

Abstract
Objective:
To determine the most critical symptoms in a national myotonic dystrophy type 1 (DM1) population and to identify the modifying factors that have the greatest effect on the severity of these symptoms.
Methods:
We performed a cross-sectional study of 278 adult patients with DM1 from the national registry of patients with DM1 between April and August 2010. We assessed the prevalence and relative significance of 221 critical DM1 symptoms and 14 disease themes. These symptoms and themes were chosen for evaluation based on prior interviews with patients with DM1. Responses were categorized by age, CTG repeat length, gender, and duration of symptoms.
Results:
Participants with DM1 provided symptom rating survey responses to address the relative frequency and importance of each DM1 symptom. The symptomatic themes with the highest prevalence in DM1 were problems with hands or arms (93.5%), fatigue (90.8%), myotonia (90.3%), and impaired sleep or daytime sleepiness (87.9%). Participants identified fatigue and limitations in mobility as the symptomatic themes that have the greatest effect on their lives. We found an association between age and the average prevalence of all themes (p < 0.01) and between CTG repeat length and the average effect of all symptomatic themes on participant lives (p < 0.01).
Conclusions:
There are a wide range of symptoms that significantly affect the lives of patients with DM1. These symptoms, some previously underrecognized, have varying levels of importance in the DM1 population and are nonlinearly dependent on patient age and CTG repeat length.
Myotonic dystrophy type 1 (DM1) is a multisystem disorder caused by an unstable trinucleotide repeat expansion on chromosome 19q13.3 in the DMPK gene.1–3 The core features of DM1 are myotonia, weakness, and early-onset cataracts (<50 years of age). Along with these core features, patients commonly report symptoms related to cognition, gastrointestinal function, sleep, fatigue, mood, ability to swallow, vision, social relations, and physical function.4 The effect of each symptom on health-related quality of life is unknown. Furthermore, given the multisystem manifestations of DM1, we hypothesize that there are additional underrecognized symptoms that are important to patients and have a critical effect on their health status.5
There is a need to identify the symptoms that are most important to patients as a means to improve studies of disease burden and therapeutic response.6–9 Interest in patient-relevant manifestations stems not only from patients and clinicians but also from insurers, government agencies, researchers, policymakers, and home care providers.10–12 In addition, the Food and Drug Administration (FDA) has established patient-centered, patient-validated, and patient-reported metrics as part of its criteria for drug approval and labeling.6,13 In preparation for the development of acceptable disease-specific patient-centered DM1 metrics, it is necessary to first determine what is most significant to the DM1 population.
In an effort to define the patient-reported disease burden in DM1, here we use data from patient interviews and a large national cross-sectional study of DM1-affected participants to determine the symptoms and themes most prevalent and important to this population.
METHODS
Study participants.
Inclusion criteria included 1) age of 21 years or older and 2) diagnosis of DM1 by genetic or clinical criteria.14,15 Patients with congenital or juvenile myotonic dystrophy were not included in this study.
Study design.
Phase 1: DM1 qualitative interviews.
We conducted interviews with 20 participants with DM1 who had various levels of disability. Using open-ended questions, we asked participants to identify the symptoms of DM1 that have the greatest effect on their lives. All interviews were audio-recorded, transcribed, coded, and analyzed using a qualitative framework technique, triangulation, and 3-investigator consensus approach.16 Three investigators with extensive clinical experience in DM1 independently and conjunctly analyzed all qualitative data. Recurring similar quotes among the interviewees were used to identify potentially relevant symptoms. Common symptoms were categorized into symptomatic themes (concepts representing a group of like symptoms) of DM1 health. Last, in accordance with the World Health Organization's framework of health, each theme was categorized as a physical, mental, social, or disease-specific component of DM1 health.
Phase 2: National cross-sectional study.
In phase 2, we used a large cross-sectional sample of participants with DM1 from the National Registry of Myotonic Dystrophy and Facioscapulohumeral Muscular Dystrophy Patients and Family Members (a national registry designed to advance research and knowledge of myotonic dystrophy and facioscapulohumeral muscular dystrophy [FSHD] through the enrolment of patients and assistance of researchers; http://www.urmc.rochester.edu/nihregistry/).17
We included each potential symptom of importance identified through phase 1 in a paper survey. When possible, the survey included the exact wording and description of symptoms obtained from participants in phase 1. For each individual symptom the survey inquired, How much does the following impact your life now? Participants were provided a 6-point Likert-type scale to record their responses. Likert scale options consisted of the following: 1 = I don't experience this; 2 = I experience this but it does not affect my life; 3 = It affects my life a little; 4 = It affects my life moderately; 5 = It affects my life very much; and 6 = It affects my life severely. Participants also provided demographic information and were asked to list any symptoms not otherwise included in the survey.
Eligible participants were mailed a recruitment/information letter, one survey, and instructions on how to complete the survey in April 2010. Surveys were collected until August 2010. There were 2 available surveys. Each survey included identical demographic and theme questions, whereas the questions regarding individual symptoms were divided between the 2 surveys to minimize participant burden. We randomly allocated each subject meeting the selection criteria to 1 of the 2 surveys. We provided patients with DM1 who have severe hand weakness, visual problems, or reading difficulty with the option to complete their survey by phone. A flow diagram summarizing the activities and analyses of phases 1 and 2 is provided in figure 1.
Figure 1. Flow diagram of activities and analyses involved in identifying the most critical symptoms and themes in myotonic dystrophy type 1 (DM1).

Standard protocol approvals, registrations, and patient consents.
All aspects of these studies were approved by the University of Rochester Institutional Review Board.
Statistical analysis.
The prevalence of each symptom and theme was calculated in our phase 2 sample. Average life impact scores (a metric with a range from 0 to 4 that measures the relative importance of a symptom to a participant) for each symptom and theme were determined. Numerical values were assigned to each response as follows: 0 = the patient experiences the issue, but it does not impact the patient's life; 1 = the issue impacts the patient's life a little; 2 = the issue impacts the patient's life moderately; 3 = the issue impacts the patient's life very much; and, 4 = the issue impacts the patient's life severely. Average life impact scores for each symptom and theme were generated based on the responses of all participants who stated that they experienced the symptom or theme. The possible range for these scores is 0 to 4 with a higher number representing a symptom that has a greater effect on patients' lives. In addition, a population impact score (frequency that an issue was experienced multiplied by the average life impact score of an issue) was calculated for each item. The possible range for this score is also 0 to 4 with a value of 4 representing a symptom that affects all patients at the highest level. Finally, composite prevalence scores and composite impact scores were computed by averaging participant responses across all themes.
Responses were further categorized based on 1) age, 2) gender, 3) CTG repeat length, and 4) duration of symptoms, each with predetermined group categorization cut points. We obtained descriptive statistics for the prevalence and impact score of each theme for the entire sample and for each subgroup. We used Fisher exact tests to compare the prevalence of each theme across the different subgroups.18 We used Kruskal-Wallis tests to compare the distributions of relative impact scores for each theme, composite prevalence scores, and composite impact scores across the different subgroups.19
RESULTS
Phase 1: DM1 qualitative interviews.
We obtained 1,165 direct DM1 quotes through qualitative interviews of 20 patients with DM1. No invited participant refused to participate in phase 1. Recurring similar quotes were grouped to identify the 221 most important symptoms and 14 most critical themes to this population (table e-1 on the Neurology® Web site at www.neurology.org).
Phase 2: National cross-sectional study.
We sent surveys to 530 eligible patients with DM1. Of these, 278 (52%) replied, addressing the relative importance and frequency of each of the symptoms and themes identified in phase 1; 135 completed survey 1 and 143 completed survey 2. The questions represented by each survey are provided in table e-1. Participants represented 43 different states. Details regarding the demographic information of the respondents are provided in table 1.
Table 1.
Clinical and demographic information of DM1 respondents from the national registry
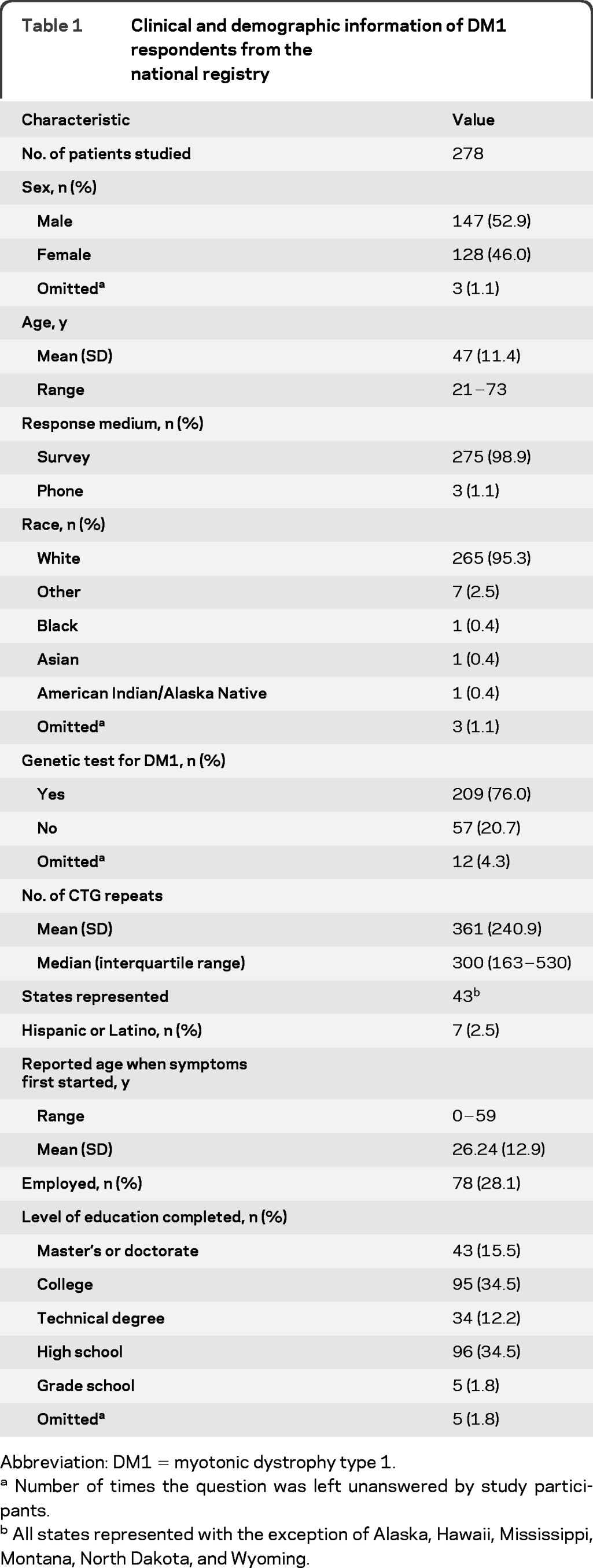
Abbreviation: DM1 = myotonic dystrophy type 1.
Number of times the question was left unanswered by study participants.
All states represented with the exception of Alaska, Hawaii, Mississippi, Montana, North Dakota, and Wyoming.
Prevalence of DM1 themes and symptoms.
The most frequently occurring themes were 1) Problems with hands or arms (93.5%), 2) Fatigue (90.8%), 3) Myotonia (90.3%), and 4) Impaired sleep or daytime sleepiness (87.9%).
Details regarding each theme's prevalence in the total study sample and by age and gender are provided in table 2. Details regarding each theme's prevalence by subgroup are provided in table e-2. A listing of all 221 symptoms (and their relative prevalence) is provided in table e-1.
Table 2.
Prevalence and relative impact of 15 critical themes as identified by patients with myotonic dystrophy type 1a


Subgroup analyses is confined to those who responded to age and gender questions. The possible range of “Relative impact on lives” is 0.0 to 4.0 with higher values representing greater theme impact.
p values are from Fisher's exact tests or Kruskal-Wallis tests, as appropriate, comparing responses among the subgroups; see text for details.
“Problems with physical health” is included as a comparative generic theme of myotonic dystrophy type 1 health.
Among the 221 symptoms evaluated individually, 6 symptoms were reported in more than 90% of the respondents. These symptoms were decreased energy (92%), daytime sleepiness (91.5%), hand myotonia (91.4%), hand weakness (91%), difficulty opening jars or bottles (91%), and tired muscles (90.7%) (table e-1).
Among themes, there were differences in prevalence among various subgroups (table e-2). Duration of symptoms was associated with a higher prevalence of Limitations with mobility or walking (p = 0.01) and an Inability to do activities (p = 0.02). CTG repeat length was associated with a higher prevalence of Problems with hands or arms (p = 0.04) and Emotional issues (p = 0.05). Aging was associated with a higher prevalence of Fatigue (p < 0.01), Limitations with mobility or walking (p < 0.01), Inability to do activities (p < 0.01), Decreased satisfaction in social situations (p < 0.01), Decreased performance in social situations (p = 0.01), Impaired sleep or daytime sleepiness (p = 0.02), Emotional issues (p = 0.04), Problems with hands or arms (p = 0.04), Problems with physical health (p = 0.05), and Problems with vision, hearing, or smell (p = 0.05). The prevalence of themes was similar between sexes with 2 exceptions: a higher prevalence of Gastrointestinal issues in women with DM1 (p = 0.01) and a higher prevalence of Communication difficulties in men with DM1 (p < 0.01).
There was a relationship between increased age and the average composite prevalence across all themes (p < 0.01). The greatest interval increase occurred between the 21- to 30-year-old group (53.8% average composite prevalence) and the 31- to 40-year-old group (76.5%). The most marked increase in prevalence of any specific theme in the subgroup analyses was seen with Limitations with mobility or walking. Between the 21- to 30-year-old age group and the 31- to 40-year-old age group, the prevalence of this manifestation rose from 33.3% to 82.7% (table 2). The associations between the prevalence of the individual themes and age groups are illustrated in figure 2.
Figure 2. Prevalence of myotonic dystrophy type 1 themes by age.

(A) First 8 themes displayed. (B) Themes 9 through 15 displayed. Association between age and the average composite prevalence over all themes: p < 0.01.
Symptoms that have the greatest effect on DM1 participant lives.
The themes with the highest average life impact scores (0–4) were 1) Fatigue (2.49), 2) Limitations with mobility or walking (2.42), 3) Inability to do specific activities (2.35), 4) Problems with hands or arms (2.27), and 5) Impaired sleep or daytime sleepiness (2.25).
Details regarding each theme's average life impact score in the total study sample and by subgroup are provided in table e-2. Details regarding the average impact score of each of the 221 symptoms are provided in table e-1.
Among the 221 individually evaluated symptoms, the following had the highest average life impact scores: difficulty having children (3.37), difficulty staying in a standing position (3.00), inability to do things that you used to do (2.81), and lack of job because of disability (2.80) (table e-1).
Among themes, there were differences in the average impact scores among various subgroups (table e-2). Duration of symptoms was associated with the burden of Problems with physical health (p = 0.04). The average life impact score associated with Myotonia worsened with longer CTG repeat length (p = 0.02). Similarly, age was associated with worsening average life impact scores in the following themes: Decreased performance in social situations (p = 0.02), Communication difficulties (p = 0.03), and Limitations with mobility or walking (p = 0.04). Compared with women with DM1, men with DM1 reported a greater effect on their lives due to Problems with hands or arms (p = 0.01) and Communication difficulties (p = 0.05) (table 2).
The average composite burden from all themes increased with greater CTG repeat length (p < 0.01). The greatest increase in average composite impact score was seen between the groups with 251–500 and 501–750 CTG repeat length with an increase from 1.70 to 2.10. The relationships between the average life impact scores of all individual themes and CTG repeat length are seen in figure 3.
Figure 3. Relative impact of myotonic dystrophy type 1 themes by CTG repeat length.

(A) First 8 themes displayed. (B) Themes 9 through 15 displayed. Association between an increasing number of CTG repeats and the average composite impact over all themes: p < 0.01.
Age was not associated with a consistent increase in composite impact score.
Population impact scores.
The 3 themes with the greatest population impact scores were Fatigue (2.26), Problems with hands or arms (2.12), and Limitations with mobility or walking (2.06) (table e-1).
The symptoms with the greatest population impact scores were difficulty opening jars or bottles (2.42), difficulty staying in a standing position (2.34), inability to do things that you used to do (2.34), limitations physically of what one can do (2.34), decreased leg energy (stamina) (2.23), difficulty walking long distances (2.17), difficulty playing sports (2.14), decreased energy (2.14), trouble with going up step ladders (2.09), difficulty with stairs (2.06), and hand weakness (2.05) (table e-1).
DISCUSSION
DM1 is one of the most variable hereditary human disorders in terms of its range of clinical manifestations, severity of illness, and age at onset.20,21 Despite the diversity and variable severity of its symptoms, our study demonstrates the feasibility of determining which aspects of the disease are most significant to affected individuals. This study fills an important gap in the present literature. It supplies information that is needed worldwide by groups who are planning clinical trials. It directly addresses a key question that is frequently asked by researchers, clinicians, industry, and regulatory agencies: what features of the illness are most important to affected individuals? This work also provides the base data necessary to develop future disease-specific, patient-relevant outcome measures for this population.
Patient Reported Impact of Symptoms in Myotonic Dystrophy Type 1 (PRISM-1) builds on previous studies of disease burden in DM1 that were limited by small sample sizes, restricted geographical range, or marginal patient input.22–24 This study shows that there are specific manifestations and symptoms that have both a common and significant effect on lives of patients with DM1. These patient-identified symptoms represent the key physical, mental, and social features of DM1 disease burden. In addition, our findings indicate the rate at which the prevalence and importance of symptoms change by patient age, duration of symptoms, and number of CTG repeats.
In this study we focused on the patient's point of view instead of prior medical reviews or opinion.24 Researchers have previously shown that noncarrier perceptions of the disability and impairment in DM1 correlate poorly with actual patient views.25 We determined the prevalence and severity of each DM1 symptom and theme using direct patient input. This is in response to the FDA and the patient-oriented research community who state that research of this type should ideally be patient-centric and antipaternalistic.13,26 The emphasis of PRISM-1 on the patient's perspective strongly enhances the content validity of the data; however, we recognize that this approach may deemphasize certain manifestations that are below patients' conscious awareness, such as asymptomatic respiratory or cardiac disease. Ultimately, correlation studies may determine the relative sensitivity of patient-reported data to traditional functional assessments in detecting subtle DM1 disease progression.
This patient-centered research demonstrates that the most prevalent (or well described) manifestations in a population are not always the manifestations that have the greatest effect on patients' lives. The medical literature does not generally report fatigue as the primary symptom of DM1; however, participants reported this symptom as having the greatest overall effect. It is likely that fatigue also has indirect effects on other key issues in DM1, such as employment status and interpersonal relations. It should be noted that “fatigue” may represent many additional issues in DM1 including tired muscles, decreased energy, prolonged recovery after exercise, daytime sleepiness, depression, impaired sexual function, problems with concentration, and decreased motivation. Interestingly, none of these issues in isolation had a population impact score as high as that for Fatigue during this study.
In some cases, there was dissociation between the prevalence of a theme and its relative importance. Whereas Problems with hands or arms had the highest theme prevalence, it did not have the highest impact score. Limitations with mobility or walking only had the sixth highest theme prevalence; however, of the patients who reported this issue, it had the second highest effect on their lives. Similarly, difficulty having children had the highest impact score of any item; however, only a relatively small portion (34.2%) of participants, consisting mainly of women, stated that they experienced this issue. Although this question was initially intended to address fertility issues, it is possible that participants instead viewed it in other terms.
The increase in composite prevalence and impact scores between the 21- to 30-year-old age group and the 31- to 40-year-old age group raises the possibility that the effect of major disease manifestations in DM1 does not progress linearly with age and that there is a period of accelerated disease progression in DM1. If confirmed in a longitudinal study, this information may provide a valuable counseling tool to be used with patients in a clinical setting and identify a critical therapeutic window in DM1.
The average composite impact score over all themes increased with CTG repeat length. Although this finding supports the often debated theory that DM1 disease severity is related to a patient's CTG repeat length, there are probably many additional genetic and environmental factors that contribute to DM1 disease severity.27–29
A notable observation was that the effect of the disease (as measured by the average composite impact score over all themes) did not universally increase with age despite increases in the prevalence of themes. This phenomenon may be due to additional unmeasured factors (e.g., experience, coping, improved social networks, or improved financial status) that help lessen the burden of disease as age increases. Another possible explanation is a survivor effect (a type of sampling bias in which an older group of participants may be healthier than the general population of people with the disease) or other sampling bias. It is possible that sicker participants with DM1 who were older were less likely to participate in the survey (or may even have failed to survive past the age of 60), leaving a healthier subset of older participants with DM1. Another possibility is that this group represents a late-onset and relatively asymptomatic subgroup of patients with DM1 who came to diagnosis through family studies and not necessarily through progression of their own disease.
The effect of DM1 manifestations is not markedly different between women and men. Although women with DM1 did have a higher prevalence of Gastrointestinal issues and men with DM1 more frequently reported Communication difficulties, these differences may be present in the general population and are not necessarily specific to DM1.30
The limitations of this study are similar to those of any cross-sectional study evaluating a rare disease that progresses over time. Although the registry used represents a wide range of ages and geographic locations, this sample does not perfectly represent the DM1 population. In addition, not every eligible patient from the registry participated in this research. To this end, there is the potential for sampling bias that could affect the generalizability of our results. Our study sample probably overrepresents patients with DM1 who seek medical attention, are interested in medical research, are less affected, and have the means, cognitive abilities, and physical function necessary to take part in research. Because participants did not report medication use, it is also possible that our sample underreported symptoms related to treated manifestations of DM1 (such as participants receiving antimyotonia therapy or stimulants for excessive daytime sleepiness).
PRISM-1 demonstrates that through rigorous qualitative and quantitative methods it is possible to obtain a patient-centered description of the most common and relevant aspects of disease burden in DM1, including the discovery of manifestations that were not as well recognized before. Systematic efforts like this may be fruitful in many other diseases. Ultimately, the information gained from this study can be used to 1) better understand the multiple systemic involvement of DM1, 2) demonstrate how DM1 disease varies with time and accordingly to CTG repeat length, and 3) better equip researchers and clinicians with the ability to identify, study, and focus on the manifestations that are most important to those affected by DM1.
Supplementary Material
GLOSSARY
- DM1
myotonic dystrophy type 1
- FDA
Food and Drug Administration
- FSHD
facioscapulohumeral muscular dystrophy
- PRISM-1
Patient Reported Impact of Symptoms in Myotonic Dystrophy Type 1.
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
C. Heatwole: drafting/revising the manuscript for content, study concept or design, analysis or interpretation of data, acquisition of data, statistical analysis, study supervision or coordination, obtaining funding. R. Bode: drafting/revising the manuscript for content, study concept or design, analysis or interpretation of data, statistical analysis. N. Johnson: drafting/revising the manuscript for content, study concept or design, analysis or interpretation of data, acquisition of data, study supervision or coordination. C. Quinn: drafting/revising the manuscript for content, analysis or interpretation of data, acquisition of data. W. Martens: drafting/revising the manuscript for content, analysis or interpretation of data, statistical analysis. M. McDermott: drafting/revising the manuscript for content, analysis or interpretation of data, statistical analysis. N. Rothrock: drafting/revising the manuscript for content, study concept or design, analysis or interpretation of data. C. Thornton: drafting/revising the manuscript for content, study concept or design, analysis or interpretation of data, obtaining funding. B. Vickrey: drafting/revising the manuscript for content, study concept or design, analysis or interpretation of data. D. Victorson: drafting/revising the manuscript for content, study concept or design, analysis or interpretation of data. R. Moxley III: drafting/revising the manuscript for content, study concept or design, analysis or interpretation of data, obtaining funding. Additional editing and manuscript consultation was provided by Mark Heatwole, BS, MBA, and Brett Kinsler, DC.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1. Fu YH, Pizzuti A, Fenwick R, Jr, et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 1992;255:1256–1258 [DOI] [PubMed] [Google Scholar]
- 2. Mahadevan M, Tsilfidis C, Sabourin L, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science 1992;255:1253–1255 [DOI] [PubMed] [Google Scholar]
- 3. Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992;68:799–808 [DOI] [PubMed] [Google Scholar]
- 4. Harper P. Myotonic Dystrophy. Philadelphia: WB Saunders; 2001 [Google Scholar]
- 5. Heatwole CR, Miller J, Martens B, Moxley RT., 3rd. Laboratory abnormalities in ambulatory patients with myotonic dystrophy type 1. Arch Neurol 2006;63:1149–1153 [DOI] [PubMed] [Google Scholar]
- 6. Patrick DL, Burke LB, Powers JH, et al. Patient-reported outcomes to support medical product labeling claims: FDA perspective. Value Health 2007;10(suppl 2):S125–S137 [DOI] [PubMed] [Google Scholar]
- 7. Turner RR, Quittner AL, Parasuraman BM, Kallich JD, Cleeland CS, Mayo/FDA Patient-Reported Outcomes Consensus Meeting Group Patient-reported outcomes: instrument development and selection issues. Value Health 2007;10(suppl 2):S86–S93 [DOI] [PubMed] [Google Scholar]
- 8. Acquadro C, Berzon R, Dubois D, et al. Incorporating the patient's perspective into drug development and communication: an ad hoc task force report of the Patient-Reported Outcomes (PRO) Harmonization Group meeting at the Food and Drug Administration, February 16, 2001. Value Health 2003;6:522–531 [DOI] [PubMed] [Google Scholar]
- 9. Frost MH, Reeve BB, Liepa AM, Stauffer JW, Hays RD, Mayo/FDA Patient-Reported Outcomes Consensus Meeting Group What is sufficient evidence for the reliability and validity of patient-reported outcome measures? Value Health 2007;10(suppl 2):S94–S105 [DOI] [PubMed] [Google Scholar]
- 10. Geigle R, Jones SB. Outcomes measurement: a report from the front. Inquiry 1990;27:7–13 [PubMed] [Google Scholar]
- 11. Mendell JR, Csimma C, McDonald CM, et al. Challenges in drug development for muscle disease: a stakeholders' meeting. Muscle Nerve 2007;35:8–16 [DOI] [PubMed] [Google Scholar]
- 12. Washington AE, Lipstein SH. The Patient-Centered Outcomes Research Institute: promoting better information, decisions, and health. N Engl J Med 2011;365:e31. [DOI] [PubMed] [Google Scholar]
- 13. Guidance for Industry Patient-Reported Outcome Measures: Use in Medical Product Development to Support Labeling Claims. Bethesda, MD: US Department of Health and Human Services Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research, and Center for Devices and Radiological Health; 2009 [Google Scholar]
- 14. New nomenclature and DNA testing guidelines for myotonic dystrophy type 1 (DM1): The International Myotonic Dystrophy Consortium (IDMC). Neurology 2000;54:1218–1221 [DOI] [PubMed] [Google Scholar]
- 15. Griggs RC, Wood DS. Criteria for establishing the validity of genetic recombination in myotonic dystrophy. Neurology 1989;39:420–421 [DOI] [PubMed] [Google Scholar]
- 16. McColl E. Developing questionnaires. In: Fayers P, Hays R. eds. Assessing Quality of Life in Clinical Trials, 2nd ed. Oxford, UK: Oxford University Press; 2005:9–23 [Google Scholar]
- 17. Hilbert J, Kissel J, Luebbe E, et al. If you build a rare disease registry, will they enroll and will they use it? Methods and data from the National Registry of Myotonic Dystrophy (DM) and Facioscapulohumeral Muscular Dystrophy (FSHD). Contemp Clin Trials 2012;33:302–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fisher RA. Statistical Methods for Research Workers, 5th ed. Edinburgh, Scotland: Oliver and Boyd; 1934 [Google Scholar]
- 19. Kruskal WH, Wallis WA. Use of ranks in one-criterion variance analysis. J Am Statist Assoc 1952;47:583–621 [Google Scholar]
- 20. Harper PS. Myotonic Dystrophy: thefacts, 2nd ed. New York: Oxford University Press; 2009 [Google Scholar]
- 21. Harley HG, Rundle SA, Reardon W, et al. Unstable DNA sequence in myotonic dystrophy. Lancet 1992;339:1125–1128 [DOI] [PubMed] [Google Scholar]
- 22. Tieleman AA, Jenks KM, Kalkman JS, Borm G, van Engelen BG. High disease impact of myotonic dystrophy type 2 on physical and mental functioning. J Neurol 2011;258:1820–1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Timman R, Tibben A, Wintzen AR. Myotonic dystrophy: the burden for patients and their partners. J Rehabil Med 2010;42:823–830 [DOI] [PubMed] [Google Scholar]
- 24. Gagnon C, Chouinard MC, Laberge L, et al. Health supervision and anticipatory guidance in adult myotonic dystrophy type 1. Neuromuscul Disord 2010;20:847–851 [DOI] [PubMed] [Google Scholar]
- 25. Laberge L, Prevost C, Perron M, et al. Clinical and genetic knowledge and attitudes of patients with myotonic dystrophy type 1. Public Health Genomics 2010;13:424–30 [DOI] [PubMed] [Google Scholar]
- 26. Washington AE, Lipstein SH. The Patient-Centered Outcomes Research Institute: promoting better information, decisions, and health. N Engl J Med 2011;365:2252–2253 [DOI] [PubMed] [Google Scholar]
- 27. Gharehbaghi-Schnell EB, Finsterer J, Korschineck I, Mamoli B, Binder BR. Genotype-phenotype correlation in myotonic dystrophy. Clin Genet 1998;53:20–26 [DOI] [PubMed] [Google Scholar]
- 28. Gennarelli M, Novelli G, Andreasi Bassi F, et al. Prediction of myotonic dystrophy clinical severity based on the number of intragenic [CTG]n trinucleotide repeats. Am J Med Genet 1996;65:342–347 [DOI] [PubMed] [Google Scholar]
- 29. Laberge L, Veillette S, Mathieu J, Auclair J, Perron M. The correlation of CTG repeat length with material and social deprivation in myotonic dystrophy. Clin Genet 2007;71:59–66 [DOI] [PubMed] [Google Scholar]
- 30. Wilson S, Roberts L, Roalfe A, Bridge P, Singh S. Prevalence of irritable bowel syndrome: a community survey. Br J Gen Pract 2004;54:495–502 [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.