SNP, single-nucleotide polymorphism.
Each PCR mixture contained 1× PCR buffer (Applied Biosystems), 1.5 mmol/L MgCl2, 5% glycerol, 0.7 mmol/L dNTP (Applied Biosystems), 2.5 U of Amplitaq (Applied Biosystems), deoxyribonuclease- and ribonuclease-free H2O, the reverse and forward primers, and 20 ng of DNA. PCR amplification was conducted using a DNA Engine Tetrad PTC-200 thermal cycler (MJ Research). The PCR profile was initiated with a denaturing step at 94 °C for 2 min followed by 30 amplification cycles. The first 10 cycles consisted of a 10-s denaturing step at 94 °C and a 60-s annealing step at 65 °C. This was followed by 20 cycles with a 10-s denaturing step at 94 °C and a 50-s annealing step at 61 °C. A final 30-s extension cycle was run at 72 °C. An ExoSAP (USB Corporation) procedure was performed on all PCR products to prevent religation of linearized plasmids and to remove unincorporated nucleotides, in preparation for DNA sequencing.
The PCR product was run on a 2% agarose gel, and bands were cut. A DNeasy Tissue kit (Qiagen) was used for DNA extraction, and sequencing was conducted by the University of Vermont core DNA facility by use of a BigDye Terminator version 3.1 (Applied Biosystems) and appropriate primers. Sequencing reaction samples were analyzed using a PRISM 3130XL genetic analyzer (Applied Biosciences).
Genotype (haplotype) construction was determined in light of published literature demonstrating 6 possible genotypes on the basis of the −221 mutation (Y vs X) and the structural mutations in exon 1 (A vs O). The 6 possible genotypes are YA/YA, YA/YO, YO/YO, YA/XA, YO/XA, and XA/XA. The X allele has been found only with the A allele, and therefore the XO/XO genotype has never been recognized [12].
Serum MBL analysis
Serum MBL levels were determined by enzyme-linked immunosorbent assay (ELISA) using the Human MBL (Lectin Assay) ELISA test kit (HyCult Biotechnology). This solid-phase ELISA uses mannan-coated plates to capture functional MBL. Samples were initially diluted 1:100 and processed in duplicate and were used only if absorbance values varied by <15% and if the concentration fell within the assay limits. MBL levels were calculated by means of a standard curve that was based on samples of known MBL concentration [3]. MBL deficiency was defined as any serum MBL level ≤500 ng/mL, as described elsewhere [14].
Statistical analysis
To evaluate the association between serum MBL levels and infections with Cryptosporidium, E. histolytica, and Giardia intestinalis, logistic regression analysis was performed using categories of serum MBL levels (<500 ng/mL and ≥500 ng/mL). Genetic associations were evaluated using logistic regression that adjusted for age, sex, and region within Mirpur. To examine trends of disease severity (single vs multiple infections), polychotomous logistic regression was performed. Individuals who never had a diagnosis of Cryptosporidium infection were compared with children for whom symptomatic infections were diagnosed once or multiple (≥2) times. An analysis of variance (ANOVA) was used to determine the effect of specific genotypes on serum levels of MBL.
Logistic regression analyses were performed using Minitab statistical software (version 15.0; Minitab) and Stata (version 10.0; StataCorp). Prism software (version 5; GraphPad Software) was used to perform ANOVA and all graphing.
RESULTS
Clinical and epidemiologic data
A total of 289 children (147 boys and 142 girls) aged 24–60 months were enrolled. Over the 3-year period, 226 children (78.2%) remained in the study. Complete MBL2 genotyping data were available for 215 children (95.1%), 105 boys (48.8%) and 110 girls (51.1%). The majority of children were in the 48–60-month age group (66%), but within each age group there were no differences in sex or distribution of disease outcomes. Throughout the study period, 56 (26%) and 57 (26.5%) of the enrolled children for whom genotyping data were available had symptomatic Cryptosporidium and E. histolytica infections, respectively. Eighty-three (39%) of the children were affected with symptomatic episodes of G. intestinalis infection. Asymptomatic episodes of E. histolytica and Giardia infections were common, being found in 187 (86.9%) and 57 (76.7%) of the enrolled children, respectively. Asymptomatic episodes of Cryptosporidium infection were less frequent, being found in 23.7% of the children.
Serum MBL level and disease outcomes
Symptomatic infection with Cryptosporidium was associated with a deficiency in serum MBL level (<500 ng/mL). Children with 1 episode of symptomatic Cryptosporidium infection (ie, diarrheal illness or cryptosporidiosis) were 4 times more likely to have a serum MBL level <500 ng/mL (odds ratio [OR], 4.11 [95% confidence interval {CI}, 1.74–9.72]; P = .001) than were children without symptomatic infections. This association nearly doubled (OR, 7.46 [95% CI, 2.43–22.9]; P < .001) among children with 2 or more symptomatic infections. A weaker association was seen for symptomatic E. histolytica infection, which initially suggested that MBL deficiency may be associated with different pathogenic diarrheal diseases (Table 2). However, there was no association between MBL deficiency and E. histolytica infection when we excluded children with E. Histolytica infection who had never had Cryptosporidium infection. On the contrary, the association between MBL deficiency and Cryptosporidium infection increased when we limited our analysis to children without a history of symptomatic E. histolytica infection (Table 2). No association between serum MBL levels and any infection with Giardia was found for (data not shown).
Table 2. Association between Serum Mannose-Binding Lectin (MBL) Deficiency and Single and Multiple Symptomatic Infections with Cryptosporidium.
| Infection history | Serum MBL level <500 ng/mL |
|
|---|---|---|
| OR (95% CI) | P | |
| Symptomatic Cryptosporidium infections | ||
| 0 (n = 83) | 1.00 (reference) | … |
| 1 (n = 38) | 4.11 (1.74–9.72) | .001 |
| ≥2 (n = 14) | 7.46 (2.43–22.9) | <.001 |
| Symptomatic E. histolytica infections | ||
| 0 (n = 92) | 1.00 (reference) | … |
| 1 (n = 29) | 1.34 (0.50–3.63) | .55 |
| ≥2 (n = 14) | 3.04 (1.1–8.34) | .03 |
| Symptomatic Cryptosporidium infections, no E. histolytica infection | ||
| 0 (n = 63) | 1.00 (reference) | … |
| 1 (n = 23) | 3.48 (1.14–10.59) | .028 |
| ≥2 (n = 6) | 10.45 (2.29–47.68) | .002 |
| Symptomatic E. histolytica infections, no Cryptosporidum infection | ||
| 0 (n = 63) | 1.00 (reference) | … |
| 1 (n = 16) | 1.65 (0.421–6.46) | .472 |
| ≥2 (n = 4) | 1.04 (0.122–8.94) | .968 |
NOTE. CI, confidence interval; OR, odds ratio.
Association between MBL2 single-nucleotide polymorphisms and disease outcomes
Polymorphisms in the exon 1 region (A/O variants) and in the promoter variant at position −221 (X/Y) of MBL2 (Table 1) were compared with outcomes of infection with Cryptosporidium and E. histolytica. Statistically significant associations were found between serum MBL levels and polymorphism at the −221 promoter site (X/Y), confirming the associations found between serum MBL levels and disease described above. As shown in Table 3, this promoter variant was associated with children who experienced multiple symptomatic infections with Cryptosporidium or multiple symptomatic infections with E. histolytica.
Table 3. Association between Cryptosporidium and E. histolytica Infections in Young Children and MBL2 −221 Promoter (X/Y) and Structural (A/O) Polymorphisms.
| Variant, infection history | Simple logistic regression |
Multiple logistic regression |
||
|---|---|---|---|---|
| OR (95% CI) | P | OR (95% CI) | P | |
| Promoter variant (XX and X/Y vs Y/Y) | ||||
| Asymptomatic Cryptosporidium infection (n = 51) | 0.62 (0.33–1.19) | .15 | 0.64 (0.33–1.24) | .19 |
| 1 symptomatic Cryptosporidium infection (n = 40) | 0.94 (0.47–1.89) | .86 | 0.89 (0.44–1.82) | .76 |
| ≥2 symptomatic Cryptosporidium infections (n = 16) | 3.81 (1.17–12.34) | .025 | 4.02 (1.19–13.5) | .025 |
| Asymptomatic E. histolytica infection (n = 187) | 2.36 (0.99–5.65) | .05 | 2.36 (0.97–5.73) | .05 |
| 1 symptomatic E. histolytica infection (n = 35) | 0.97 (0.46–2.03) | .93 | 0.93 (0.44–1.97) | .85 |
| ≥2 symptomatic E. histolytica infections (n = 22) | 2.76 (1.06–7.15) | .036 | 2.70 (1.01–7.19) | .046 |
| Structural variant (A/O and O/O vs A/A) | ||||
| Asymptomatic Cryptosporidium infection (n = 51) | 1.27 (0.68–2.40) | .45 | 1.27 (0.67–2.40) | .46 |
| 1 symptomatic Cryptosporidium infection (n = 40) | 3.18 (1.49–6.81) | .003 | 3.15 (1.47–6.77) | .003 |
| ≥2 symptomatic Cryptosporidium infections (n = 16) | 0.94 (0.33–2.64) | .91 | 0.877 (0.30–2.51) | .81 |
| Asymptomatic E. histolytica infection (n = 187) | 0.86 (0.39–1.90) | .70 | 0.85 (0.37–1.91) | .69 |
| 1 symptomatic E. histolytica infection (n = 35) | 0.95 (0.39–2.31) | .91 | 0.78 (0.37–1.64) | .52 |
| ≥2 symptomatic E. histolytica infections (n = 22) | 0.85 (0.47–1.56) | .61 | 0.82 (0.44–1.51) | .52 |
NOTE. CI, confidence interval; OR, odds ratio.
An additional structural polymorphism was associated with children having a single symptomatic Cryptosporidium infection (OR, 3.18 [95% CI, 1.49–6.81]) but not with multiple infections. Asymptomatic Cryptosporidium infections were not found to be associated with either structural or promoter variants.
No association was found between structural variants and infections with E. histolytica. These associations remained after controlling for the potential confounders of age, sex, and region within Mirpur. There were no associations between either promoter or structural polymorphism and any (symptomatic, asymptomatic, single, or multiple) infection with Giardia intestinalis (data not shown).
Haplotype of MBL2 and disease
As shown in Table 4, the YO/XA haplotype was strongly associated with symptomatic Cryptosporidium infections. Associations were not found between disease and any other haplotype (YA/YA or XA/XO; data not shown). Analyses of serum MBL levels and haplotype are shown in Figure 1. The YO/XA haplotype produced statistically significantly lower serum MBL levels (median, 223.5 ng/mL; interquartile range [IQR], 71.5–2078 ng/mL; P < .001) than did the YA/YA haplotype representing no polymorphisms (median, 2162 ng/mL; IQR, 1795–3639 ng/mL).
Table 4.
Haplotype of MBL2 and Disease
| Pathogen, haplotype, infection history | Simple logistic regression |
Multiple logistic regression |
||
|---|---|---|---|---|
| OR (95% CI) | P | OR (95% CI) | P | |
| Cryptosporidium | ||||
| YO/XA | ||||
| 0 symptomatic infections (n = 159) | 1.00 (reference) | … | 1.00 (reference) | … |
| 1 symptomatic infections (n = 40) | 3.15 (1.37–7.23) | .007 | 3.14 (1.33–7.41) | .009 |
| ≥2 symptomatic infections (n = 16) | 4.42 (1.44–13.5) | .009 | 4.91 (1.49–16.1) | .009 |
| YO/YA | ||||
| 0 symptomatic infections (n = 159) | 1.00 (reference) | … | 1.00 (reference) | … |
| 1 symptomatic infections (n = 40) | 1.54 (0.75–3.16) | .24 | 1.52 (0.72–3.22) | .26 |
| ≥2 symptomatic infections (n = 16) | 0.15 (0.02–1.20) | .07 | 0.15 (0.02–1.19) | .07 |
| E. Histolytica | ||||
| YO/XA | ||||
| 0 symptomatic infections (n = 158) | 1.00 (reference) | … | 1.00 (reference) | … |
| 1 symptomatic infections (n = 35) | 1.05 (0.40–2.78) | .92 | 1.00 (0.37–2.68) | .99 |
| ≥2 symptomatic infections (n = 22) | 1.49 (0.50–4.41) | .47 | 1.40 (0.46–3.27) | .55 |
| YO/YA | ||||
| 0 symptomatic infections (n = 158) | 1.00 (reference) | … | 1.00 (reference) | … |
| 1 symptomatic infections (n = 35) | 0.89 (0.40–1.99) | .78 | 0.81 (0.40–2.04) | .81 |
| ≥2 symptomatic infections (n = 22) | 0.83 (0.31–2.26) | .72 | 0.82 (0.29–2.26) | .70 |
NOTE. The MBL2 haplotype YO/XA demonstrates statistically significant associations with single and multiple Cryptosporidium infections in young children. Other genotypes (YA/YA, YO/YO, YA/XA, and XA/XA) had no statistically significant association with infection. CI, confidence interval; OR, odds ratio.
Figure 1.
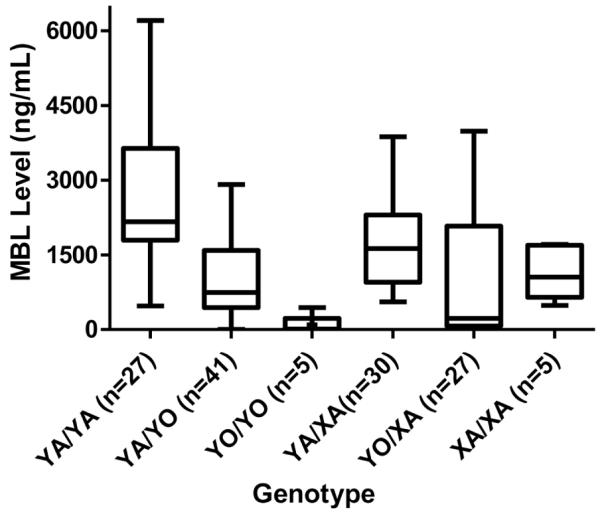
Serum mannose-binding lectin (MBL) levels in genotype groups. In a population of young children, MBL2 genotypes with either structural mutations or X/Y promoter mutations produced statistically significantly lower serum MBL levels than did the wild-type MBL2 genotype YA/YA (no polymorphisms). The wild-type genotype YA/YA produced a median serum MBL level of 2162 ng/mL (interquartile range [IQR], 1795-3639 ng/mL), versus a median serum MBL level of 223.5 ng/mL (IQR, 71.5-2078 ng/mL) for the YO/XA genotype (P < .001). Significant associations were also present between wild-type YA/YA and YO/YO or YA/YO (P < .001) and between YA/YA and XA/XA or YA/XA (P = .05).
DISCUSSION
Our study of a large, closely monitored cohort of preschool children from Dhaka, Bangladesh, demonstrates that polymorphisms in the MBL2 gene (and corresponding haplotypes) and deficient serum levels of MBL are associated with increased susceptibility to infection with the intestinal protozoa Cryptosporidium. These observations were statistically significant for any symptomatic infection with Cryptosporidium, but they were most striking for children with recurrent symptomatic infections. Although early associations were also seen between deficient serum MBL levels, MBL2 gene polymorphisms, and E. histolytica infections, these associations appeared only in children who also had been infected with Cryptosporidium during the study period and disappeared when these children were removed from the analysis. MBL level was not found to be associated with infections with Giardia. Additionally, to our knowledge this work is the first to describe the importance of the MBL2 polymorphisms and the −221 promoter variant in infections with an enteric pathogen.
To demonstrate the association between MBL and infections with clinically important enteric protozoa, our study asked whether serum MBL deficiency was associated with clinical outcomes of infections and disease, whether genetic polymorphisms in MBL2 were associated with the same infection and disease outcomes, and, to close the loop, whether any statistically significant polymorphisms and haplotypes found were themselves associated with serum MBL deficiency. All 3 observations proved statistically significant. Our data confirm and expand on the prior observation that decreased serum MBL levels are associated with single episodes of cryptosporidiosis [17]. In our prospectively followed-up cohort, we demonstrate that MBL deficiency of <500 ng/mL is associated with single and multiple symptomatic episodes of Cryptosporidium infection, with an OR of 7.6 for children with multiple symptomatic infections with Cryptosporidium.
Furthermore, the genetic analysis of structural and promoter polymorphisms of MBL2 demonstrated a strong association between MBL2 X/Y promoter variants and Cryptosporidium infections. Although there are several known promoter variants, only the polymorphism at the −221 location was studied because the literature demonstrated that other promoter polymorphisms play minor roles or no role in disease or serum levels of MBL [20, 21]. Our finding that polymorphisms at the X/Y promoter site were associated with children experiencing multiple episodes of symptomatic Cryptosporidium infection (OR, 4.02) add to the above-noted associations between serum MBL level and infection.
Haplotypes were constructed on the basis of the structural and X/Y promoter polymorphisms to determine whether a predominant MBL2 haplotype was contributing to the associations found between infection/disease and the X/Y promoter variant. We demonstrate that the YO/XA haplotype alone was statistically significantly associated with single and recurrent Cryptosporidium infections. No associations were found between haplotype and E. histolytica infection. Overall, our data demonstrate that polymorphisms in the MBL2 promoter, particularly as part of the YO/XA haplotype, are strongly associated with both serum MBL deficiency and infections with Cryptosporidium. These associations were most significant for children with recurrent symptomatic infections with Cryptosporidium (cryptosporidiosis).
Interestingly, single episodes of symptomatic cryptosporidiosis were also associated with a structural polymorphism (exon 1). Apart from this, there were no other associations in our study population between structural mutations and infection. Because MBL2 polymorphisms are likely inherited together, the current study cannot further delineate which polymorphism (the −221 promoter or the structural mutation) is directly linked to the clinical findings. Of note, the allelic frequencies found in our study population were similar to those found in other studies of South Asian populations [34, 35]. In several other infections, MBL2 structural mutations (particularly the O/O genotype) are strongly linked to decreased serum levels [2, 14]. Although the YO/XA genotype found in our work is clearly associated with lower serum MBL levels, the immunologic role that MBL plays in protection from recurrent disease due to Cryptosporidium and E. histolytica may not be completely reflected by serum level. Previous work on hepatitis B virus has similarly demonstrated that individuals carrying the −221 promoter polymorphism and XA genotype do not recover from infection [36]. The mechanism by which promoter variants cause modification in serum MBL levels is also unclear. Because the sequences of MBL promoter variants associated with lower serum levels (particularly −221) are located close to transcriptional regulatory elements (including glucocorticoid regulatory elements), these regulatory elements may modify expression of MBL independently of structural variants [37].
The mechanism by which MBL controls Cryptosporidium infection and protects children from it is an avenue for future investigation. Although MBL is proposed to act within the gut lumen during diarrheal illnesses, it has been shown to bind to Cryptosporidium sporozoites but not to oocysts [8]. MBL has been detected in fluid from the small intestines in persons with AIDS and Cryptosporidium infections [8]. After binding to sugar moieties on the infecting organisms, MBL serves as a direct opsonin to promote phagocytosis and also can trigger the lectin arm of the complement system via MBL-associated serine proteases, leading to opsonization and cell lysis [38]. However, in the absence of systemic or extracellular disease (as in Cryptosporidium infections), the interaction between MBL and cells infected with Cryptosporidium and its mechanism of action deserve further evaluation.
MBL also appears to have immunomodulatory properties and a role in leukocyte adhesion in some infections [1, 39]. In studies of Neisseria meningitidis, for example, MBL has been shown to profoundly decrease the production of tumor necrosis factor α, interleukin 6, and interleukin 1β, perhaps through mechanisms involving Toll-like receptors [39]. Since gut inflammation predisposes children to morbidity and repeated infection with enteric protozoa, further work should evaluate the effect that MBL has on disease and on intestinal inflammation caused by Cryptosporidium [40, 41].
Our work has several limitations. First, despite the excellent clinical follow-up of children in this trial, serum analysis for MBL levels was not available for all children and was not uniformly performed at the time when children were experiencing acute symptomatic infections. Thus, we were unable to evaluate whether serum MBL levels statistically significantly changed during acute disease. Additionally, although it was unlikely in our homogenous population, there may also have been a potential for population stratification in our analysis. Finally, the trial was not designed to investigate very young children (<24 months of age), who are the most susceptible to cryptosporidiosis and also represent the window of 6-18 months of age (after weaning) in which MBL may be the most clinically significant. We anticipate that our findings would be more striking in this younger population.
Future work is needed to evaluate MBL and its contribution to infection and clinically evident disease with enteric protozoa. First, a large cohort of children that includes infants should be studied, and serum MBL levels should be evaluated before, during, and after symptomatic infection with Cryptosporidium and/or E. histolytica. The effect that nutritional status has on serum MBL levels should also be determined. Evaluation of a large cohort, ideally drawn from 2 or more populations, would permit genetic confirmation of this work and evaluate the role of the MBL-associated serine proteases and similar collectins (eg, surfactant protein D). Finally, the mechanisms by which MBL and its genetic polymorphisms might modify pathogen binding, disease, or inflammation in Cryptosporidium infection deserve further study.
In conclusion, we have identified MBL as a potentially important component of the innate immune system that may protect, either independently or as part of an orchestrated response, small children from diarrhea due to recurrent infections with an intestinal protozoan, particularly Cryptosporidium. Because MBL appears to play a clinically large role at mucosal surfaces in the respiratory and intestinal tracts, future work confirming the effect that MBL has on other enteric pathogens (ie, bacterial and viral causes of diarrhea) is warranted.
Acknowledgments
Financial support:National Institutes of Health (grants U54 AI57168 and K08 AIO54563-05 to B.D.K. and grant AI0143596 to R.H. and W.A.P.).
Footnotes
Potential conflicts of interest: none reported.
References
- 1.Turner MW. The role of mannose-binding lectin in health and disease. Mol Immunol. 2003;40:423–9. doi: 10.1016/s0161-5890(03)00155-x. [DOI] [PubMed] [Google Scholar]
- 2.Eisen DP, Minchinton RM. Impact of mannose-binding lectin on susceptibility to infectious diseases. Clin Infect Dis. 2003;37:1496–505. doi: 10.1086/379324. [DOI] [PubMed] [Google Scholar]
- 3.Cambi A, Koopman M, Figdor CG. How C-type lectins detect pathogens. Cell Microbiol. 2005;7:481–8. doi: 10.1111/j.1462-5822.2005.00506.x. [DOI] [PubMed] [Google Scholar]
- 4.Miller ME. A familial, plasma-associated defect of phagocytosis: a new cause of recurrent bacterial infections. Lancet. 1968;292:60–3. [Google Scholar]
- 5.Super M, Thiel S, Lu J, Levinsky RJ, Turner MW. Association of low levels of mannan-binding protein with a common defect of opsonisation. Lancet. 1989;2:1236–9. doi: 10.1016/s0140-6736(89)91849-7. [DOI] [PubMed] [Google Scholar]
- 6.Neth O, Jack DL, Dodds AW, Holzel H, Klein NJ, Turner MW. Mannose-binding lectin binds to a range of clinically relevant microorganisms and promotes complement deposition. Infect Immun. 2000;68:688–93. doi: 10.1128/iai.68.2.688-693.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Capparelli R, Iannaccone M, Palumbo D, et al. Role played by human mannose-binding lectin polymorphisms in pulmonary tuberculosis. J Infect Dis. 2009;199:666–72. doi: 10.1086/596658. [DOI] [PubMed] [Google Scholar]
- 8.Kelly P, Jack DL, Naeem A, et al. Mannose-binding lectin is a component of innate mucosal defense against Cryptosporidium parvum in AIDS. Gastroenterology. 2000;119:1236–42. doi: 10.1053/gast.2000.19573. [DOI] [PubMed] [Google Scholar]
- 9.Ezekowitz RA. Role of the mannose-binding lectin in innate immunity. J Infect Dis. 2003;187:S335–9. doi: 10.1086/374746. [DOI] [PubMed] [Google Scholar]
- 10.Super M, Ezekowitz RA. The role of mannose-binding proteins in host defense. Infect Agents Dis. 1992;1:194–9. [PubMed] [Google Scholar]
- 11.Turner MW. Mannose-binding lectin: the pluripotent molecule of the innate immune system. Immunol Today. 1996;17:532–40. doi: 10.1016/0167-5699(96)10062-1. [DOI] [PubMed] [Google Scholar]
- 12.Garred P, Larsen F, Madsen HO, Koch C. Mannose-binding lectin deficiency—revisited. Mol Immunol. 2003;40:73–84. doi: 10.1016/s0161-5890(03)00104-4. [DOI] [PubMed] [Google Scholar]
- 13.Ohlenschlaeger T, Garred P, Madsen HO, Jacobsen S. Mannose-binding lectin variant alleles and the risk of arterial thrombosis in systemic lupus erythematosus. N Engl J Med. 2004;351:260–7. doi: 10.1056/NEJMoa033122. [DOI] [PubMed] [Google Scholar]
- 14.Eisen DP, Dean MM, Boermeester MA, et al. Low serum mannosebinding lectin level increases the risk of death due to pneumococcal infection. Clin Infect Dis. 2008;47:510–6. doi: 10.1086/590006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fidler KJ, Wilson P, Davies JC, Turner MW, Peters MJ, Klein NJ. Increased incidence and severity of the systemic inflammatory response syndrome in patients deficient in mannose-binding lectin. Intensive Care Med. 2004;30:1438–45. doi: 10.1007/s00134-004-2303-8. [DOI] [PubMed] [Google Scholar]
- 16.Rantala A, Lajunen T, Juvonen R, et al. Mannose-binding lectin concentrations, MBL2 polymorphisms, and susceptibility to respiratory tract infections in young men. J Infect Dis. 2008;198:1247–53. doi: 10.1086/591912. [DOI] [PubMed] [Google Scholar]
- 17.Kirkpatrick BD, Huston CD, Wagner D, et al. Serum mannose-binding lectin deficiency is associated with cryptosporidiosis in young Haitian children. Clin Infect Dis. 2006;43:289–94. doi: 10.1086/505396. [DOI] [PubMed] [Google Scholar]
- 18.Casanova JL, Abel L. Human mannose-binding lectin in immunity: friend, foe, or both? J Exp Med. 2004;199:1295–9. doi: 10.1084/jem.20040537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garred P, Larsen F, Seyfarth J, Fujita R, Madsen HO. Mannose-binding lectin and its genetic variants. Genes Immun. 2006;7:85–94. doi: 10.1038/sj.gene.6364283. [DOI] [PubMed] [Google Scholar]
- 20.Madsen HO, Satz ML, Hogh B, Svejgaard A, Garred P. Different molecular events result in low protein levels of mannan-binding lectin in populations from southeast Africa and South America. J Immunol. 1998;161:3169–75. [PubMed] [Google Scholar]
- 21.Juliger S, Luckner D, Mordmuller B, et al. Promoter variants of the human mannose-binding lectin gene show different binding. Biochem Biophys Res Commun. 2000;275:617–22. doi: 10.1006/bbrc.2000.3343. [DOI] [PubMed] [Google Scholar]
- 22.Summerfield JA, Sumiya M, Levin M, Turner MW. Association of mutations in mannose binding protein gene with childhood infection in consecutive hospital series. BMJ. 1997;314:1229–32. doi: 10.1136/bmj.314.7089.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hilgendorff A, Heidinger K, Pfeiffer A, et al. Association of polymorphisms in the mannose-binding lectin gene and pulmonary morbidity in preterm infants. Genes Immun. 2007;8:671–7. doi: 10.1038/sj.gene.6364432. [DOI] [PubMed] [Google Scholar]
- 24.Koch A, Melbye M, Sorensen P, et al. Acute respiratory tract infections and mannose-binding lectin insufficiency during early childhood. JAMA. 2001;285:1316–21. doi: 10.1001/jama.285.10.1316. [DOI] [PubMed] [Google Scholar]
- 25.Thorarinsdottir HK, Ludviksson BR, Vikingsdottir T, et al. Childhood levels of immunoglobulins and mannan-binding lectin in relation to infections and allergy. Scand J Immunol. 2005;61:466–74. doi: 10.1111/j.1365-3083.2005.01588.x. [DOI] [PubMed] [Google Scholar]
- 26.Hibberd ML, Sumiya M, Summerfield JA, Booy R, Levin M, Meningococcal Research Group Association of variants of the gene for mannose-binding lectin with susceptibility to meningococcal disease. Lancet. 1999;353:1049–53. doi: 10.1016/s0140-6736(98)08350-0. [DOI] [PubMed] [Google Scholar]
- 27.Luty AJ, Kun JF, Kremsner PG. Mannose-binding lectin plasma levels and gene polymorphisms in Plasmodium falciparum malaria. J Infect Dis. 1998;178:1221–4. doi: 10.1086/515690. [DOI] [PubMed] [Google Scholar]
- 28.Lima AA, Moore SR, Barboza MS, Jr, et al. Persistent diarrhea signals a critical period of increased diarrhea burdens and nutritional shortfalls: a prospective cohort study among children in northeastern Brazil. J Infect Dis. 2000;181:1643–51. doi: 10.1086/315423. [DOI] [PubMed] [Google Scholar]
- 29.Newman RD, Sears CL, Moore SR, et al. Longitudinal study of Cryptosporidium infection in children in northeastern Brazil. J Infect Dis. 1999;180:167–75. doi: 10.1086/314820. [DOI] [PubMed] [Google Scholar]
- 30.Haque R, Duggal P, Ali IM, et al. Innate and acquired resistance to amebiasis in Bangladeshi children. J Infect Dis. 2002;186:547–52. doi: 10.1086/341566. [DOI] [PubMed] [Google Scholar]
- 31.Rossignol JF, Ayoub A, Ayers MS. Treatment of diarrhea caused by Cryptosporidium parvum: a prospective randomized, double-blind, placebo-controlled study of nitazoxanide. J Infect Dis. 2001;184:103–6. doi: 10.1086/321008. [DOI] [PubMed] [Google Scholar]
- 32.Haque R, Mondal D, Kirkpatrick BD, et al. Epidemiologic and clinical characteristics of acute diarrhea with emphasis on Entamoeba histolytica infections in preschool children in an urban slum of Dhaka, Bangladesh. Am J Trop Med Hyg. 2003;69:398–405. [PubMed] [Google Scholar]
- 33.Steffensen R, Thiel S, Varming K, Jersild C, Jensenius JC. Detection of structural gene mutations and promoter polymorphisms in the mannan-binding lectin (MBL) gene by polymerase chain reaction with sequence-specific primers. J Immunol Methods. 2000;241:33–42. doi: 10.1016/s0022-1759(00)00198-8. [DOI] [PubMed] [Google Scholar]
- 34.Alagarasu K, Selvaraj P, Swaminathan S, Raghavan S, Narendran G, Narayanan PR. Mannose binding lectin gene variants and susceptibility to tuberculosis in HIV-1 infected patients of South India. Tuberculosis (Edinb) 2007;87:535–43. doi: 10.1016/j.tube.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 35.Catano G, Agan BK, Kulkarni H, et al. Independent effects of genetic variations in mannose-binding lectin influence the course of HIV disease: the advantage of heterozygosity for coding mutations. J Infect Dis. 2008;198:72–80. doi: 10.1086/588712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thio CL, Mosbruger T, Astemborski J, et al. Mannose binding lectin genotypes influence recovery from hepatitis B virus infection. J Virol. 2005;79:9192–6. doi: 10.1128/JVI.79.14.9192-9196.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Naito H, Ikeda A, Hasegawa K, et al. Characterization of human serum mannan-binding protein promoter. J Biochem. 1999;126:1004–12. doi: 10.1093/oxfordjournals.jbchem.a022543. [DOI] [PubMed] [Google Scholar]
- 38.Arnold JN, Dwek RA, Rudd PM, Sim RB. Mannan binding lectin and its interaction with immunoglobulins in health and in disease. Immunol Lett. 2006;106:103–10. doi: 10.1016/j.imlet.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 39.Jack DL, Read RC, Tenner AJ, Frosch M, Turner MW, Klein NJ. Mannose-binding lectin regulates the inflammatory response of human professional phagocytes to Neisseria meningitidis serogroup B. J Infect Dis. 2001;184:1152–62. doi: 10.1086/323803. [DOI] [PubMed] [Google Scholar]
- 40.Agnew DG, Lima AA, Newman RD, et al. Cryptosporidiosis in northeastern Brazilian children: association with increased diarrhea morbidity. J Infect Dis. 1998;177:754–60. doi: 10.1086/514247. [DOI] [PubMed] [Google Scholar]
- 41.Kirkpatrick BD, Daniels MM, Jean SS, et al. Cryptosporidiosis stimulates an inflammatory intestinal response in malnourished Haitian children. J Infect Dis. 2002;186:94–101. doi: 10.1086/341296. [DOI] [PubMed] [Google Scholar]