

Abstract
We have examined 46 tetramethylrosamine/rhodamine derivatives with structural diversity in the heteroatom of the xanthylium core, the amino substituents of the 3- and 6-positions, and the alkyl, aryl, or heteroaryl group at the 9-substituent. These compounds were examined for affinity and ATPase stimulation in isolated MDR3 CL P-gp and human P-gp-His10, for their ability to promote uptake of calcein AM and vinblastine in multidrug-resistant MDCKII-MDR1 cells, and for transport in monolayers of MDCKII-MDR1 cells. Thioamide 31-S gave KM of 0.087 μM in human P-gp. Small changes in structure among this set of compounds affected affinity as well as transport rate (or flux) even though all derivatives examined were substrates for P-gp. With isolated protein, tertiary amide groups dictate high affinity and high stimulation while tertiary thioamide groups give high affinity and inhibition of ATPase activity. In MDCKII-MDR1 cells, the tertiary thioamide-containing derivatives promote uptake of calcein AM and have very slow passive, absorptive, and secretory rates of transport relative to transport rates for tertiary amide-containing derivatives. Thioamide 31-S promoted uptake of calcein AM and inhibited efflux of vinblastine with IC50’s of ~2 μM in MDCKII-MDR1 cells.
Keywords: Multidrug resistance, P-glycoprotein, rhodamines, rosamines, inhibition
Introduction
The treatment of cancer with chemotherapeutic agents is often thwarted by the emergence of multidrug resistance (MDR) in the transformed cells. P-glycoprotein (P-gp, also known as MDR1 or ABCB1)1-4 is a member of the ATP-binding cassette (ABC) superfamily and was the first efflux protein identified and associated with multidrug resistance in cancer chemotherapy. The onset of P-gp expression and drug resistance can be quite rapid, with mdr gene expression being observed within an hour of treatment.5 Since the discovery of verapamil (VER) as an inhibitor of P-gp,6 many approaches to the development of inhibitors/modulators of P-gp have been examined.7,8 Although a large number of compounds, possessing diverse chemical structures and biological activities, are able to reverse MDR, there are currently no approved reversal agents available in the clinic.9,10
P-gp is able to transport a diverse array of anticancer drugs including anthracyclines, vinca alkaloids, taxanes, epipodophyllotoxins, and agents such as mitomycin C, dactinomycim, and trimetrexate.9-12 This varied set of chemical structures as well as other substrates for P-gp are transported with a wide range of rates yet mechanistic studies suggest that transport of these structural classes involves a common transition state in the transporter.13 Related chemical structures within a single class can have markedly divergent rates of transport. Among the rhodamines, tetramethylrosamine (TMR, Chart 1) is the best transport substrate for P-gp both in viable MDR cells and in reconstituted P-gp.14-16 The transport of TMR is 5- to 10-fold faster than the reported transport of other rhodamine derivatives.16
Chart 1.

Structures of tetramethylrosamine analogues.
An obvious starting point to circumvent MDR in P-gp expressing cells is to design chemotherapeutic agents that are not recognized/transported by the pump.9 This approach led to two new agents (irinotecan and imatinib) that were thought to be non-substrates for P-gp. However, both are now recognized to be P-gp substrates.17,18 In a corollary to this approach, one might ask what structural features in a given class of compounds are responsible for recognition by the pump and what structural features are required for transport – either fast or slow? If critical structural features could be identified, could one then design either more effective drugs in known classes or, perhaps, identify additional classes of inhibitors/modulators for the pump?
Identifying the pharmacophore that enables drug transport is not a trivial issue especially since it is recognized that both competitive and noncompetitive interactions exist within the drug pocket19-21 and that P-gp also has allosteric sites that appear to reside outside the common pocket.22,23 Two non-overlapping binding sites were first identified on P-gp and were called the “H” and “R” sites for binding by Hoechst 33342 and rhodamine 123, respectively.24,25 The H-site is well studied and includes many of the known inhibitors of P-gp.26,27 The Clarke laboratory has reported that methanethiosulfonate derivatives of rhodamine and VER crosslink human P-gp at different sites.29 In contrast, Pajeva and Wiese in their pharmacophore model for rhodamines27 suggest that VER and rhodamines have structural features that can adopt comparable spatial orientations. Seelig discusses the importance of tight-binding between a substrate and its transporter and the importance of both π-electron interactions with a cation and hydrogen-bonding interactions between a substrate and its transporter.30-33 However, there is no clear algorithm for predicting either substrates or inhibitors.27,34
Rhodamine dyes have been used to assay P-gp-mediated transport. Efflux of rhodamine 123 from cells was used to define P-gp transport substrates/antagonists in a cross-correlation of drug resistance patterns in the NCI 60 set of cells with the NCI Drug Screen Database of compounds.35,36 It seemed plausible to design rhodamine-related structures that would behave as modulators/inhibitors of P-gp since small changes in rhodamine structure had large impacts on rates of transport by P-gp. The Tombline/Detty lab has recently described the synthesis of a small library of rhodamine-related structures and the impact of these molecules on ATPase activity in mouse cys-less P-gp (MDR3 CL).37,38 The initial library of rhodamines appeared to validate the presence of the “R” binding site in P-gp first described by Shapiro and Ling24,25 and also suggested that the pocket may be pliable since roughly a 1000-fold range of ATPase activities was observed. Based on structures in this initial library, we characterized a smaller set of TMR analogues which initially displayed a relatively negligible stimulation of ATPase suggesting that they were not good transport substrates.39 However, their ability to promote ATP occlusion, inhibit VER-dependent ATPase activity competitively, and be transported in cells revealed a high affinity for P-gp at a legitimate transport-competent site albeit at a slower rate.
Building on these initial data, we examined a series of 46 rhodamine derivatives to identify structural features that promote a high affinity for P-gp with minimal stimulation of ATPase activity with the expectation that these derivatives would be inhibitors of P-gp-mediated efflux. The rhodamine derivatives were evaluated for their ability to stimulate ATPase activity not only in MDR3 CL but also in wild-type human P-gp, their ability to be transported in multidrug-resistant cells, and their ability to inhibit/modulate P-gp in multidrug-resistant cells. This library of rhodamine derivatives contains structural diversity at the heteroatom atom of the xanthylium core (S, Se, Te) and the 3,6-diamino substituents (-NH2, -NMe2, julolidyl) in addition to systematic variation of the 9-substituent (aryl, alkyl, heteroaryl) to introduce hydrogen-bond donors and acceptors with a variety of spatial orientations. Structural features were identified that gave high affinity for P-gp with limited stimulation of ATPase activity in isolated protein and inhibited/modulated P-gp in multidrug-resistant cells to increase uptake of calcein AM (CAM) and vinblastine (VIN).
Results and Discussion
Synthesis of Rhodamine Analogues
The rhodamine analogues investigated in this study fall into two categories. The first category shown in Chart 1 includes analogues of TMR, which lacks the carboxylic acid oxidation state that characterizes most rhodamine derivatives. The derivatives 1-E with S, Se, and Te replacing O in the xanthylium core help delineate the role of the chalcogen atom while derivatives 1-E, 2-E, 3-S, and 4-S were prepared to vary the character of the amino group. Derivatives 5-E through 12-E examine the role of hydrogen-bond acceptors on 9-aryl substituents while derivative 13-S has a hydrogen-bond acceptor on a more flexible 9-alkyl substituent.
Chalcogenorhodamines 1-E, 2-E, and 5-E through 13-S38-40 were synthesized via the addition of the appropriate Grignard reagent (prepared from the corresponding bromoalkane or arylbromide) to chalcogenoxanthones 14-E41or 15-E42 (Chart 2). The initial addition products were dehydrated in aqueous HPF6 or HBr to give the indicated salts. The chloride salt of 7-S was obtained using an ion exchange resin to exchange chloride for the PF6−.
Chart 2.
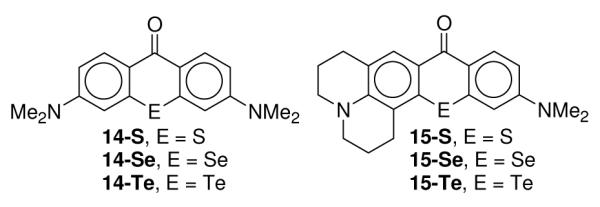
Chalcogenoxanthone precursors to tetramethylrosamine and rhodamine analogues.
The TMR analogues 3-S and 4-S were synthesized as shown in Scheme 1. 3-Aminothioxanthone 16-S43 was converted to 3-dimethylaminothioxanthone 17-S with excess iodomethane, which resulted in the formation of some quaternary salt. Heating the reaction mixture with 1,4-diaza[2.2.2]bicyclooctane (DABCO) demethylated any quaternary product that was formed and thioxanthone 17-S was isolated in 72% yield overall. The nitro group of 17-S was reduced to the primary amino group with SnCl2 in ethanol to give thioxanthone 18-S in 81% yield. The primary amino group was protected with trityl chloride and triethylamine to give thioxanthone 19-S in 75% yield. The addition of excess phenylmagnesium bromide to 19-S followed by treatment with aqueous HPF6 gave thioxanthylium dye 3-S in 68% isolated yield. The reduction of 16-S with SnCl2 in ethanol gave diaminothioxanthone 20-S43 in 98% isolated yield. Both amino groups were protected with trityl chloride and triethylamine to give thioxanthone 21-S in 59% isolated yield. The addition of excess phenylmagnesium bromide to 21-S followed by treatment with aqueous HPF6 gave thioxanthylium dye 4-S in 46% isolated yield.
Scheme 1.

Synthesis of tetramethylrosamine analogues 3-S and 4-S.
Compounds in the second category are shown in Chart 3 and introduce the carboxylic acid oxidation state into the 9-aryl or heteroaryl substituent. Groups that can be either a hydrogen-bond donor or acceptor on the 9-substituent (carboxylic acid or primary carboxamide) or groups that can only function as a hydrogen-bond acceptor (ester, tertiary amide, or tertiary thioamide) are found in this series. Carboxylic acid groups in different spatial orientations on a 9-thienyl substituent are found in 22-E and 23-E. The carboxylic acid group of 22-S is replaced with an ethyl ester and a primary carboxamide group in 24-S and 25-S, respectively, and with tertiary N,N-diethylamide substituents in 26-E. Compound 27-S is the thioxanthylium analogue of rhodamine II with a 9-2-carboxyphenyl substituent,40 while 28-S and 29-S are the methyl ester and N,N-diethylamide analogues, respectively. Compounds 30-E, 31-E, and 32-E incorporate tertiary thioamide functionality.
Chart 3.

Structures of rhodamine analogues with carboxylic acid oxidation states in the 9-substituents.
The addition of lithium 3-lithiothiophene-2-carboxylate (33)44 to xanthones 14-E followed by treatment with aqueous HPF6 gave rhodamines 22-S and 22-Se (Scheme 2). The addition of lithium 5-lithiothiophene-2-carboxylate (34) followed by treatment with aqueous HPF6 gave rhodamines 23-E.44 The addition of lithium 2-lithiobenzoate (35) to thioxanthone 15-S followed by treatment with aqueous HPF6 gave thiorhodamine 27-S in 75% isolated yield (Scheme 2).40 Carboxylic acid derivative 23-S was converted to ethyl ester 24-S in 85% isolated yield with carbonyl diimidazole (CDI) and ethanol and to carboxamide derivative 25-S in 83% isolated yield with CDI and ammonia.45 Similarly, carboxylic acid derivative 27-S was converted to methyl ester 28-S with CDI and methanol.
Scheme 2.

Synthesis of 9-thienyl carboxylic acid derivatives 22-E, 23-E, 24-S, 25-S, 27-S, and 28-S.
The addition of N,N-diethyl 5-lithiothiophene-2-carboxamide (36) to chalcogenoxanthones 14-E gave chalcogenorhodamines 26-S and 26-Se (Scheme 3).38 Similarly, the addition of N,N-diethyl 2-lithiobenzamide (37) to 14-S gave chalcogenorhodamine 29-S in 43% isolated yield (Scheme 3).
Scheme 3.

Synthesis of tertiary amide containing derivatives 26-E and 29-S.
Thioamide derivatives 30-E-32-E were prepared as shown in Scheme 4. Thiophene-2-carboxaldehyde was oxidized under Willgerodt-Kindler conditions with elemental sulfur and piperidine to give thioamide 3846 in 81% isolated yield and with elemental sulfur and morpholine to give thioamide 3947 in 99% isolated yield. Both thioamides were deprotonated with lithium diisopropylamide to give 5-lithiothiophenes 40 and 41. The addition of a solution of 40 to solutions of chalcogenoxanthones 14-E gave 30-S, 30-Se, and 30-Te in 75, 79, and 94% isolated yields, respectively, following a workup with aqueous HPF6. Similarly, the addition of a solution of 40 to chalcogenoxanthones 15-E followed by an acidic workup gave 31-S, 31-Se, and 31-Te in 62, 31, and 54% isolated yields, respectively. Chalcogenorhodamines 32-S and 32-Se were prepared in 57 and 70% isolated yields, respectively, following the addition of a solution of 41 to chalcogenoxanthones 15-E with an acidic workup. Unlike the tertiary amide group,48 which is highly directing, the thioamide functionality does not direct lithiation in thiophenes. Only the more acidic α-proton was removed and none of the corresponding 2,3-disubstituted thiophenes were detected in the product mixtures.
Scheme 4.

Synthesis of thioamide derivatives 30-E – 32-E.
P-gp ATPase Activity of TMR Analogues of Chart 1
We examined the ATPase activity of the TMR analogues of Chart 1 using MDR3 CL P-gp,49 which is 87% identical to human MDR1 P-gp in sequence, and using human P-gp-His10 activated with sheep brain phosphatidylethanolamine.29,50,51 Many of the thio- and selenoxanthylium analogues of Chart 1 have shown activity as photosensitizers.52,53 Consequently all interactions of these derivatives with P-gp were conducted in the dark. Results from the mouse MDR3 Cl P-gp are found in the Supporting Information (Table S1).
ATPase Activity with Human P-gp
We determined the apparent Michaelis-Menten constant (KM) for the stimulating rosamine/rhodamine compound of Charts 1 and 3, and the drug-induced stimulation of maximal ATPase activity (Vmax) using human P-gp-His10 (Table 1). VER was included as a control compound (generally considered relatively robust and one of the most stimulating drugs known for P-gp).54,55 ATPase activity was determined from the release of inorganic phosphate.55 For several compounds, which were weakly stimulating for P-gp ATPase activity, we also determined an IC50, the concentration of compound required for 50% inhibition of VER-stimulated (400 μM) ATPase activity.
Table 1.
Stimulation of human P-gp-His10 ATPase activity by the rosamine and rhodamine analogues of Charts 1 and 3.
| Human P-gp-His10 | |||
|---|---|---|---|
| Compd |
Vmax, a Fold |
KM, b μM |
IC50,c μM |
| Ver | 17.9 ± 2.0 | 24 ± 2.6 | |
| 1-S | 7.7 ± 1.0 | 8.5 ± 1.2 | |
| 1-Se | 7.2 ± 1.1 | 9.1 ± 1.1 | |
| 1-Te | 8.4 ± 1.9 | 8.3 ± 0.8 | |
| 2-S | 2.2 ± 0.3 | 1.3 ± 0.5 | |
| 2-Se | < 1.5 | N.D.d | 2.3 ± 0.4 |
| 2-Te | 3.0 ± 0.7 | 4.4 ± 0.8 | |
| 3-S | 7.5 ± 1.5 | 9.3 ± 1.0 | |
| 4-S | 6.4 ± 0.6 | 32 ± 3 | |
| 5-S | 9.0 ± 0.5 | 8.3 ± 1.2 | |
| 5-Se | 6.8 ± 1.0 | 10.1 ± 0.7 | |
| 5-Te | 4.5 ± 0.3 | 9.0 ± 0.8 | |
| 6-Se | < 1.5 | N.D.d | 6.8 ± 0.2 |
| 7-S | 7.7 ± 0.5 | 3.4 ± 0.6 | |
| 7-Se | 7.6 ± 0.6 | 3.5 ± 0.2 | |
| 7-Te | 5.5 ± 0.3 | 2.0 ± 0.1 | |
| 8-Se | < 1.5 | N.D.d | 23.6 ± 3.3 |
| 10-S | 5.9 ± 1.3 | 3.9 ± 0.1 | |
| 10-Se | 2.5 ± 0.2 | 3.3 ± 0.6 | |
| 10-Te | 5.7 ± 0.7 | 3.2 ± 1.0 | |
| 11-S | 8.2 ± 0.4 | 4.9 ± 0.8 | |
| 11-Se | 7.5 ± 0.9 | 8.3 ± 0.5 | |
| 12-S | 4.8 ± 0.3 | 10.0 ± 1.3 | |
| 12-Se | 3.3 ± 0.6 | 10.4 ± 1.0 | |
| 13-S | 6.2 ± 0.8 | 3.2 ± 0.4 | |
| 22-S | 4.3 ± 0.3 | 111 ± 4 | |
| 22-Se | 2.1 ± 0.3 | 56 ± 11 | |
| 23-S | 2.7 ± 0.2 | 140 ± 11 | |
| 23-Se | 2.8 ± 0.6 | 89 ± 7 | |
| 23-Te | 1.8 ± 0.1 | 9.6 ± 1.1 | |
| 26-Se | 10.5 ± 1.6 | 0.68 ± 0.14 | |
| 30-S | 9.0 ± 0.5 | 0.36 ± 0.08 | 7.9 ± 1.1 |
| 30-Se | 5.0 ± 0.6 | 0.35 ± 0.02 | 9.0 ± 0.2 |
| 30-Te | 7.0 ± 0.4 | 0.41 ± 0.03 | 5.3 ± 1.2 |
| 31-S | 3.1 ± 0.3 | 0.087 ± 0.012 | 0.67 ± 0.14 |
Vmax is the ratio of the maximum stimulation in the presence of compound relative to that in the absence of compound (the basal activity).
KM is the apparent Michaelis-Menten constant or the concentration of compound required for half maximal stimulation of ATPase activity.
IC50 is the concentration of compound required for 50% inhibition of verapamil-stimulated (400 μM in human P-gp-His10) ATPase activity.
N.D., not determined because of low stimulation of ATPase activity.
Rosamine Analogues of Chart 1
Compounds 2-Se, 6-Se, and 8-Se with the julolidyl fragments as one amino group gave very little stimulation of P-gp (Vmax <1.5-fold stimulation), which precluded accurate determination of KM. Values of IC50 for 50% inhibition of VER-stimulated (400 μM) ATPase activity for 2-Se, 6-Se, and 8-Se were 2.3, 6.8, and 23.6 μM (Table 1), respectively, indicating that these compounds could inhibit Ver-induced stimulation even if they were not stimulating ATPase activity. For the remaining compounds investigated, Vmax ranged from 2.2-fold stimulation for 2-S to 9-fold stimulation for 5-S, which is roughly 50% of the 17.9-fold stimulation observed for VER. Values of KM with human P-gp ranged from 1.3 μM for 2-S to 32 μM for 4-S (Table 1).
In general for the rosamine analogues of Chart 1, the identity of the heteroatom (S, Se, Te) in the chalcogenoxanthylium core had little impact on either Vmax or KM. Replacing one dimethylamino group with an amino group had no impact on either Vmax or KM (1-S vs. 3-S). Replacing both dimethylamino substituents with amino substituents in 4-S had little impact on Vmax but gave a four-fold higher value of KM (Table S1). A hydrogen-bond acceptor in the 3-position of the 9-aryl substituent gave lower values of KM (7-E and 10-E) relative to compounds with a hydrogen-bond acceptor in the 4-position (5-E). The flexible 9-(3-phenoxy)propyl substituent of 13-S also gave a KM of 3 μM. The incorporation of two additional rings in the julolidyl fragment of 2-E, 6-Se, and 8-Se gave reduced values of Vmax. For compounds 2-E, values of KM and IC50 were lower (1.3-4.4 μM) than values of KM observed for compounds 1-E (8.3-9.1 μM) again suggesting higher affinity for P-gp from the two additional rings.
Rhodamine Analogues of Chart 3
Values of Vmax, KM, and IC50 (Table 1) were determined for selected rhodamine analogues in Chart 3 with human P-gp-His10 as described above. In this set of compounds, the carboxylic acid groups of 22-E, 23-E, and 27-S and the primary carboxamide of 25-S can act as either a hydrogen-bond donor or acceptor while the esters 24-S and 28-S and tertiary amides 26-E and 29-S can only act as hydrogen-bond acceptors. Dye 26-Se with a tertiary amide was more stimulating (Vmax of 10.5-fold stimulation) with respect to ATPase activity and had higher affinity (KM aof 0.68 μM) than carboxylic acid-containing derivatives 22-E and 23-E (Vmax ≤ 4.3-fold; KM > 9 μM). Within the carboxylic acid-containing derivatives 22-E and 23-E, KM decreased as the heteroatom became larger. Tertiary Amide-containing derivatives 26-E and 29-S also showed high affinity and high ATPase stimulation with mouse MDR3 CL with Vmax >10-fold above basal levels and with values of KM in the 3-6 μM range (Table S1).
Thioamide-containing Rhodamine Derivatives of chart 3
The tertiary amide-containing derivatives 26-E and 29-S were highly stimulating toward ATPase activity with high affinity for P-gp. Are both attributes due to the strength of the amide carbonyl as a hydrogen-bond acceptor or due to the increased hydrophobicity from the two alkyl substituents on the amide nitrogen? To probe this question, the affinity of tertiary thioamides 30-E and 31-S (Chart 3) for P-gp was determined as well as the impact of the tertiary thioamides on ATPase activity. Values of Vmax, KM, and IC50 are compiled in Table 1. All four compounds displayed sub-micromolar values of KM toward human P-gp with 31-S having the highest affinity for human P-gp (KM of 0.087 μM). The substitution of the julolidyl fragment for a dimethylamino group gave reduced stimulation of ATPase activity (Figure 1 comparing 30-S and 31-S). Rhodamines 30-E displayed values of Vmax between 5- and 9-fold stimulation for derivatives 30-E and 3.1-fold stimulation for 31-S, as one might expect from the trends observed in the other julolidyl-containing compounds of this study. This comparison also held with respect to values of IC50 for VER-induced stimulation of ATPase activity in human P-gp as shown in Figure 2. Compounds 30-E gave values of IC50 of 5.3-9.0 μM toward human P-gp while 31-S displayed an IC50 of 0.67 μM (Table 1).
Figure 1.

Stimulation of human P-gp ATPase activity by compounds 30-S and 31-S. Histidine-tagged P-glycoprotein was expressed in BHK cells, isolated by nickel-chelate chromatography and mixed with lipid. P-glycoprotein ATPase activity was then measured in the presence of various concentrations of rhodamine derivatives 30-S or 31-S. Error bars represent one standard deviation from the mean.
Figure 2.

Inhibition of human P-gp VER-stimulated ATPase activity. Histidine-tagged P-glycoprotein was expressed in BHK cells, isolated by nickel-chelate chromatography and mixed with lipid. P-glycoprotein ATPase activity was then measured in the presence of 0.4 mM VER with various concentrations of rhodamine derivatives 30-S, 30-Se, 30-Te, or 31-S. Data represent the average for triplicate measurements and error bars represent the standard deviation.
Transport Across Monolayers of MDCKII-MDR1 Cells
The transport of selected rosamine and rhodamine dyes was examined in monolayers of MDCKII-MDR1 transfected cells, which over-express P-gp (Table 2).56 MDCKII-MDR1 monolayers display apical and basolateral polarized membranes and are considered to be a near-physiological model for studying P-gp drug transport. In the monolayer, P-gp is asymmetrically distributed being solely present at the apical membrane. In order to evaluate the role of P-gp in the transport of the compounds of this series, transport was measured in the absorptive (apical to basolateral or AB) and secretory (basolateral to apical or BA) transport direction of the cell monolayer, and then an efflux ratio (PBA/AB) was calculated.57 With the exception of carboxylic acid-containing derivative 23-Se, bovine serum albumin (BSA) addition to the buffer was required because a marked fraction of mass added to the donor equilibrated with the cell monolayer for some of the compounds and this resulted in gross underestimation of the permeability coefficient.58 The assay was repeated in the presence of 2.5 μM (R)-4-[(1a,6,10b)-1,1-dichloro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cyclohepten-6-yl]-[(5-quinolinyloxy)methyl]-1-piperazineethanol (LSN335984, 42), which is structurally related to the P-gp-specific inhibitor (R)-4-[(1a,6,10b)-1,1-difluro-1,1a,6,10b-tetrahydrodibenzo[a,e]cyclopropa[c]cyclohepten-6-yl]-[(5-quinolinyloxy)methyl]-1-piperazineethanol (LY335979 or zosuquidar),59 to measure transport when P-gp was fully inhibited. Compound 42 completely inhibits P-gp with an IC50 of 0.4 μM. The ratio PBA/AB reported in Table 2 is normalized by dividing PBA/AB in the absence of inhibitor by the secretory/basal ratio observed in the fully inhibited system. Large efflux ratios are assumed to be due to efficient P-gp-mediated efflux of the compound. Values of passive transport (PPassive) in the presence of inhibitor, transport in the absorptive (PAB) and secretory (PBA) mode in the absence of inhibitor, the normalized ratio [PBA/AB (no inhibitor)/PBA/AB (with inhibitor)], and the % cell-associated dye in the AB direction in the absence or presence of inhibitor are compiled in Table 2. Of the 14 compounds examined in the MDCKII-MDR1 cell monolayer system, all were shown to be P-gp substrates as indicated by net efflux defined by normalized PBA/AB ratios of >2, with TMR having the highest ratio (217) and carboxylic acid-containing derivative 23-Se, the lowest (2.6).
Table 2.
Transport and cell association studies of rosamine and rhodamine analogues with MDCKMDR1 cells.a
| Compd |
PAB nm s−1 |
PBA nm s−1 |
P BA/AB | Normalized Ratio, PBA/AB (−/+ inh)b |
PPassivec nm s−1 |
% Cell Associatedd |
Ratio (+/− inh)e |
log P |
|---|---|---|---|---|---|---|---|---|
|
TMR (+ inh) |
3.0 ± 0.1 14 ± 1 |
1010 ± 10 21 ± 2 |
338 1.6 |
217 | 17 ± 5 | 14.5 ± 0.9 63 ± 2 |
4.3 | 1.1 |
|
1-S (+ inh) |
6.0 ± 0.1 30 ± 1 |
900 ± 120 39 ± 2 |
149 1.3 |
114 | 34 ± 7 | 14 ± 1 71 ± 2 |
5.1 | 1.2 |
|
2-S (+ inh) |
0.8 ± 0.1 3.3 ± 0.1 |
360 ± 50 11 ± 3 |
450 3.4 |
130 | 7 ± 6 | 30 ± 2 64 ± 3 |
2.1 | 2.3 |
|
2-Se (+ inh) |
16.0 ± 0.1 13.0 ± 0.8 |
250 ± 40 12 ± 1 |
15 0.9 |
17 | 12.3 ± 0.8 | 32 ± 1 64 ± 4 |
2.0 | 2.4 |
|
9-S (+ inh) |
116 ± 3.4 39.0 ± 0.2 |
530 ± 33 18.0 ± 0.3 |
4.6 0.5 |
10 | 29 ± 14 | 47 ± 26 66 ± 18 |
1.4 | −0.09 |
|
10-S (+ inh) |
169 ± 7 59 ± 4 |
693 ± 155 30.0 ± 0.4 |
4.1 0.5 |
8.2 | 45 ± 21 | 13 ± 3 56 ± 18 |
4.3 | 0.14 |
|
11-S (+ inh) |
118 ± 1 48 ± 2.6 |
653 ± 39 26 ± 0.1 |
5.5 0.6 |
10 | 37 ± 3 | 16 ± 12 53 ± 3.1 |
3.3 | 0.28 |
|
11-Se (+ inh) |
131 ± 7 31 ± 2 |
340 ± 100 15 ± 0.3 |
2.6 0.5 |
5.3 | 23 ± 11 | 37 ± 24 57 ± 9.7 |
1.5 | 0.18 |
|
23-Sef (+ inh) |
45 ± 19 16 ± 4 |
200 ± 24 28 ± 0.1 |
4.4 1.7 |
2.6 | 22 ± 6 | 5.6 ± 0.2 4.8 ± 0.8 |
0.9 | -- |
|
25-S (+ inh) |
20 ± 1 12 ± 0 |
73 ± 8 9 ± 0.1 |
3.7 0.7 |
5.1 | 11 ± 2.3 | 12 ± 9 6.7 ± 2.9 |
0.6 | -- |
|
26-Se (+ inh) |
101 ± 1.6 63 ± 0.7 |
424 ± 9 29 ± 0.03 |
4.2 0.5 |
9.2 | 46 ± 24 | 11 ± 5.5 24 ± 0.73 |
2.2 | −0.39 |
|
30-S (+ inh) |
1.5 ± 0.1 1.8 ± 0.5 |
80 ± 19 3.9 ± 0.1 |
54 2.1 |
25 | 2.8 ± 1 | 16 ± 1 29 ± 1.5 |
1.8 | 1.7 |
|
30-Se (+ inh) |
1.1 ± 0.2 1.3 ± 0.1 |
41 ± 3 2.9 ± 0.1 |
37 2.3 |
16 | 2.1 ± 0.8 | 8.6 ± 0.3 21 ± 3.3 |
2.4 | -- |
|
30-Te (+ inh) |
0.9 ± 0.1 1.2 ± 0.1 |
22 ± 8 2.0 ± 0.1 |
24 1.7 |
14 | 1.6 ± 0.4 | 12 ± 3 23 ± 1.5 |
1.9 | 0.9 |
|
31-S (+ inh) |
0.8 ± 0.1 0.8 ± 0.1 |
50 ± 1 1.6 ± 0.1 |
61 2.1 |
29 | 1.2 ± 0.4 | 23 ± 3 30 ± 1.5 |
1.3 | 2.7 |
Experiments were run with 5 μM dye and 4.3 mg mL−1 BSA. Values of transport in the absorptive (PAB) and secretory (PBA) mode in the absence or presence of inhibitor, the ratio of secretory to absorptive transport (PBA/AB) in the absence or presence of inhibitor, the normalized ratio [PBA/AB (no inhibitor)/PBA/AB (with inhibitor)], the % cell associated rhodamine analogue in the absence or presence of inhibitor, the ratio of cell associated rhodamine in the presence or absence of inhibitor, and values of log P (the n-octanol/water partition coefficient). Details for methods are provided in Experimental Procedures. Error limits represent ± standard deviation.
The normalized ratio represents the PBA/AB ratio in the absence of inhibitor divided by the PBA/AB ratio in the presence of inhibitor.
PPassive represents the mean of PAB and PBA in the fully inhibited system.
% Cell Associated is the fraction of mass extracted from the cell monolayer by methanol wash after 1-h flux in the AB direction.
For % cell associated dye.
No added BSA.
Rates of transport in the absorptive direction were comparable for compounds with no hydrogen-bond acceptor in the 9-aryl substituent (TMR, 1-S, 2-S, 2-Se) and fell in the PAB range of 0.8-16.0 nm s−1. Compounds with weak hydrogen-bond acceptors (thioamide-containing derivatives 30-S, 30-Se, 30-Te, and 31-S) had even slower transport in AB direction with values of PAB in the range of 0.8-1.5 nm s−1. The addition of a stronger hydrogen-bond acceptor on the 9-aryl or heteroaryl group gave an order of magnitude or greater increase in absorptive transport (PAB of 116-169 nm s−1 for methoxy-containing derivatives 9-S, 10-S, 11-S, and 11-Se and PAB of 101 nm s−1 for tertiary amide-containing derivative 26-Se). Substituents that function as both a hydrogen-bond donor and acceptor gave intermediate values of absorptive transport (PAB of 45 nm s−1 for 23-Se with a carboxylic acid group and PAB of 20 nm s−1 for 25-S with a primary amide group). Rates of transport in the secretory direction were maximal for TMR and the structurally related 1-S (PBA of 1010 and 900 nm s−1, respectively), were minimal for primary amide derivative 25-S and the thioamide-containing 30-E and 31-S (PBA of 22-80 nm s−1), and were intermediate for the remaining 10 derivatives (PBA of 200-693 nm s−1). The normalized PBA/AB ratios were smallest for those derivatives with a hydrogen-bond acceptor in the 9-substituent (normalized PBA/AB ratios of 2.6-11) and much larger for derivatives with either a 9-phenyl group (normalized PBA/AB ratios of 17-217) or a tertiary thioamide group on the 9-substituent (normalized PBA/AB ratios of 14-29).
Passive transport, PPassive, in the presence of 42 was markedly lower for derivatives 30-E and 31-S with tertiary thioamide groups (1.2-2.8 nm s−1). For compounds 2-S and 2-Se, PPassive was 7 and 12.3 nm s−1, respectively (Table 3). For the remaining compounds, PPassive fell in the range of 11-56 nm s−1.
Table 3.
IC50 for the uptake of CAM in rhodamine-treated MDCKII-MDR1 cells.
| Compd | CAM uptake IC50, μMa |
|---|---|
| 1-S | 11 ± 3 |
| 1-Se | 11 ± 3 |
| 2-S | 4.7 ± 2.0 |
| 2-Se | 4.9 ± 0.6 |
| 30-S | 19 ± 2 |
| 30-Se | 9 ± 1 |
| 30-Te | 13 ± 1 |
| 31-S | 2.1 ± 1.2 |
| 31-Se | 5.3 ± 1.2 |
| 31-Te | 14 ± 1 |
| 32-S | 20 ± 1 |
| 32-Se | 14 ± 1 |
Details for methods are provided in Experimental Procedures. Error limits represent ± standard deviation.
The % cell-associated dye for derivatives with a 9-phenyl substituent (TMR, 1-S, 2-S, 2-Se) or a 9-3-methoxyaryl substituent (10-S, 11-S, 11-Se) was high in the inhibited system (54-71%) but was significantly lower when the pump was active (7.5-32%) giving ratios between the inhibited and uninhibited systems from ~2 for 2-S and 2-Se to 7.2 for 11-S. Compound 9-S with a 9-4-methoxyphenyl substituent was an anomaly with high cell association in both the uninhibited (47%) and inhibited (66%) systems. Amide-containing derivative 25-S, diethylamide-containing derivative 26-Se, and the thioamide-containing derivatives 30-E and 31-S showed less cell-associated dye in both inhibited (6.7-30%) and uninhibited (12-23%) systems. Compound 31-S had essentially identical values of % cell-associated dye in the inhibited (30%) and uninhibited (23%) systems [ratio (+/− inhibitor) of 1.3].
Enhancement of CAM uptake into MDCKII-MDR1 cells
Rosamine analogues 1-S, 1-Se, 2-S, and 2-Se and rhodamine analogues 30-E, 31-E, and 32-E were next evaluated for their ability to facilitate uptake of CAM into MDCKII-MDR1 cells.56 Values of IC50 for CAM uptake (Table 3) were determined by measuring relative fluorescence values obtained after 20-min incubation with CAM at 37 °C. Typical curves are shown in Figure 3 for CAM uptake in the presence of various concentrations of 30-E and 31-S. Substitution of the julolidyl fragment reduced IC50 from 11 μM for 1-S and 1-Se to an IC50 of 4-5 μM for 2-S and 2-Se.. Similarly, values of IC50 were lowered from 19 and 9 μM for 30-S and 30-Se, respectively, to 2.1 and 5.3 μM for 31-S and 31-Se, respectively, by introduction of the julolidyl fragment. Replacing a methylene group of the piperidine-containing thioamides 31-E with the O atom of morpholino thioamides 32-S and 32-Se gave higher values of IC50 (20 and 14 M, respectively).
Figure 3.

Uptake of CAM into MDCKII-MDR1 cells as a function of concentration of rhodamine dyes 30-S, 30-Se, 30-Te, and 31-S. Values of IC50 were determined by a sigmoidal dose-response (variable slope) analysis and are compiled in Table 3. Data represent the average and standard error mean for duplicate measurements.
Inhibition of VIN Efflux by 31-S in MDCKII-MDR1 Cells
Since VIN is an actual chemotherapeutic drug used clinically, inhibition of VIN transport by the rhodamines is likely to indicate translational value. [3H]-Vinblastine in an appropriate dilution series with 31-S and BSA was introduced to the basolateral chamber of a monolayer of MDCKII-MDR1 transfected cells. The appearance of [3H]-VIN in the apical chamber was monitored by scintillation counting and gave an IC50 of 2.4 ± 0.4 μM (Figure S1, Supporting Information), which is essentially identical to the IC50 observed for CAM uptake.
Inhibition of VIN transport in a membrane vesicle model
Compound 31-S was evaluated for its ability to inhibit VIN transport by human P-gp in an inside-out membrane vesicle model, prepared from human embryonic kidney (PEAKSTABLE) cells over expressing human Pgp.60,61 The accumulation of [3H]-VIN into P-gp membrane vesicles in the presence of 31-S (0.01 to 10 μM) was monitored, using 5 μM 42 as a standard for complete P-gp inhibition. Previously, the inhibition of [3H]-VIN accumulation in P-gp membrane vesicles by 42 gave an IC50 value of 6.5 ± 1.1 nM and, thus, complete inhibition of P-gp was assumed at 5 μM.60 Mixtures of [3H]-VIN and 31-S were added to the vesicles and, following a reaction time of 2.5 min, the vesicles were removed by filtration and the residual radiation on the filter was measured by scintillation counting to give an IC50 of 0.13 ± 0.03 μM as shown in Figure 4. The IC50 measured in this model is similar to the 0.67 μM value of IC50 observed with isolated human P-gp-His10 (Table 1).
Figure 4.

Analyses for inhibition of VIN transport by wild-type, human Pgp using 31-S in membrane vesicles from PEAK-MDR1 cells. The line represents the non-linear least-squares regression fits of the data. Data represent the average and standard error mean for duplicate measurements.
Discussion
Interactions of the rosamines/rhodamines with P-gp
Within a library of closely related molecules that all bind to the same protein, the assumption is often made that binding occurs at a common site. P-glycoprotein and other ABC transporters appear to have multiple drug-binding sites that allow a diverse array of structures to bind to the protein. Early studies by Shapiro and Ling revealed the presence of at least two binding sites on P-gp described as “H” and “R” sites for Hoechst 33342 and rhodamine 123, respectively.24,25 However, it is not at all clear that these sites in P-gp are “rigid.” The Clarke laboratory described an “induced fit” model for binding of small molecules to P-gp in which the shape of the drug-binding site changes upon binding of compounds with different shape.62 The binding pocket also may have many different ways of binding to a single molecule. QacR, a member of the TetR family of suppressors, binds to many of the same molecules as QacA, the MDR pump that QacR regulates.63 Structural studies with QacR suggested that 5 glutamate residues were critical for charge neutralization in the binding of cationic molecules to the protein.64 However, mutations of QacR showed that binding of cations to the protein was still observed when several of the glutamate residues had been replaced.65 In the case of one cationic compound, the positive charge was neutralized by side-chain oxygen atoms and by aromatic residues. Consequently, the drug-binding pocket of P-gp may not have well-defined binding sites for molecules of diverse structure, but may be a highly flexible domain in which conformational changes can have pronounced impact on binding. A model of human P-gp generated by comparison to the crystal structures of two bacterial transporters featured a large cavity within the transmembrane domains into which drug substrates such as rhodamine B, VER, and VIN could be successfully docked.66 The docked positions of rhodamine and other drug substrates were found to interact with residues that alter substrate specificity upon mutation.
The rhodamines of this study were selected to have similar shapes and similar non-covalent interactions with P-gp in the xanthylium core in the hope that one might examine the impact of subtle structural changes in the substrate to a single binding region of P-gp. The rosamines of Chart 1 and rhodamines of Chart 3 share certain structural similarities: 1) All derivatives have two N-atoms as hydrogen-bond acceptors at the 3- and 6-positions. In most of the derivatives, the N-atoms are part of dimethylamino substituents. Compounds 3-S and 4-S have NH2 substituents while compounds 2-E, 6-Se, 8-Se, 31-E, and 32-E incorporate one N-atom as part of a julolidyl fragment with the other as a dimethylamino substituent. 2) All derivatives have a chalcogen atom (O, S, Se, or Te) in the xanthylium core, which can also function as an H-bond acceptor. 3) All derivatives bear a positive charge, which is delocalized over the xanthyllium π-framework. Seelig has discussed the importance of π-electron interactions with a cation and hydrogen-bonding interactions between a substrate and its transporter for tight binding and substrate recognition to occur.30-32 The rosamines and rhodamines of this study are all delocalized aromatic cations and they each contain two of the so-called Seelig Type II hydrogen bond acceptor units, with two hydrogen bond-accepting atoms 4.6 ± 0.6 Å apart – the chalcogen atom-nitrogen atom arrays, whose separation is on the order of 5.0-5.15 Å, as determined by x-ray crystallographic studies.38,40
Most of the significant structural variation within these compounds comes at the 9-position where a variety of aliphatic, aromatic, and heteroaromatic substituents have been incorporated. Among these, compounds 11-E, 12-E, and 22-E through 32-E add a Seelig Type I unit to the 9-aryl or heteroaryl substituent. The Seelig Type I units separate the hydrogen bond-accepting atoms by 2.5 ± 0.5 Å.30-32 This latter group of compounds varies in the relative spatial orientation of the Type I unit as well as in the strength of the hydrogen-bond acceptor.
We can also examine the affinity of the rhodamine molecules for P-gp as well as the impact of the small molecule on ATPase activity (Vmax). For derivatives that stimulate ATPase activity, affinity can be approximated by KM. In systems that do not stimulate ATPase activity, affinity can be measured with respect to IC50 for the inhibition of VER-induced ATPase activity. Affinity for P-gp measured by binding in the transmembrane domains and impact upon ATPase activity driven by the nucleotide binding domains (NBDs) can be separated. The Clarke laboratory has shown that a truncation mutant of P-gp lacking both NBDs could still interact with rhodamine-type compounds.67
Certain structural features appear critical for affinity and ATPase activity. Comparison of KM and Vmax for compounds 1-E and 2-E show significant increases in affinity (p < 0.05) upon replacement of one dimethylamino substituent with the julolidyl fragment and significant decreases in ATPase activity in human P-gp-His10. Similarly, comparison of KM and Vmax for compounds 30-E and 31-E show significant increases in affinity (p < 0.05) upon introduction of the julolidyl fragment and significant decreases in ATPase activity.
Simple hydrogen-bond acceptors in the 9-substituents of the rosamine analogues of Chart 1 had little impact on either affinity (KM) or ATPase stimulation (Vmax) in human P-gp. The introduction of Seelig Type I hydrogen-bond acceptors on the 9-substituents had a much greater impact. The addition of a carboxylic acid group, which is also a hydrogen-bond donor as well as a Seelig Type I acceptor, gave a decrease in ATPase stimulation relative to the 9-phenyl substituent and affinity was either reduced or unchanged. The tertiary amide substituents in compounds 26-E and 29-S impart high affinity (KM on the order of 3-6 μM) and high ATPase activity (>10-fold stimulation above basal) in both human and mouse P-gp (Tables 1 and S1). Replacing the carbonyl of the amide with the thiocarbonyl group of the tertiary thioamides 30-E gave slightly increased affinity and reduced ATPase activity. However, incorporation of the julolidyl fragment and the thioamide functionality in 31-S gave the highest affinity observed in this study (KM of 0.087 μM) with low ATPase activity (Vmax of 3-fold stimulation above basal in human P-gp-His10) and inhibition of Ver-induced stimulation with an IC50 of 0.67 μM.
Cell Studies
The rosamine and rhodamine dyes of this study enter MDCKII-MDR1 cells as indicated by their active transport by P-gp (normalized PBA/AB > 2, Table 2) and by their facilitation of CAM uptake (Table 3). Thus, all of the compounds of Table 2 bind to transport sites including those that have Vmax values at or near basal levels. With respect to inhibition of P-gp in whole cells, the same structural features that gave higher affinity and reduced ATPase activity in the isolated protein also gave lower values of IC50 for CAM uptake in the MDCK-MDR1 cells. The incorporation of the julolidyl fragment in 2-S and 2-Se lowered IC50 for CAM uptake to 4.7 and 4.9 M, respectively, relative to 1-S and 1-Se with two dimethylamino substituents at the 3- and 6-positions (IC50’s of 11 μM for both). Addition of the thioamide functionality in 31-S lowered IC50 to 2.1 μM. Interestingly, the addition of another hydrogen bond-accepting atom in the morpholino thioamides 32-E gave increased values of IC50 for CAM uptake (20 μM for 32-S vs. 2.1 μM for 31-S and 14 μM for 32-Se vs. 5.3 μM for 31-Se, Table 3).
Digoxin has been used as a default substrate for P-gp because of the clinical implications of recognizing a drug-drug transporter (P-gp) interaction where digoxin with a very narrow margin of safety is unusually affected.68 However, CAM uptake can be utilized as a higher throughput and equally sensitive primary assay and was deemed more efficient and practical for these initial studies using fluorescence as a method of detection. While most of the rhodamines of Table 3 were comparable to VER with respect to IC50 for CAM uptake in the MDCKII-MDR1 cells, compound 31-S was more potent with an IC50 more similar to those of quinidine, cyclosporin A, zosuquidar, and 42 (Table 3). Compound 31-S also inhibited the efflux of VIN, a clinically used chemotherapeutic drug, with a similar IC50 of 2.4 μM in MDCKII-MDR1 cells. In this assay as well as in the vescicle assay, the more passively permeable VIN gave a more robust signal relative to digoxin.
The compound TMR has been recognized as one of the best transport substrates for P-gp14-16 with, in our hands, KM of 77 μM and ATPase stimulation 3.7-fold above basal (Table S1) and with a rate of efflux, PBA, of 1010 nm s−1 in MDCKII-MDR1 cells (Table 2). One might conclude from this work that a series of closely related rhodamine and rosamine structures can be tuned for high affinity and high ATPase activity (for 26-Se, KM of 0.68 μM and Vmax of 10.5-fold stimulation in human Pgp-His10) or for high affinity and high inhibitory activity (for 31-S, KM of 0.087 μM and IC50 of 0.67 μM for inhibition of VER-induced stimulation of ATPase activity in human P-gp-His10). The highly stimulating 26-Se has a secretory rate of transport of 424 nm s−1 for PBA while the highly inhibitory 31-S has a slower secretory rate of 50 nm s−1.
Earlier studies demonstrated that TMR is transported by P-gp 5- to 10-times more rapidly than other rhodamine derivatives.16 TMR was transported the fastest among the 15 compounds examined in Table 3. For rosamine analogues 1-S, 2-S, 2-Se, 9-S, 10-S, 11-S, and 11-Se PBA was 250-900 nm s−1, which is on the same order of magnitude as PBA for TMR. The rhodamine analogues 23-Se, 25-S, 26-Se, 30-E, and 31-S displayed more varied transport, but values of PBA reflected affinity and ATPase stimulation. TMR was transported 5-fold faster than 5-carboxy-2-thienyl derivative 23-Se, which displayed low affinity for P-gp and low stimulation of ATPase activity. TMR was transported 13- to 46-times more rapidly than thioamides 30-E and 31-S, which displayed high affinity and low stimulation of ATPase activity. In contrast, the high affinity, highly stimulating tertiary amide derivative 26-Se was transported with a rate more comparable to that of TMR (424 nm s−1). Again, the amide/thioamide “switch” changed the fundamental interaction of the small molecule with P-gp.
One would anticipate that the performance of 31-S as an inhibitor would be similar in isolated protein or in whole cells. Compound 31-S effectively interacts with P-gp in a native membrane environment and facilitates the uptake of CAM and inhibits the efflux of VIN. However, the values of IC50 of ~2 μM are significantly higher than values IC50 for inhibition of VER-induced stimulation of P-gp (0.68 μM) in human P-gp-His10 activated with sheep brain phosphatidylethanolamine and IC for inhibition of [350 H]-VIN uptake (0.13 μM) in an inside-out P-gp membrane vesicle model.
There are several reasons why effective concentrations in whole cells might be different than those in isolated protein. It is known that lipid composition and fluidity greatly influence drug interactions with P-gp,20,31,69 and differences between experimental conditions or Pgp environment (i.e., membrane composition) may account for differences in behavior between ATPase and [3H]-VIN experiments with isolated protein and CAM and [3H]-VIN experiments in whole cells.
An alternative explanation is that 31-S has limited access to the pump in the whole cell experiments. This may be because PPassive into the favored membrane microenvironment is rate limiting, or other efflux transporters assist in escorting 31-S from the cell,70 or because 31-S has high binding affinity, specific or non-specific, to the membrane and the unbound fraction at the pump is very low. Compounds 30-E and 31-S show particularly slow PPassive rates (1.2-2.8 nm s−1) that correlate with relatively slower efflux rates in the uninhibited system (PBA ≤ 80 nm s−1). This is consistent with passive replenishing of the driving membrane concentration lagging behind active removal. Complete inhibition of efflux by inhibitor 42 implies that P-gp is solely responsible; however, involvement of one or more additional unknown efflux transporters cannot be excluded unequivocally if 42 also happens to inhibit these putative transporters. One important comparison is between 31-S with tertiary thioamide functionality, where PPassive is 1.2 nm s−1 and PAB is 50 nm s−1 in the uninhibited system, and 26-Se with tertiary amide functionality, where PPassive is 46 nm s−1 and PAB is 101 nm s−1 in the uninhibited system. The former is inhibitory toward P-gp while the latter is highly stimulating. The 40-fold difference in PPassive suggests that membrane concentrations of the former will be replenished much more slowly than the latter. If the available membrane concentrations are indeed low, then more will be required to be added to the whole cell system to raise these membrane concentrations effectively to levels that result in pump inhibition. For the membrane vesicle systems, this limitation is unlikely to exist since the favored membrane microenvironment should be more readily accessible due to inside-out nature of the membrane vesicle.
The role of the carboxylic acid oxidation state in the 9-position of the rosamine and rhodamine derivatives is important. For the “stimulating” drugs TMR, 1-S, 9-S, 10-S, 11-S, and 11-Se and the “inhibitory” compounds 2-S and 2-Se, which lack the carboxylic acid oxidation state, the % cell associated dye (% cell) in the uninhibited system is 14-47% and in the inhibited system is 53-71% with ratios between 1.5 and 5.1 for the uninhibited to inhibited systems (Table 2). The addition of a substituent with a carboxylic acid oxidation state in compounds 23-S, 25-S, 26-Se, 30-E, and 31-S gives a lower cell association in both the uninhibited (5.6-23%) and inhibited (4.8-30%) systems with ratios between 0.6 and 2.4 for the uninhibited to inhibited systems. These differences are not a simple function of lipophilicity since values of log P, the n-octanol/water partition coefficient, fall in the range of −0.09-2.4 for TMR, 1-S, 9-S, 10-S, 11-S, and 11-Se and −0.39-2.7 for compounds 23-S, 25-S, 26-Se, 30-E, and 31-S (Table 2). All compounds should have access to aqueous and lipophilic environments. For 31-S in particular, the % cell is essentially unchanged in the presence or absence of inhibitor with a ratio of 1.3 for the uninhibited (23% cell associated) to inhibited (30% cell associated) systems. Again, these results suggest that the thioamides and 31-S, in particular, have limited access to the pump.
Conclusions
We have demonstrated that simple structural changes in a series of rosamine/rhodamine structures can give molecules with high affinity for P-gp and that are highly stimulating for ATPase activity or molecules with high affinity and high inhibitory activity toward P-gp. Tertiary amide and thioamide groups on the 9-substituent, in particular, dictate ATPase stimulation for the former and inhibition for the latter. The amide/thioamide “switch” markedly decreases ATPase stimulation with an increase in affinity for the thioamides. The amide/thioamide “switch” also slows the rate of transport of the rhodamine derivatives in both absorptive and secretory directions in the cell. The substitution of a julolidyl fragment for a dimethylamino substituent in the xanthylium core also gives higher affinity for P-gp in both isolated protein and in whole cells.
Compound 31-S represents an interesting lead compound for the development of more potent inhibitors of P-gp. In particular, understanding the factors that limit the availability of 31-S to the pump in the native membrane environment is critical for the development of more potent rhodamine inhibitors of P-gp.
Experimental
General Methods
Dyes 1-E, 2-E, and 5-E through 13-E were prepared from xanthones 14-E41or 15-E42 as described in references 38-40. Dyes 22-E and 23-E were prepared according to reference 44. Compound 27-S was prepared according to reference 40. Tetrahydrofuran was distilled from sodium benzophenone ketyl prior to use. Reactions were run under Ar. Concentration in vacuo was performed on a Büchi rotary evaporator. NMR spectra were recorded on an Inova 500 instrument (500 MHz for 1H, 125 MHz for 13C) with residual solvent signal as internal standard. Infrared spectra were recorded on a Perkin-Elmer FTIR instrument. UV-vis near-IR spectra were recorded on a Perkin-Elmer Lambda 12 spectrophotometer or on a Shimadzu UV-3600 spectrophotometer in quartz cuvettes with a 1-cm path length. Melting points were determined with a Büchi capillary melting point apparatus and are uncorrected. New compounds have a purity of ≥ 95% as determined by elemental analyses for C, H, and N, which were performed by Atlantic Microlab, Inc., Norcross, GA. Experimental values of C, H, and N are within 0.4% of theoretical values.
3,6-Bis(N,N-dimethylamino)-9-(2-thienyl-5-carboxamido)thioxanthylium Hexafluorophosphate (25-S)
Dye 27-S40 (0.055 g, 0.10 mmol) and CDI (50 mg, 0.31 mmol) in 4.5 mL CH2Cl2 were stirred for 4 h. Gaseous NH3, generated by heating an NH4OH solution under a stream of N2, was bubbled through the reaction mixture for 1.0 h. Water (3 mL) was added to the reaction mixture and the products were extracted with CH2Cl2 (2 × 10 mL). The combined organic extracts were washed with 20 mL of saturated NaHCO3 solution (2×), and concentrated in vacuo. The crude dye was recrystallized from CH3CN/Et2O (2×) to give 0.045 g (83%) of 25-S as a purple solid, mp 239-242 °C: 1H NMR (CD3CN) δ 7.75 (d, 1H, J = 3.6 Hz), 7.58 (d, 2H, J = 9.6 Hz), 7.23 (d, 1H, J = 3.6 Hz), 7.19 (d, 2H, J = 2.8 Hz), 7.03 (d×d, 2H, J = 2.8, 9.6 Hz), 3.23 (s, 12H); 13C NMR (CD2Cl2) δ 163.5, 154.5, 152.0, 144.6, 143.3, 140.7, 136.4, 132.4, 129.5, 120.2, 116.7, 106.6, 41.0; λmax (H2O) 595 nm (ε 6.1 × 10−4 M−1 cm−1); HRMS-ES m/z 408.1202 (calcd for C22H22ON3S2: 408.1199).
3,6-Bis(N,N-dimethylamino)-9-(2-methylcarboxyphenyl)thioxanthylium Hexafluorophosphate (28-S)
Dye 27-S40 (0.055 g, 0.10 mmol) and CDI (50 mg, 0.31 mmol) in 4.5 mL CH2Cl2 were stirred for 4 h. Methanol (30 μL, 24 mg, 0.75 mmol) was added and the resulting mixture was stirred for 24 h. Water (3 mL) was added and products were extracted with CH2Cl2 (2 × 10 mL). The organic extracts were washed with saturated NaHCO3 (2 × 20 mL), dried, and concentrated in vacuo. The residue was recrystallized from CH3CN/Et2O to give 0.041 g (80%) of 28-S, mp 263-264 °C; 1H NMR (CD2Cl2) δ 8.32 (d×d, 1H, J = 1.6, 7.8 Hz), 7.82 (t×d, 1H, 1.6, 7.8 Hz), 7.76 (t×d, 1H, 1.6, 7.8 Hz), 7.31 (d×d, 1H, J = 1.6, 7.8 Hz), 7.22 (d, 2H, J = 9.6 Hz), 7.10 (d, 2H, J = 2.4 Hz), 6.88 (d×d, 2H, J = 2.4, 9.6 Hz), 3.59 (s, 3H), 3.26 (s, 12H); λmax (CH3OH) 571 nm (ε 74,000 M−1 cm−1); HRMS (ESI) m/z 417.1635 (Calcd for C25H25N2O2S2+: 417.1637).
3,6-Bis(N,N-dimethylamino)-9-(2-N,N-diethylcarboxamidophenyl)thioxanthylium Hexafluorophosphate (29-S)
tert-Butyllithium (2.2 mL of a 1.5 M solution, 3.3 mmol) was added dropwise to N,N-diethylbenzamide (0.53 g, 3.0 mmol) and tetramethylethylenediamine (TMEDA, 0.5 mL, 3.0 mmol) in THF (5 mL) cooled to −78 °C to generate a solution of N,N-diethyl 2-lithiobenzamide (37). Xanthone 14-S (0.10 g, 0.34 mmol) in 1 mL of THF was added immediately. The reaction mixture was stirred at ambient temperature for 14 h and then was cooled to 0 °C. Acetic acid (3 mL) was added followed by 10% aqueous HPF6 (10 mL) until a deep purple color was observed. Ice water (50 mL) was added, the purple precipitate was collected by filtration, the resulting solid was recrystallized from CH3CN/ether to yield 85 mg (42%) of 29-S as a green crystalline solid, mp >300°C; 1H NMR (CD3CN) δ 7.74 (m, 2H), 7.61 (m, 1H), 7.54 (m, 1H), 7.47 (m, 1H), 7.38 (d, 2H, J = 9.9 Hz), 7.24 (d, 2H, J = 2.8), 7.02 (d×d, 2H, J = 9.8, 2.8 Hz), 3.27 (s, 12H), 3.13 (q, 2H, J = 7.0Hz), 3.01 (q, 2H, J = 7.0 Hz) 1.08 (t, 3H, J = 6.7Hz), 0.507 (t, 3H, J = 6.7Hz); 13C NMR (CD2Cl2) δ 167.9, 158.9, 154.0, 144.4, 137.2, 133.8, 130.4, 130.0, 129.4, 129.4, 126.7, 119.7, 115.2, 105.7, 43.6, 40.9, 38.4, 14.1, 11.8, 2.0; λmax (EtOH) 578 nm (ε = 6.2 × 104 M−1 cm−1); HRMS (ESI) m/z 458.2261 (calcd for C28H32N3S+: 458.2266).
3,6-Bis(dimethylamino)-9-(N-piperidyl-2-thienyl-5-thiocarboxamido)thioxanthylium Hexafluorophosphate (30-S)
n-Butyllithium (2.5 M in hexane, 0.30 mL, 0.78 mmol) was added to a stirred solution of diisopropylamine (0.11 mL, 0.78 mmol) in 2.0 mL of anhydrous THF at 0 °C. After 15 min, the reaction mixture was cooled to −78°C and 38 (0.15 g, 0.71 mmol) in 1 mL of anhydrous THF was added. The resulting mixture was stirred for 0.5 h at −78 °C. An aliquot (1.0 mL, 0.22 mmol) of the 1-(5-lithio-2-thienyl)piperidin-1-ylmethanethione (40) solution was added to a solution of xanthone 14-S (50 mg, 0.17 mmol) in 1 mL of dry THF and the resulting mixture was warmed at 35 °C for 0.5 h, and then cooled to ambient temperature. Acetic acid (0.25 mL) was added, and the resulting mixture was then poured into a 10% wt./wt. solution of aqueous HPF6. After 1 h of stirring, the precipitate was collected by filtration, washed with water and ether, and recrystallized from CH3CN/Et2O to give 0.080 g (75%) of 30-S as a green solid, mp 264-266 °C; 1H NMR (CD3CN) δ 7.68 (d, 2H, J = 10.0 Hz), 7.45 (d, 1H, J = 4.0 Hz), 7.20 (d, 2H, J = 2.0 Hz), 7.12 (d, 1H, J = 3.5 Hz), 7.07 (d×d, 2H, J = 2.5, 9.5 Hz), 4.28 (br s, 2H), 4.01 (br s, 2H), 3.24 (s, 12H), 1.79 (m, 6H); 13C NMR (CD3CN) δ 188.7, 154.6, 152.2, 149.5, 144.7, 138.8, 136.6, 131.1, 126.1, 120.4, 116.7, 106.7, 41.1, 24.8; λmax (H2O) 595 nm (ε = 4.5 × 10-4 M−1 cm−1); HRMS (ESI) m/z 492.1586 (Calcd for C27H30N3S3+: 492.1596).
3,6-Bis(dimethylamino)-9-(N-piperidyl-2-thienyl-5-thiocarboxamido)selenoxanthylium Hexafluorophosphate (30-Se)
From xanthone 14-Se (50.0 mg, 0.145 mmol) and 0.22 M 40 (1.00 mL, 0.22 mmol), 30-Se was obtained as described for the preparation of 30-S. Yield: 80 mg (79%), mp 215-217 °C; 1H NMR (CD2Cl2) δ 7.76 (d, 2H, J = 9.5 Hz), 7.26 (d, 2H, J = 2.5 Hz), 7.21 (d, 1H, J = 3.5 Hz), 7.07 (d, 1H, J = 3.5 Hz), 6.95 (d×d, 2 H, J = 2.5, 9.8 Hz), 4.29 (br s, 2H), 4.02 (br s, 2H), 3.28 (s, 12H), 1.81 (br s, 6H); 13C NMR (DMSO-d6) δ 186.1, 152.2, 150.5, 146.9, 143.7, 139.0, 136.4, 129.6, 124.8, 118.7, 115.2, 109.1, 39.8, 23.0; λmax (H2O) 606 nm (ε = 4.8 × 104 M−1 cm−1); HRMS (ESI) m/z 540.1034 (Calcd for C27H30N3S280Se+: 540.1041).
3,6-Bisa(dimethylamino)-9-(N-piperidyl-2-thienyl-5-thiocarboxamido)telluroxanthylium Hexafluorophosphate (30-Te)
From xanthone 15-Te (50.0 mg, 0.127 mmol) and 0.19 M 40 (1.00 mL, 0.19 mmol), 30-Te was obtained as described for the preparation of 30-S. Yield: 0.090 g (94%), mp 156-158 °C; 1H NMR (CD2Cl2) δ 7.85 (d, 2H, J = 10.0 Hz), 7.51 (d, 2H, J = 2.5 Hz), 7.19 (d, 1H, J = 2.5 Hz), 7.02 (d, 1H, J = 3.5 Hz), 6.86 (d×d, 2H, J = 2.5, 10.0 Hz), 4.28 (br s, 2H), 4.04 (br s, 2H), 3.22 (s, 12H), 1.81 (br s, 6H); 13C NMR (DMSO-d6) δ 186.7, 153.5, 151.6, 147.0, 142.5, 139.2, 138.4, 129.6, 125.2, 121.3, 116.3, 115.5, 23.5; λmax (H2O) 621 nm (ε = 4.0 × 104 M−1 cm−1); HRMS (ESI) m/z 590.0918 (Calcd for C27H30N3S2130Te+: 590.0938).
12-(Dimethylamino)-2,3,6,7-tetrahydro-9-(N-Piperidyl-2-thienyl-5-thiocarboxamido)-1H,5H-thioxantheno[2,3,4-ij]quinolizin-14-ium Hexafluorophosphate (31-S)
From xanthone 15-S (50.0 mg, 0.143 mmol) and 0.19 M 40 (1.1 mL, 0.21 mmol), 31-S was obtained as described above for the preparation of 30-S. Yield: 0.060 g (62%), mp 169-171 °C; 1H (CD2Cl2) δ 7.65 (d, 1H, J = 9.5 Hz), 7.40 (s, 1H), 7.22 (d, 1H, J = 3.5 Hz), 7.09 (d, 1H, J = 2.5 Hz), 7.07 (d, 1H, J = 3.5 Hz), 6.98 (d×d, 1H, J = 2.5, 9.5 Hz), 4.28 (br s, 2H), 4.04 (br s, 2H), 3.56 (t, 4H, J = 5.5 Hz), 3.25 (s, 6H), 2.86 (t, 2H, J = 6.0 Hz), 2.78 (t, 2H, J = 6.0 Hz), 2.17 (quintet, 2H, J = 6.0 Hz), 2.01 (quintet, 2H, J = 6.0 Hz), 1.82 (s, 6H); 13C NMR (DMSO-d6) δ 186.1, 152.0, 148.3, 147.1, 147.0, 140.0, 137.9, 137.6, 133.6, 130.9, 129.8, 125.4, 125.0, 118.2, 117.1, 114.9, 113.4, 105.6, 50.5, 49.4, 26.7, 23.0, 19.2, 18.7; λmax (H2O) 607 nm (ε = 3.4 × 104 M−1 cm−1); HRMS (ESI) m/z 544.1905 (Calcd for C31H34N3S3+: 544.1909).
12-(Dimethylamino)-2,3,6,7-tetrahydro-9-(N-Piperidyl-2-thienyl-5-thiocarboxamido)-1H,5H-selenoxantheno[2,3,4-ij]quinolizin-14-ium Hexafluorophosphate (31-Se)
From xanthone 15-Se (50.0 mg, 0.126 mmol) and 1.9 M 40 (1.0 mL, 0.19 mmol), 31-Se was obtained as described above for the preparation of 30-S. Yield: 70 mg (31%), mp 175-176 °C; 1H (CD2Cl2) δ 7.64 (d, 1H, J = 9.5 Hz), 7.40 (s, 1H), 7.24 (s, 1H), 7.20 (d, 1H, J = 2.5 Hz), 7.03 (d, 1H, J = 3.0 Hz), 6.90 (d, 1H, J = 9.5 Hz), 4.27 (br s, 2H), 4.07 (br s, 2H), 3.54 (m, 4H), 3.23 (s, 6H), 2.75 (br s, 4H), 2.19 (br s, 2H), 2.00 (br s, 2H), 1.82 (br s, 6H); 13C NMR (CD2Cl2) δ 188.6, 152.6, 150.8, 149.6, 148.6, 143.4, 142.4, 140.8, 137.2, 135.2, 130.1, 125.9, 125.3, 121.0, 120.0, 117.3, 114.9, 108.8, 52.0, 51.1, 40.6, 28.1, 26.2, 24.5, 20.7, 20.3; λmax (H2O) 621 nm (ε = 2.9 × 104 M−1 cm−1); HRMS (ESI) m/z 592.1349 (Calcd for C31H34N3S280Se+: 592.1354).
12-(Dimethylamino)-2,3,6,7-tetrahydro-9-(N-Piperidyl-2-thienyl-5-thiocarboxamido)-1H,5H-telluroxantheno[2,3,4-ij]quinolizin-14-ium Hexafluorophosphate (31-Te)
From xanthone 15-Te (50.0 mg, 0.112 mmol) and 0.19 M 40 (0.80 mL, 0.15 mmol), 31-Te was obtained as described above for the preparation of 30-S. Yield: 50 mg (54%), mp 172-174 °C; 1H (CD2Cl2) δ 7.45 (d, 1H, J = 9.8 Hz), 7.51 (s, 1H), 7.47 (d, 1H, J = 2.5 Hz), 7.18 (d, 1H, J = 4.0 Hz), 7.00 (d, 1H, J = 3.5 Hz), 6.84 (d×d, 1H, J = 2.5, 9.8 Hz), 4.27 (br s, 2H), 4.07 (br s, 2H), 3.50 (m, 4H), 3.20 (s, 6H), 2.73 (t, 2H, J = 6.0 Hz), 2.61 (t, 2H, J = 6.0 Hz), 2.21 (quintet, 2H, J = 6.0 Hz), 2.00 (m, 2H), 1.82 (s, 6H); 13C NMR (DMSO-d6) δ 186.8, 151.5, 151.1, 148.1, 146.8, 143.0, 138.3, 136.5, 136.3, 134.1, 129.7, 125.3, 125.0, 121.9, 121.3, 120.6, 115.7, 114.9, 51.0, 50.3, 39.9, 29.6, 27.0, 23.5, 19.9; λmax (H2O) 635 nm (ε = 1.9 × 104 M−1 cm−1); HRMS (ESI) m/z 642.1231 (Calcd for C31H34N3S2130Te+: 642.1256).
12-(Dimethylamino)-2,3,6,7-tetrahydro-9-(N-Morpholino-2-thienyl-5-thiocarboxamido)-1H,5H-thioxantheno[2,3,4-ij]quinolizin-14-ium Hexafluorophosphate 32-S
n-Butyllithium (2.4 M in hexane, 300 μL, 0.72 mmol) was added to a stirred solution of diisopropylamine (109 μL, 0.774 mmol) in 2.0 mL of anhydrous THF at 0 °C. The solution was stirred for 15 min before cooling to −78 °C. Thioamide 39 (150 mg, 0.703 mmol) in 1 mL of anhydrous THF was added. The reaction mixture was stirred for 0.5 h to give a 0.22 M solution of 1-N-morpholino-1-(5-lithio-2-thienyl)methanethione 41. An aliquot (1.0 mL, 0.22 mmol) of this solution was added to xanthone 15-S (50.0 mg, 0.143 mmol) in 1 mL THF. The resulting mixture was heated at 35 °C for 0.5 h and cooled to room temperature. Workup and purification as described for the preparation of 30-S gave 32-S. Yield: 60 mg (57 %), mp 194-196 °C; 1H (CD2Cl2) δ 7.62 (d, 1H, J = 9.5 Hz), 7.38 (s, 1H), 7.24 (d, 1H, J = 3.5 Hz), 7.09 (m, 2 H), 6.98 (d×d, 1H, J = 2.5, 9.5 Hz), 4.26 (br s, 2H), 3.86 (br s, 2H), 3.56 (t, 4H, J = 5.5 Hz), 3.25 (s, 6H), 2.86 (t, 2H, J = 6.5 Hz), 2.78 (t, 2H, J = 6.5 Hz), 2.17 (quintet, 2H, J = 6.5 Hz), 2.01 (quintet, 2H, J = 6.5 Hz); 13C NMR (CD2Cl2) δ 189.9, 153.2, 149.8, 149.3, 148.2, 142.4, 140.1, 139.9, 135.3, 132.9, 130.5, 126.6, 126.1, 120.3, 119.2, 115.4, 114.5, 106.0, 100.4, 66.9, 52.0, 51.0, 40.7, 28.2, 24.4, 20.7, 20.2; λmax (H2O) 612 nm (ε = 2.6 × 104 M−1 cm−1); HRMS (ESI) m/z 546.1704 (Calcd for C30H32N3OS3+: 546.1702).
12-(Dimethylamino)-2,3,6,7-tetrahydro-9-(N-Morpholino-2-thienyl-5-thiocarboxamido)-1H,5H-selenoxantheno[2,3,4-ij]quinolizin-14-ium Hexafluorophosphate (32-Se)
From xanthone 15-Se (50.0 mg, 0.126 mmol) and a 0.20 M solution 41 (0.10 mL, 0.20 mmol), 32-Se was obtained as described above for the preparation of 32-S. Yield: 70 mg (70%), mp 171-172 °C; 1H (CD2Cl2) δ 7.62 (d, 1H, J = 9.5 Hz), 7.40 (s, 1H), 7.24 (d, 1H, J = 2.5 Hz), 7.22 (d, 1H, J = 4.0 Hz), 7.06 (d, 1H, J = 4.0 Hz), 6.91 (d×d, 1H, J = 2.5, 9.8 Hz), 4.25 (br s, 2H), 3.86 (br s, 2H), 3.55-3.52 (m, 4H), 3.23 (s, 6H), 2.77-2.75 (m, 4H), 2.19 (quintet, 2H, J = 6.5 Hz), 2.00 (quintet, 2H, J = 6.5 Hz); 13C NMR (CD2Cl2) δ 190.1, 152.8, 150.8, 149.7, 148.0, 143.5, 142.5, 141.7, 137.2, 135.3, 130.3, 126.1, 126.0, 121.0, 120.0, 117.4, 115.1, 108.9, 67.0, 52.1, 51.2, 40.7, 28.1, 26.3, 20.7, 20.4; λmax (H2O) 621 nm (ε = 2.9 × 104 M−1 cm−1); HRMS (ESI) m/z 594.1158 (Calcd for C30H32N3OS280Se+: 594.1147).
1-N-Piperidinyl-1-(2-thienyl)methanethione (38)
Piperidine (5.30 mL, 53.5 mmol), thiophene-2-carboxyaldehyde (1.40 mL, 17.8 mmol) and elemental sulfur (1.40 g, 44.6 mmol) in 12.0 mL of dry DMF were stirred at 110 °C for 0.5 h, cooled to ambient temperature, and poured into 60 mL of CH2Cl2. The resulting solution was washed with water (2 × 15 mL), brine (2 × 15 mL), dried over Na2SO4, and concentrated in vacuo. Chromatography (1:9 EtOAc/hexanes, then 1:1 EtOAc/hexanes) on SiO2 followed by recrystallization from EtOAc/hexanes gave 3.0 g (81 %) of 38 as yellow crystals, mp 83-84 °C (lit.71 mp 85-86 °C), 1H NMR (CDCl3) δ 7.35 (d×d, 1H, J = 1.5, 5.3 Hz), 7.03 (d×d, 1H, J = 1.5, 3.5 Hz), 6.93 (d×d, 1H, J = 3.5, 4.8 Hz), 4.26 (br s, 2H), 3.82 (br s, 2H), 1.75 (br m, 6H); LRMS (ESI) m/z 212.1 (calcd for C10H13NS2 + H+: 212.0).
1-N-Morpholino-1-(2-thienyl)methanethione (39)
From thiophene-2-carboxyaldehyde (1.90 mL, 20.0 mmol), elemental sulfur (1.60 g, 50.0 mmol), and morpholine (5.2 mL, 60 mmol), 3972 was obtained as described for the preparation of 38. Yield: 4.5 g (99%), mp 64-66 °C, 1H NMR (CDCl3) δ 7.35 (d×d, 1H, J = 1.5, 5.3 Hz), 7.03 (d×d, 1H, J = 1.5, 3.5 Hz), 6.93 (d×d, 1H, J = 3.5, 4.8 Hz), 4.26 (br s, 2H), 3.82 (br s, 2H), 1.75 (br m, 6H); LRMS (ESI) m/z 212.1 (calcd for C10H13NS2 + H+: 212.0).
Expression of P-gp, Purification and Measurement of ATPase Activity
The cDNA for human P-glycoprotein (P-gp)50 was modified to contain a 10-histidine tag at the COOH-terminal end (P-gp-His10) to facilitate purification by metal-chelate chromatography.51 Baby hamster kidney (BHK) cells were co-transfected with P-gp-His10 and pWL-neo (Stratagene, Cedar Creek, TX) and the transfected cells treated with the cytotoxic agent G418 as described previously.73 G418-resistant colonies were selected and clones over-expressing P-gp were identified by subjecting whole cell extracts to immunoblot analysis with a rabbit polyclonal antibody to P-gp.74 BHK cells over-expressing P-gp-His10 were then expanded and then used to purify the protein.
Histidine-tagged P-gp was isolated by nickel-chelate chromatography as described previously.51 A sample of the isolated histidine-tagged P-gp was mixed with an equal volume of 10 mg/ml sheep brain phosphatidylethanolamine (Type II-S, Sigma) that had been washed and suspended in TBS (Tris-buffered saline: 10 mM Tris/HCl, pH 7.4 and 150 mM NaCl). The sample was sonicated and ATPase activity measured in the absence of drug substrate or in the presence of various concentrations of rhodamine compounds. The samples were incubated for 30 min at 37 °C and the amount of inorganic phosphate released was determined.55
To test for inhibition of P-gp drug-stimulated ATPase activity, samples of P-gp-His10 in lipid were pre-incubated with various concentrations of the rhodamine compounds for 15 min at 20 °C. The ATPase reactions were then started by addition of ATPase reaction mix containing verapamil (final concentration of 0.4 mM) and ATPase activity determined as described above. Verapamil was used as a substrate to test for inhibition because it is one of the most potent activators of P-gp ATPase activity.54
Enhancement of Calcein-AM Uptake into MDCK-MDR1 Cells by Rhodamine Analogues
MDCK cells transfected with wild-type MDR1 (ABCB1) were obtained at passage number 12 from Dr. Piet Borst at The Netherlands Cancer Institute. Cell growth was maintained in Dulbecco’s Modified Eagle’s Medium (Gibco) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin and 100 g/ml streptomycin in 75-cm2 flasks. Cultures were passaged by trypsinization 1:10 twice a week and used at passage number 16-42. Cells were seeded at 40,000 cells/well in 96-well flat bottom plates (Falcon) using a medium volume of 200 μL, which was replaced on day 3 prior to their use on day 4.
Cells were washed once with Dulbecco’s phosphate-buffered saline containing 10 mM Hepes buffer at pH 7.4 (DPBSH) (Gibco) and incubated with solutions of the rhodamine analogue in DPBSH at 37 °C in room atmosphere. EC50 values were calculated from 1:1 serial dilution series. After 30 min, the test compound was replaced to include 0.5ug mL−1 CAM and incubated an additional 20 min. Calcein fluorescence was read on a Cytofluor series 4000 Multi Well Plate Reader (PerSeptive Biosystems) with λEX and λEM set at 485 nm and 530 nm, respectively. Negative (0.25% DMSO in DPBSH), and positive [2.5 M 42] controls were included in each plate. EC50 values were calculated from the serial dilution curves using GraphPad PRISM version 4.03 software. Briefly, compound concentration was plotted as log M concentration versus relative fluorescence units (rfu) and a sigmoidal dose-response (variable slope) analysis with no weighting or restrictions was applied.
Pgp-transport Studies Across MDCK-MDR1 Monolayers
MDCK-MDR1 cells that were seeded at 50,000 cells cm−2 onto 12-well (1.13 cm2 surface area) Transwell polycarbonate filters (Costar) were fed on days 3 and 5, and used on day 6. The upper and lower chamber volumes were 0.5 mL and 1.0 mL, respectively. Cells were rinsed 10 min in DPBSH at 37 °C with mixing on a nutator (Clay Adams). Cells were pre-incubated with 4.3 mg mL−1 bovine serum albumin (BSA) in DPBSH alone or containing 2.5 M 42. After 30 min, 5 M test compound in BSA/DPBSH with or without inhibitor was added to the donor chamber (0.5 mL upper or apical, 1.0 mL lower or basolateral). Initial donor samples were taken at t = 0. For apical-to-basolateral (AB) flux, D0 was taken from the mixing tube before addition to the cell monolayer. For basolateral-to-apical (BA) flux this sample was taken from the 12-well plate 10 min after transfer, but before cell wells were added. Samples were taken from both the donor and receiver chambers following a 1-h incubation at 37 °C with constant mixing by nutation. Cell monolayers were rinsed briefly two times using cold DPBS and extracted with 500 L methanol for 3 min. All samples were 200 L and transferred directly to a black-wall, flat bottom 96-well plate (Costar). The plate was centrifuged 5 min at 1500 rpm to reduce bubbles, then analyzed using a Spectromax M2 Fluorescent Plate reader (Molecular Devices) with λEX and λEM set at 584 nm and 612 nm, respectively.
Inhibition of Vinblastine Efflux in MDCKII-MDR1 Cells by 31-S
MDCKII-MDR1 cells were seeded onto Costar Transwell polycarbonate membranes and maintained as previously described. On day 6, cells were rinsed 1 × 10 min in DPBSH with nutation at 37 °C, and then conditioned in 0.0001, 0.01, 0.05, 0.1, 0.5, 1 or 10 M 31-S in BSA/DPBSH. After 30 min at 37 °C, 1.0 mL [3H]-VIN (0.25 Ci mL−1 from 0.1 mCi mL−1 EtOH stock) in appropriate 31-S solution was introduced to the basolateral chamber and 0.5 mL fresh 31-S was added to the apical chamber. Initial donor samples were taken from the basolateral chamber at t = 0. The apical solution was replaced every 10 min with fresh buffer and appearance of [3H]-VIN in the apical chamber was measured by scintillation counting. An IC50 was calculated using XLfit software.
Inhibition of Vinblastine Efflux in an Inside-out P-gp Membrane Vesicle Model by 31-S
The inhibition assay was conducted using 0.1 M [3H]-VIN (0.25 μCi mL−1) as a P-gp substrate. The accumulation of [3H]-VIN into P-gp membrane vesicles was inhibited by 17-S (0.01-10 μM), using 5 M 42 as a standard for complete P-gp inhibition. Previously, the inhibition of [3H]-VIN accumulation in P-gp membrane vesicles by 42 was shown to exhibit an IC50 value of (6.5 ± 1.1) nM and thus a complete inhibition of P-gp was assumed at 5 μM. The reaction mixtures contained 4 mM ATP, ATP with different concentrations of 17-S, ATP with 5 M 42, or 4 mM AMP-PNP. All reaction mixtures contained 0.1 M [3H]-VIN, the regenerating system including 100 μg mL−1 creatine kinase, 0.010 M creatine phosphate, and 0.010 M MgCl2. The reaction was started by addition of 30 μL reaction mixtures to vesicles (20 μg/20 μL) or 20 μL sucrose buffer. The reactions were incubated for 2.5 min at 37 °C with gentle agitation. The reactions were stopped by the addition of ice-cold sucrose buffer to wells and filtered onto a membrane filter (GF/B glass fiber plate soaked overnight in 10% FBS/0.25 M sucrose buffer, v/v) with a 96-channel cell harvester (PerkinElmer, Boston, MA). Radioactivity retained on the filter was determined by liquid scintillation counting.
Supplementary Material
Acknowledgments
This research was supported in part by NIH Grant T32 CA09363 (Postdoctoral Training Grant) to G.T. G.T. gratefully acknowledges Alan Senior, Robert Bambara, and Barbara Iglewski for encouragement and the use of their laboratory space and supplies. We thank Dr. Piet Borst at The Netherlands Cancer Institute for supplying the MDCKII-MDR1 cells, William Perry at Eli Lilly and Company for the human embryonic kidney (PEAKSTABLE) cells, and Anastasiya Sokolova at the University at Buffalo for technical assistance in the preparation of intermediates.
Footnotes
Abbreviations list: ABC, ATP-binding cassette; BSA, bovine serum albumin; CDI, carbonyl diimidazole; DABCO, 1,4-diaza[2.2.2]bicyclooctane; DMSO, dimethylsulfoxide; DPBSH, Dulbecco’s Hepes-containing phosphate-buffered saline; MDR, multidrug resistance; NBD, nucleotide binding domain; P-gp, P-glycoprotein; TMEDA, tetramethylethylenediamine
Supporting Information Available: Experimental procedures for the preparation of 3-S, 4-S, 5-Te, 6-Se, 7-Se, 7-Te, 8Se, 9-Te, 12-S, 17-S, 18-S, 19-S, 20-S, and 21-S and for the preparation of vesicles from human embryonic kidney (PEAKSTABLE) cells; calculations for vesicle inhibition studies; Table S1 for mouse MDR3 CL P-gp data; and Figure S1 showing the inhibition of [3H]-VIN efflux from MDCK-MDR1 with 31-S. These materials are available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Gottesman MM, Ling V. The molecular basis of multidrug resistance in cancer: The early years of P-glycoprotein research. FEBS Lett. 2006;580:998–1009. doi: 10.1016/j.febslet.2005.12.060. [DOI] [PubMed] [Google Scholar]
- (2).Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Ann. Rev. Pharmacol. Tox. 1999;39:361–368. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- (3).Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat. Rev. Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- (4).Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discovery. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- (5).Abolhoda A, Wilson AI, Ross H, Danenberg PV, Burt M, Scotto KW. Rapid activiation of MDR1 gene expression in human metastatic sarcoma after in vivo exposure to doxorubicin. Clinical Cancer Res. 1999;5:3352–3356. [PubMed] [Google Scholar]
- (6).Tsuruo T, Iida H, Tsukagoshi S, Sakurai Y. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cyto-toxicity of vincristine and vinblastine by verapamil. Cancer Res. 1981;41:1967–1972. [PubMed] [Google Scholar]
- (7).Loo TW, Clarke DM. Do drug substrates enter the common drug-binding pocket of P-glycoprotein through “gates”? Biochem. Biophys. Res. Commun. 2005;329:419–422. doi: 10.1016/j.bbrc.2005.01.134. [DOI] [PubMed] [Google Scholar]
- (8).Raub TJ. P-Glycoprotein recognition of substrates and circumvention through rational drug design. Mol. Pharmaceutics. 2006;3:3–25. doi: 10.1021/mp0500871. [DOI] [PubMed] [Google Scholar]
- (9).Dantzig AH, de Alwis DP, Burgess M. Considerations in the design and development of transport inhibitors as adjuncts to drug therapy. Adv. Drug Delivery Rev. 2003;55:133–150. doi: 10.1016/s0169-409x(02)00175-8. [DOI] [PubMed] [Google Scholar]
- (10).Sandor V, Fojo T, Bates SE. Future perspectives for the development of P-glycoprotein modulators. Drug Resistance Updates. 1998;1:190–200. doi: 10.1016/s1368-7646(98)80039-3. [DOI] [PubMed] [Google Scholar]
- (11).Mahon FX, Deininger MWN, Schultheis B, Chabrol J, Reiffers J, Goldman JM, Melo JV. Selection and characterization of BCR-ABL positive cell lines with differential sensitivity to the tyrosine kinase inhibitor STI571: diverse mechanisms of resistance. Blood. 2000;96:1070–1079. [PubMed] [Google Scholar]
- (12).de Jong MC, Slootstra JW, Scheffer GL, Schroeijers AB, Puijk WC, Dinkelberg R, Icool M, Broxterman HJ, Meloen RH, Scheper RJ. Peptide transport by the multidrug resistance protein MRP1. Cancer Res. 2001;61:2552–2557. [PubMed] [Google Scholar]
- (13).Al-Shawi MK, Polar MK, Omote H, Figler RA. Transition state analysis of the coupling of drug transport to ATP hydrolysis by P-glycoprotein. J. Biol. Chem. 2003;278:52629–52640. doi: 10.1074/jbc.M308175200. [DOI] [PubMed] [Google Scholar]
- (14).Eytan GD, Regev R, Hurwitz CD, Assaraf YG. Efficiency of P-glycoprotein-mediated exclusion of rhodamine dyes from multidrug-resistant cells is determined by their passive transmembrane movement rate. Eur. J. Biochem. 1997;248:104–112. doi: 10.1111/j.1432-1033.1997.00104.x. [DOI] [PubMed] [Google Scholar]
- (15).Lu P, Liu R, Sharom FJ. Drug transport by reconstituted P-glycoprotein in proteoliposomes. Effect of substrates and modulators, and dependence on bilayer phase state. Eur. J. Biochem. 2001;268:1687–1695. [PubMed] [Google Scholar]
- (16).Loetchutinat C, Saengkhae C, Marbeuf-Gueye C, Garnier-Suillerot A. New insights into the P-glycoprotein-mediated effluxes of rhodamines. Eur. J. Biochem. 2003;270:476–485. doi: 10.1046/j.1432-1033.2003.03403.x. [DOI] [PubMed] [Google Scholar]
- (17).Jansen WJM, Hulscher TM, van Ark-Otte J, Giacone G, Pinedo HM, Boven E. CPT-11 sensitivity in relation to the expression of P170-glycoprotein and multidrug resistance-associated protein. Br. J. Cancer. 1998;77:359–365. doi: 10.1038/bjc.1998.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Mahon FX, Deininger MWN, Schultheis B, Chabrol J, Reiffers J, Goldman JM, Melo JV. Selection and characterization of BCR-ABL positive cell lines with differential sensitivity to the tyrosine kinase inhibitor STI571: diverse mechanisms of resistance. Blood. 2000;96:1070–1079. [PubMed] [Google Scholar]
- (19).Litman T, Zeuthen T, Skovsgaard T, Stein WD. Competitive, non-competitive and cooperative interactions between substrates of P-glycoprotein as measured by its ATPase activity. Biochim. Biophys. Acta. 1997;1361:169–176. doi: 10.1016/s0925-4439(97)00027-6. [DOI] [PubMed] [Google Scholar]
- (20).Stein WD. Kinetics of the P-glycoprotein, the multidrug transporter. Exp. Physiol. 1998;83:221–232. doi: 10.1113/expphysiol.1998.sp004106. [DOI] [PubMed] [Google Scholar]
- (21).Martin C, Berridge G, Higgins CF, Mistry P, Charlton P, Callaghan R. Communication between multiple drug binding sites on P-glycoprotein. Mol. Pharm. 2000;58:624–632. doi: 10.1124/mol.58.3.624. [DOI] [PubMed] [Google Scholar]
- (22).Dey S, Ramachandra M, Pastan I, Gottesman MM, Ambudkar SV. Evidence for two nonidentical drug interaction sites in the human P-glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 1997;94:10594–10599. doi: 10.1073/pnas.94.20.10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Maki N, Hafkemeyer P, Dey S. Allosteric modulation of human P-glycoprotein. Inhibition of transport by preventing substrate translocation and dissociation. J. Biol. Chem. 2003;278:18132–18139. doi: 10.1074/jbc.M210413200. [DOI] [PubMed] [Google Scholar]
- (24).Shapiro AB, Ling V. Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. Eur. J. Biochem. 1997;250:130–137. doi: 10.1111/j.1432-1033.1997.00130.x. [DOI] [PubMed] [Google Scholar]
- (25).Shapiro AB, Ling V. Stoichiometry of rhodamine 123 transport to ATP hydrolysis by Pglycoprotein. Eur. J. Biochem. 1998;254:189–193. doi: 10.1046/j.1432-1327.1998.2540189.x. [DOI] [PubMed] [Google Scholar]
- (26).Pajeva IK, Wiese M. Human P-glycoprotein pseudoreceptor modeling: 3D-QSAR study on thioxanthene type multidrug resistance modulators. Quant. Struct.-Act. Relat. 2001;20:130–138. [Google Scholar]
- (27).Pajeva IK, Wiese M. Pharmacophore model of drugs involved in P-glycoprotein multidrug resistance: explanation of structural variety (hypothesis) J. Med. Chem. 2002;45:5671–5686. doi: 10.1021/jm020941h. [DOI] [PubMed] [Google Scholar]
- (28).Pajeva IK, Globisch C, Wiese M. Structure-function relationships of multidrug resistance P-glycoprotein. J. Med. Chem. 2004;47:2523–2533. doi: 10.1021/jm031009p. [DOI] [PubMed] [Google Scholar]
- (29).Loo TW, Bartlett MC, Clarke DM. Methanethiosulfonate derivatives of rhodamine and verapamil activate human P-glycoprotein at different sites. J. Biol. Chem. 2003;278:50136–50141. doi: 10.1074/jbc.M310448200. [DOI] [PubMed] [Google Scholar]
- (30).Seelig A. A general pattern for substrate recognition by P-glycoprotein. Eur. J. Biochem. 1998;251:252–261. doi: 10.1046/j.1432-1327.1998.2510252.x. [DOI] [PubMed] [Google Scholar]
- (31).Seelig A, Gatlik-Landwojtowicz E. Inhibitors of multidrug efflux transporters: their membrane and protein interactions. Mini-Rev. Med. Chem. 2005;5:135–151. doi: 10.2174/1389557053402693. [DOI] [PubMed] [Google Scholar]
- (32).Gatlik-Landwojtowicz E, Äänismaa P, Seelig A. Quantification and characterization of P-glycoprotein-substrate interactions. Biochemistry. 2006;45:3020–3032. doi: 10.1021/bi051380+. [DOI] [PubMed] [Google Scholar]
- (33).Äänismaa P, Gatlik-Landwojtowicz E, Seelig A. P-Glycoprotein senses its substrates and the lateral membrane packing density: consequences for the catalytic cycle. Biochemistry. 2008;47:10197–10207. doi: 10.1021/bi800209h. [DOI] [PubMed] [Google Scholar]
- (34).Crivori P, Reinach B, Pezzetta D, Poggesi I. Computational models for identifying potential P-glycoprotein substrates and inhibitors. Mol. Pharmaceutics. 2006;3:33–44. doi: 10.1021/mp050071a. [DOI] [PubMed] [Google Scholar]
- (35).Scala S, Akhmed N, Rao US, Paull K, Lan L-B, Dickstein B, Lee J-S, Elgemeie GH, Stein WD, Bates SE. P-glycoprotein substrates and antagonists cluster into two distinct groups. Mol. Pharmacol. 1997;51:1024–1033. doi: 10.1124/mol.51.6.1024. [DOI] [PubMed] [Google Scholar]
- (36).Lee JS, Paull K, Alvarez M, Hose C, Monks A, Grever M, Fojo AT, Bates SE. Rhodamine efflux patterns predict P-glycoprotein substrates in the National Cancer Institute drug screen. Mol. Pharmacol. 1994;46:627–638. [PubMed] [Google Scholar]
- (37).Tombline G, Urbatsch IL, Virk N, Muharemagic A, White LB, Senior AE. Expression, purification, and characterization of cysteine-free mouse P-glycoprotein. Arch. Biochem. Biophys. 2006;445:124–128. doi: 10.1016/j.abb.2005.11.004. [DOI] [PubMed] [Google Scholar]
- (38).Tombline G, Donnelly DJ, Holt JJ, You Y, Ye M, Gannon MK, Nygren CL, Detty MR. Stimulation of P-glycoprotein ATPase by analogues of tetramethylrosamine: coupling of drug binding at the “R” site to the ATP hydrolysis transition state. Biochemistry. 2006;45:8034–8047. doi: 10.1021/bi0603470. [DOI] [PubMed] [Google Scholar]
- (39).Tombline G, Holt JJ, Gannon MK, Donnelly DJ, Wetzel B, Sawada GA, Raub TJ, Detty MR. ATP occlusion by P-glycoprotein as a surrogate measure of drug coupling. Biochemistry. 2008;47:3294–3307. doi: 10.1021/bi7021393. [DOI] [PubMed] [Google Scholar]
- (40).Calitree BD, Donnelly DJ, Holt JJ, Gannon MK, II, Nygren C, Sukumaran DK, Autschbach J, Detty MR. Tellurium analogues of rosamine and rhodamine dyes: synthesis, structure, 125Te NMR, and heteroatom contributions to excitation energies. Organometallics. 2007;26:6248–6257. [Google Scholar]
- (41).Del Valle DJ, Donnelly DJ, Holt JJ, Detty MR. 2,7-Bis-N,N-dimethylaminochalcogenoxanthen-9-ones via electrophilic cyclization with phosphorus oxychloride. Organometallics. 2005;24:3807–3810. [Google Scholar]
- (42).Holt JJ, Calitree BD, Vincek J, Gannon MK, II, Detty MR. A microwave-assisted synthesis of julolidine-9-carboxamide derivatives and their conversion to chalcogenoxanthones via directed metalation. J. Org. Chem. 2007;72:2690–2693. doi: 10.1021/jo070086f. [DOI] [PubMed] [Google Scholar]
- (43).Ahn Y-H, Lee JS, Chang Y-T. Combinatorial rosamine library and application to in vivo glutathione probe. J. Am. Chem. Soc. 2007;129:4510–4511. doi: 10.1021/ja068230m. [DOI] [PubMed] [Google Scholar]
- (44).Gannon MK, II, Detty MR. Generation of 3- and 5-lithiothiophene-2-carboxylates via metal-halogen exchange and their addition reactions to chalcogenoxanthones. J. Org. Chem. 2007;72:2647–2650. doi: 10.1021/jo062370x. [DOI] [PubMed] [Google Scholar]
- (45).Brooks DW, Lu LD-L, Masamune S. C-Acylation under virtually neutral conditions. Angew. Chem., Int. Ed. Engl. 1979;18:72–74. [Google Scholar]
- (46).Katritzky AR, Witek RM, Rodriguez-Garcia V, Mohapatra PP, Rogers JW, Cusido J, Abdel-Fattah AAA, Steel PJ. Benzotriazole-assisted thioacylation. J. Org. Chem. 2005;70:7866–7881. doi: 10.1021/jo050670t. [DOI] [PubMed] [Google Scholar]
- (47).(a) Aghapoor K, Mohsenzadeh F, Kharalizader G, Darabi HR. The Willgerodt-Kindler reaction in water. High chemoselectivity of benzaldehydes over acetophenones. Monat. Chem. 2007;138:61–65. [Google Scholar]; Shibuya I, Taguchi Y, Tsuchiya T, Oishi A, Katoh E. Silver(I) ion-mediated desulfurization-condensation of thiocarbonyl compounds with several nucleophiles. Bull. Chem. Soc. Jpn. 1994;67:3048–3052. [Google Scholar]
- (48).Beak P, Brown RA. The tertiary amide as an effective director of ortho lithiation. J. Org. Chem. 1982;47:34–46. [Google Scholar]
- (49).Tombline GL, Urbatsch IL, Virk N, Muharemagic A, Bartholomew White L, Senior AE. Expression, purification, and characterization of cysteine-free mouse P-glycoprotein. Arch. Biochem. Biophys. 2006;445:124–128. doi: 10.1016/j.abb.2005.11.004. [DOI] [PubMed] [Google Scholar]
- (50).Loo TW, Clarke DM. Functional consequences of proline mutations in the predicted transmembrane domain of P-glycoprotein. J. Biol. Chem. 1993;268:3143–3149. [PubMed] [Google Scholar]
- (51).Loo TW, Clarke DM. Rapid purification of human P-glycoprotein mutants expressed transiently in HEK-293 cells by nickel-chelate chromatography and characterization of their drug-stimulated ATPase activities. J. Biol. Chem. 1995;270:21449–21452. doi: 10.1074/jbc.270.37.21449. [DOI] [PubMed] [Google Scholar]
- (52).Gibson SL, Holt JJ, Ye M, Donnelly DJ, Ohulchanskyy TY, You Y, Detty MR. Structure-activity studies of uptake and phototoxicity with heavy-chalcogen analogues of tetramethylrosamine in vitro in chemosensitive and multidrug-resistant cells. Biorg. Med. Chem. 2005;13:6394–6403. doi: 10.1016/j.bmc.2005.06.056. [DOI] [PubMed] [Google Scholar]
- (53).Wagner SJ, Skripchenko A, Donnelly DJ, Ramaswamy K, Detty MR. Chalcogenoxanthylium photosensitizers for the photodynamic purging of blood-borne viral and bacterial pathogens. Biorg. Med. Chem. 2005;13:5927–5935. doi: 10.1016/j.bmc.2005.07.035. [DOI] [PubMed] [Google Scholar]
- (54).Loo TW, Clarke DM. Identification of residues in the drug-binding domain of human P-glycoprotein – Analysis of transmembrane segment 11 by cysteine-scanning mutagenesis and inhibition by dibromobimane. J. Biol. Chem. 1999;274:35388–35392. doi: 10.1074/jbc.274.50.35388. [DOI] [PubMed] [Google Scholar]
- (55).Chifflet S, Torriglia A, Chiesa R, Tolosa S. A method for the determination of inorganic-phosphate in the presence of labile organic phosphate and high-concentrations of protein – application to LENS ATPases. Anal. Biochem. 1988;168:1–4. doi: 10.1016/0003-2697(88)90002-4. [DOI] [PubMed] [Google Scholar]
- (56).Evers R, Kool M, Smith AJ, van Deemter L, de Haas M, Borst P. Inhibitory effect of the reversal agents V-104, GF120918 and Pluronic L61 on MDR1Pgp-, MRP1- and MRP2-mediated transport. Br. J. Cancer. 2000;83:366–374. doi: 10.1054/bjoc.2000.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Troutman MD, Thakker DR. Rhodamine 123 requires carrier-mediated influx for its activity as a P-glycoprotein substrate in Caco-2 Cells. Pharm. Res. 2003;20:1192–1199. doi: 10.1023/a:1025096930604. [DOI] [PubMed] [Google Scholar]
- (58).Sawada GA, Barsuhn CL, Lutzke BS, Houghton ME, Padbury GE, Ho NFH, Raub TJ. Increased lipophilicity and subsequent cell partitioning decrease passive transcellular diffusion of novel, highly lipophilic antioxidants. J. Pharm. Exper. Therap. 1999;288:1317–1326. [PubMed] [Google Scholar]
- (59).Dantzig AH, Shepard RL, Law KL, Tabas L, Pratt S, Gillespie JS, Binkley SN, Kuhfeld MT, Starling JJ, Wrighton SA. Selectivity of the multidrug resistance modulator, LY335979, for P-glycoprotein and effect on cytochrome P450 activities. JPET. 1999;290:854–890. [PubMed] [Google Scholar]
- (60).Dantzig AH, Shepard RL, Cao J, Law KL, Ehlhardt WJ, Baughman TM, Bumol TF, Starling JJ. Reversal of P-glycoprotein-mediated multidrug resistance by a potent cyclopropyldibenzosuberane modulator, LY335979. Cancer Res. 1996;56:4171–4179. [PubMed] [Google Scholar]
- (61).Lever JE. An effect of ATP on the permeability of transport-competent plasma membrane vesicles from mouse fibroblasts. Biochem. Biophys. Res. Commun. 1977;79:1051–1058. doi: 10.1016/0006-291x(77)91111-1. [DOI] [PubMed] [Google Scholar]
- (62).Loo TW, Bartlett MC, Clarke DM. Substrate-induced conformational changes in the transmembrane segments of human P-glycoprotein. Direct evidence for the substrate-induced fit mechanism for drug binding. J. Biol. Chem. 2003;278:13603–13606. doi: 10.1074/jbc.C300073200. [DOI] [PubMed] [Google Scholar]
- (63).Grkovic S, Brown MH, Roberts NJ, Paulsen IT, Skurray RA. QacR is a repressor protein that regulates expression of the Staphylococcus aureus multidrug efflux pump QacA. J. Biol. Chem. 1998;273:18665–18673. doi: 10.1074/jbc.273.29.18665. [DOI] [PubMed] [Google Scholar]
- (64).Schumacher MA, Miller MC, Grkovic S, Brown MH, Skurray RA, Brennan RG. Structural mechanisms of QacR induction and multidrug recognition. Science. 2001;294:2158–2163. doi: 10.1126/science.1066020. [DOI] [PubMed] [Google Scholar]
- (65).Peters KM, Schuman JT, Skurray RA, Brown MH, Brennan RG, Schumacher MA. QacR-cation recognition is mediated by a redundancy of residues capable of charge neutralization. Biochemistry. 2008;47:8122–8129. doi: 10.1021/bi8008246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Becker J-P, Depret G, Van Bambeke F, Tulkens PM, Prévost M. Molecular models of human P-glycoprotein in two different catalytic states. BMC Structural Biology. 2009;9 doi: 10.1186/1472-6807-9-3. doi:10.1186/1472-6807-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Loo TW, Clarke DM. Location of the rhodamine-binding site in the human multidrug resistance P-glycoprotein. J. Biol. Chem. 2002;277:44332–44338. doi: 10.1074/jbc.M208433200. [DOI] [PubMed] [Google Scholar]
- (68).Rautio J, Humphreys JE, Webster LO, Balakrishnan A, Keogh JP, Kunta JR, Serabjit-Singh CJ, Polli JW. In vitro P-glycoprotein inhibition assays for assessment of clinical drug interaction potential for new drug candidates: A recommendation for probe substrates. Drug Metab. Dispos. 2006;34:786–792. doi: 10.1124/dmd.105.008615. [DOI] [PubMed] [Google Scholar]
- (69).Omote H, Al-Shawi MK. Interaction of transported drugs with the lipid bilayer and P-glycoprotein through a solvation exchange mechanism. Biophys. J. 2006;90:4046–4059. doi: 10.1529/biophysj.105.077743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Acharya P, O’Connor MP, Polli JW, Ayrton A, Ellens H, Bentz JP. Kinetic identification of membrane transporters that assist P-glycoprotein-mediated transport of digoxin and loperamide through a confluent monolayer of MDCKII-hMDR1 Cells. Drug Metab. Dispos. 2008;36:452–460. doi: 10.1124/dmd.107.017301. [DOI] [PubMed] [Google Scholar]
- (71).Katritzky AR, Witek RM, Rodriguez-Garcia V, Mohapatra PP, Rogers JW, Cusido J, Abdel-Fattah AAA, Steel PJ. Benzotriazole-assisted thioacylation. J. Org. Chem. 2005;70:7866–7881. doi: 10.1021/jo050670t. [DOI] [PubMed] [Google Scholar]
- (72).(a) Aghapoor K, Mohsenzadeh F, Kharalizader G, Darabi HR. The Willgerodt-Kindler reaction in water. High chemoselectivity of benzaldehydes over acetophenones. Monat. Chem. 2007;138:61–65. [Google Scholar]; (b) Shibuya I, Taguchi Y, Tsuchiya T, Oishi A, Katoh E. Silver(I) ion-mediated desulfurization-condensation of thiocarbonyl compounds with several nucleophiles. Bull. Chem. Soc. Jpn. 1994;67:3048–3052. [Google Scholar]
- (73).Loo TW, Bartlett MC, Clarke DM. Rescue of F508 and other misprocessed CFTR mutants by a novel quinazoline compound. Mol. Pharmaceutics. 2005;2:407–413. doi: 10.1021/mp0500521. [DOI] [PubMed] [Google Scholar]
- (74).Loo TW, Clarke DM. P-glycoprotein – associations between domains and molecular chaperones. J. Biol. Chem. 1995;270:21839–21844. doi: 10.1074/jbc.270.37.21839. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.