

Abstract
Introduction
Mutations in the gene that encodes filamin C, FLNC, represent a rare cause of a distinct type of myofibrillar myopathy (MFM).
Methods
We investigated an Italian patient by means of muscle biopsy, muscle and brain imaging and molecular analysis of MFM genes.
Results
The patient harbored a novel 7256C>T, p.Thr2419Met mutation in exon 44 of FLNC. Clinical, pathological and muscle MRI findings were similar to the previously described filaminopathy cases. This patient had, in addition, cerebellar ataxia with atrophy of cerebellum and vermis evident on brain MRI scan. Extensive screening failed to establish a cause of cerebellar atrophy.
Discussion
We report an Italian filaminopathy patient, with a novel mutation in a highly conserved region. This case raises the possibility that the disease spectrum caused by FLNC may include cerebellar dysfunction.
Keywords: filaminopathy, FLNC, myofibrillar myopathy, cerebellar ataxia, muscle MRI
1. Introduction
Myofibrillar myopathies (MFM) are a group of muscle diseases defined by the presence of distinctive muscle biopsy findings that include focal areas of myofibrillar disorganization, accumulation of myofibrillar degradation products, and ectopic expression of several proteins inside the muscle fibers1. Although the histological findings in MFM are by definition rather homogenous, the genetic causes as well as the phenotype are heterogeneous. At present, mutations in six genes that encode Z-disk associated proteins (desmin, alpha-B crystallin, myotilin, ZASP, filamin C, and Bag-3) are known to account for less than one-half of the MFM cases2. Some authors broaden the genetic spectrum of MFM to include myopathies caused by mutations in FHL1, PLEC and SEPN1, although specific clinical and histological features often distinguish these diseases. From a clinical point of view, MFMs can show predominantly distal or proximal muscle involvement, or mixed patterns. Sometimes associated findings include cardiac involvement or extramuscular manifestations such as peripheral neuropathy. Filamin-C mutations cause an MFM that typically manifests with late onset proximal weakness3, 4. In the majority of known cases of filaminopathy the onset of illness was in the early 40s (range 24 to 60)3, 5.
Filamins are a family of proteins that crosslink actin filaments into networks and anchor membrane proteins to the cytoskeleton. Three isoforms are encoded by the human genome: filamin A, B and C. Filamin C (or gamma-filamin) is a Z-disk protein whose expression is largely restricted to skeletal and cardiac muscle6. Only three mutations in the gene encoding filamin C, FLNC, associated with autosomal dominantly inherited MFM have been described so far. The first mutation, p.W2710X (c.8130G>A), was identified in the last FLNC exon7 in four German families who likely share a common founder3, and subsequently in three other MFM families at the Mayo Clinic8. Two other mutations have been reported in the seventh immunoglobulin-like repeat of the FLNC gene, a p.Val903_Thr933del (c.2997_3008del) and a p.Val899_Cys900ins/del in a German and a Chinese family, respectively5, 9. Recently two additional mutations have been described in the actin binding domain of FLNC in the N-terminal region of the protein, which are associated with a different phenotype, i.e. distal myopathy without MFM features10. While this manuscript was under review a frameshift deletion (c.5160delC) was reported to cause a distal myopathy with upper limb predominance and minor myofibrillar changes on muscle biopsy11.
Here we describe clinical, histological and muscle MRI data in an Italian patient with a complex phenotype characterized by pathologically confirmed MFM and concomitant ataxia with cerebellar atrophy. The molecular culprit in this case was a novel 7256C>T, p.Thr2419Met FLNC mutation.
2. Materials and methods
2.1. The affected family
A 70-year-old man was admitted to the Neurology Department of the Catholic University in Rome for investigations of slowly progressive walking difficulties that began 10 years previously. His parents were first cousins and were reported to be unaffected (they died at ages 95 and 92 years). His sister who died at age 78 did not show symptoms of a neuromuscular or cerebellar disorder. His 44-year-old son, who suffered from idiopathic epilepsy with generalized seizures, and his 75-year-old brother were examined and found not to show any of the signs and symptoms detected in the index case.
2.2. Muscle biopsy
10-μm thick cryostat sections were stained by routine histochemical techniques: hematoxylineosin, nicotinamide adenine dinucleotide dehydrogenase tetrazolium reductase (NADH-TR), succinate dehydrogenase, cytochrome-C oxidase, modified Gomori trichrome, alkaline phosphatase, periodic acid-Schiff, Congo red and ATPase pH 4.3, 4.6 and 9.4. Immunohistochemistry was performed on 6-μm thick sections using FITC-conjugated phalloidin (Sigma, St. Louis, MO, USA) and the following primary antibodies: monoclonal anti desmin (Chemicon, Temecula, CA, USA) diluted 1/100, monoclonal anti myotilin (Novocastra, Newcastle, UK) diluted 1/20, anti alpha-dystroglycan clone IIH6C4 (Upstate, Lake Placid, NY, USA) diluted 1/100, and monoclonal anti slow- and fast-myosin (Novocastra, Newcastle, UK) diluted 1/20. For electron microscopy, a specimen was fixed in 2.5% glutaraldehyde and processed by standard methods. Ultrathin skeletal muscle sections were analyzed using a Zeiss Libra 120 electron microscope.
2.3. Mutation detection
The strategy of genetic analysis in this family was based on direct sequencing of candidate genes selected with consideration of the complicated phenotype that included muscle pathology reminiscent of MFM and cerebellar dysfunction. The following myopathy-associated genes were analyzed: LMNA (lamin A/C), the (CCTG)n expansion in ZNF9 (zinc finger protein 9), DES (desmin), CRYAB (alpha-B crystallin), TTID (myotilin), LDB3 (ZASP), BAG3 (BCL2-associated athanogene 3), FLNC, ACTA1 (actin alpha 1), TPM2 and TPM3 (two tropomyosin genes), and MYH7 (myosin heavy chain 7, exons 30 to 40). In addition, a series of tests was performed to investigate the possible cause of cerebellar disease including analysis of the genes for spinocerebellar ataxia types 1, 2, 6, 7 and 17, POLG (mitochondrial polymerase gamma), and the CGG expansion in FMR-1 (FXTAS). Genomic DNA was extracted from anticoagulated blood of the index patient, his brother and his son and used for amplification of each coding exon with intronic primers constructed to amplify the exons and intron-exon boundaries of the listed genes (primer sequences are available on request). Resulting fragments were purified by MinElute™ 96 UF PCR purification kit (Qiagen Sciences, Gaithersburg, MD, USA) and sequenced in both directions using DyeTerminator™ Sequencing Protocol on an ABI 3100 Prism Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Mutation detection was accomplished by aligning results with the database sequences utilizing the Mutation Surveyor Software (SoftGenetics, State College, PA, USA). Negative controls were included as part of each run. Total mRNA was extracted from muscle biopsy tissue of the index patient using Trizol® Reagent (Gibco BRL, Grand Island, NY) and DNA contaminants were removed using Turbo DNA-free™ (Ambion, Austin, TX, USA). 0.5 microgram of total RNA was used for reverse transcription (RT) with SuperScript™ First-Strand Synthesis system (Invitrogen, Carlsbad, CA). The exon 44 region of FLNC was amplified with primers 44Rf (TCCAGCTCAGAGACGTAGCA) and 44Rr (ATTCACCACAAAGGGGCTCT) and sequenced.
Genetic studies were approved by the institutional review boards of each collaborating institution and were performed in accordance with the Declaration of Helsinki. All subjects gave written informed consent prior to participation.
3. Results
3.1. Clinical manifestations
At age 70 years, the patient was able to walk only with support and for short distances. His difficulties were due to both muscle weakness and trunk and gait ataxia. He could rise from a chair only with assistance. He had lumbar hyperlordosis, mild pes cavus and moderate calf hypertrophy. Examination also disclosed limb dysmetria and slightly dysarthric and hypernasal speech. The Romberg maneuver was negative. There was mild facial weakness. Eye movements were normal. Manual muscle strength testing revealed weakness of neck flexors, biceps brachialis and triceps muscles (MRC 4). Axial weakness was also evident. Muscle stretch reflexes were absent, and plantar responses were normal.
The patient's serum CK level was normal. EMG examination disclosed myopathic abnormalities and spontaneous activity characterized by fibrillation potentials and rare myotonic discharges. Motor and sensory nerve conduction studies and motor and sensory evoked potentials were normal. ECG disclosed a complete right bundle branch block, and echocardiogram documented mild left ventricle hypertrophy with normal ejection fraction that could be ascribed to hypertension. His forced vital capacity was 67% of the predicted value.
Lower limb muscle MRI showed marked involvement of paraspinal muscles (erector spinae in particular), vastus lateralis (with a thin peripheral rim of relative sparing), followed by less severe involvement of vastus intermedius and medialis (Figure 1 A–C). In the anterior compartment of the thigh, rectus femoris, sartorius and gracilis were relatively spared. In the posterior compartment, semimembranosus was more involved than semitendinosus, and biceps femoris short head was relatively spared. At calf level, soleus and gastrocnemius medialis were severely involved. Brain and spine MRI were performed and showed bilateral cerebellar hemisphere and vermis atrophy (Figure 1D).
Figure 1.

T1-weighted MRI images of pelvic and lower limb muscles (A–C) and sagittal T1-weighted brain MRI image (D). Note the prominent involvement of erector spinae at the pelvic level (A), vastus lateralis and intermedius, semimembranosus and biceps femoris long head at the thigh level (B), gastrocnemius medialis and soleus at the calf level (C), and the evident cerebellar atrophy (D).
There was no history of chronic alcohol use, exposure to toxic agents, celiac disease or autonomic failure. There were no vitamin deficiencies, and paraneoplastic onconeural antibodies (anti Hu, Yo, Ri, amphiphysin) were absent.
Clinical examination 3 years later showed deterioration of walking ability mainly due to worsening imbalance. Muscle weakness had extended to scapular and upper limb proximal (supra and infraspinatus MRC 3, deltoid MRC 4), and lower limb distal muscles (tibialis anterior and extensor hallucis longus MRC 4).
3.2. Muscle pathology
Biopsy of the deltoid muscle showed myopathic features with increased fiber size variation and internal nuclei. Infrequently, fibers contained a few rimmed vacuoles. Some fibers harbored small subsarcolemmal cytoplasmic bodies or small collections of rod bodies. On NADH-TR and oxidative enzyme staining, many fibers showed uneven staining. Rare COX-negative ragged red fibers and ring fibers were evident, and type 1 fibers were predominant. Immunoreaction with the anti alpha-dystroglycan antibody was normal. Abnormal accumulations of desmin and myotilin were present in many fibers; some of the inclusions displayed congophilia with a dotted pattern. In some cases, the desmin reaction was positive in a rim, surrounding subsarcolemmal, roughly triangular structures that could morphologically resemble caps. However, unlike cap structures, the subsarcolemmal areas which appeared hyaline-like on hematoxylin-eosin and blue with NADH-TR contained slow-myosin as judged by their positive staining with anti slow-myosin heavy chain antibody, but they lacked myosin-ATPase activity (Figure 2).
Figure 2.

Light microscopy of a muscle biopsy sample from the index patient. (A) Hyaline-like peripheral inclusion with a basophilic rim (asterisk); (B) Arrows indicate small pleomorphic aggregates that stain dark green with Gomori Trichrome; (C) NADH-TR showing fibers with unevenness of stain; (D) Desmin immunoreactivity in central regions of some fibers, often with a serpiginous appearance (arrows); some subsarcolemmal structures are diffusely stained (asterisk), and some appear as surrounded by a positive rim (“cap-like” appearance - arrowhead); (E) The same structures are visible with double immunolabeling for desmin (red) and phalloidin (green); (F) Myotilin accumulations are present in a number of fibers; (G) ATPase pH 4.3 showing type 1 fiber predominance and focal absence of the reaction in some subsarcolemmal regions, corresponding to areas that still maintain immunoreactivity with the anti slow-myosin heavy chain antibody (H, arrowhead); (I) Congo red under Texas Red optics showing a round inclusion with dotted congophilia.
Electron microscopy (EM) on the available transverse sections (Figure 3) showed subsarcolemmal regions composed of dissolved myofilaments, including thick filaments and sarcomeric remnants, which likely correspond to the areas that were reactive for slow-myosin without ATPase activity. Subsarcolemmal accumulations of mitochondria, Z-disk streaming (that in some regions constituted true minicores) and focal accumulations of dense and filamentous material were present as well. One nucleus displayed a deposit of thin filaments and Z-disk density material (Figure 3F).
Figure 3.

Ultrastructural findings of a muscle biopsy sample from the index patient. (A) A subsarcolemmal triangular area of dissolved myofilaments; some sarcomeric remnants are still visible at a higher magnification in the insert; (B) Focal sarcomeric disruption and deposits of dense and filamentous material; (C) Small focus of Z-line streaming; (D) Myeloid structures among apparently intact myofibers; (F) Nuclear accumulation of filamentous and Z-disk density material (higher magnification in E), possibly deriving from the sarcoplasm through nuclear membrane leakage (arrowhead).
3.3. Analysis of gene sequences and mutation screening
Analysis of DNA sequences of coding exons in studied genes resulted in the identification of a heterozygous C>T substitution at the c.7256 position within exon 44 of FLNC corresponding to a predicted p.Thr2419Met mutation (Figure 4). No other known or unknown variations in genes associated with myopathy or cerebellar dysfunction were encountered. The mutation was found in the genomic DNA of the patient and his unaffected son, but not the unaffected brother. Sequencing of the patient's RT-PCR fragment of exon 44 that was amplified from mRNA extracted from a biopsied muscle confirmed the presence of the c.7256 C>T mutation at the RNA level. No sequence irregularities in FLNC exon 44 were identified in 232 control individuals of the same ethnic background and 100 additional controls from other European countries (664 chromosomes). In addition, this change was not observed in the latest 1000 genome data release which contains data from 629 control individuals 12. The Thr2419 residue is located within the FLNC immunoglobulin-like repeat 22 that is highly conserved in evolution (Figure 4). The variant is located outside the region of homology of the transcribed FLNC with the recently identified FLNC-like pseudogene 13.
Figure 4.
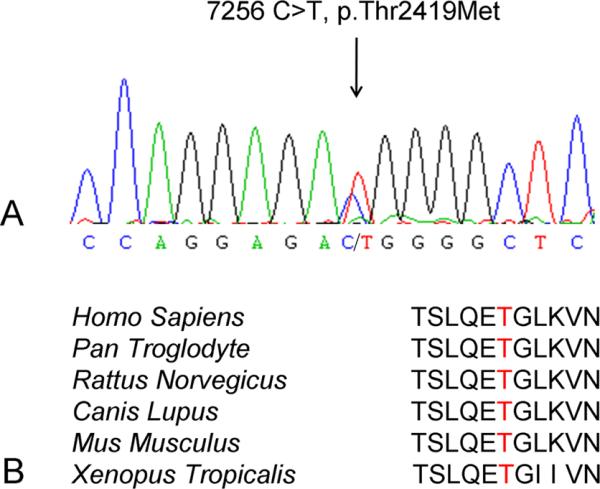
Sequence analysis of the FLNC gene fragments. (A) Chromatogram of a nucleotide sequence demonstrating the c.7256 C>T missense mutation in exon 44 of the FLNC gene. (B) Amino acid sequence alignment of a fragment of filamin C 22nd Ig-like repeat showing that the threonine (highlighted) is conserved through evolution from Xenopus to humans.
4. Discussion
This patient who harbors the novel FLNC mutation presented with a late onset slowly progressive axial and proximal myopathy, mild nasal speech, mild restrictive respiratory insufficiency, arrhythmogenic heart disease with a right bundle branch block and ataxia. Muscle biopsy findings were suggestive of MFM, with evidence of abnormal protein aggregation detectable with histochemical and immunohistochemical stainings, and myofibrillar disorganization observed in many areas on EM. Typical granulofilamentous material was not detected on EM, however, its absence could be due to the availability of only a small muscle fragment for EM analysis. Unusually for an MFM, along with the classical intracellular inclusions, some areas of myofibrillar degradation assumed a subsarcolemmal, triangular “cap-like” appearance. Unlike caps, these areas also displayed slow-myosin immunoreactivity, which has been reported in hyaline bodies. All of the genes known to be associated with cap myopathy (TPM2, TPM3, ACTA1) 14–16 and hyaline body myopathy (MYH7) 17 have been screened with negative results in our patient. Another interesting feature is the peculiar intranuclear accumulation of filamentous and Z-disk density material. Whereas the presence of filamentous or tubulofilamentous material inside the nuclei has been described in various diseases (such as inclusion body myositis, oculopharyngeal muscular dystrophy 18 and myotilinopathy 19), the occurrence of intranuclear Z-disk derived material (in the shape of rods) is generally considered a distinctive feature of nemaline myopathies with ACTA1 mutations 20. Interestingly however, small intranuclear rods can be found occasionally in MFMs 19, thus giving rise to interesting theories concerning their origin. While intranuclear rods associated with ACTA1 mutations are known to originate from abnormal aggregation of nuclear actin 20, the detected accumulation in our case could derive from the cytoplasm through a nuclear membrane rupture (or a physiological opening). This perturbation in the nuclear structure and eventual function may represent a mechanism that contributes to muscle damage.
MFM is known to be associated with mutations in one of six genes: DES, MYOT, ZASP, CRYAB, BAG3, or FLNC 1. The overall phenotypic profile of a late-onset myopathy involving predominantly proximal muscles, the suggestive muscle imaging pattern and the identification of a non-synonymous mutation in the highly conserved through evolution rod domain of the patient's FLNC gene at the genomic and RNA levels, together with the absence of this mutation in healthy unrelated controls, strongly indicates that the patient is affected with filaminopathy. Filaminopathy is a rare subtype of MFM worldwide, although the disease frequency is likely to be higher in Germany due to a founder effect 3. The distribution of muscle weakness in the filaminopathy cases published so far is strikingly homogenous as compared with the other MFMs. There is early involvement of proximal lower and upper limb muscles, followed by distal lower limb muscles, and there is often associated axial and abdominal weakness 3, 5, 9. Our patient showed a typical limb-girdle phenotype and a consistent muscle imaging pattern of involvement that perfectly matches the previously described filaminopathy characteristics 21. In the lower limbs, muscle weakness was not very pronounced, although the involvement was clearly seen on muscle imaging. Muscle imaging findings are very homogeneous in the reported filaminopathy cases, and the pattern of fatty replacement helps to distinguish filaminopathy from other MFMs 22. At variance with other MFMs, peripheral neuropathy is uncommon, while myotonic discharges on EMG and cardiomyopathy, both arrhythmogenic and dilated, are frequent features 3. The patient's son harbors the same mutation, but he belongs to an age group below the maximal age of disease onset. Periodic follow-up may reveal emergence of the disease.
Almost concomitantly with the skeletal and cardiac muscle condition, the patient developed progressive gait and trunk ataxia with a brain MRI scan indicating cerebellar atrophy, which is not explained by the investigated acquired or genetic etiologies. Cerebellar atrophy is a finding that has never been reported to accompany filaminopathy or other subtypes of MFM. The possibility that the patient is affected by some other myopathy known to be associated with central nervous system involvement, such as mitochondrial myopathies or alpha-dystroglycanopathies, has been ruled out by histopathological analysis. In addition, our patient was screened and found to be negative for the most prevalent spinocerebellar ataxias in the Italian population, Fragile X-associated Tremor/Ataxia Syndrome, and ataxia due to mitochondrial polymerase gamma mutations. Other causes of late-onset ataxia including alcoholism, vitamin deficiencies, celiac disease and paraneoplastic syndromes have been excluded as well. We could not exclude that our patient is affected by an autosomal recessive cerebellar ataxia (ARCA), which can be a possibility considering that the patient's parents were reported to be first cousins. ARCAs are a genetically heterogeneous group of rare disorders that generally present with a complicated phenotype and an earlier onset than occurred in our patient 23. However, some of them can have onset in the early forties, and a relatively pure phenotype has been described recently for ARCA1 due to mutations in the giant nuclear gene SYNE1 24 in only Quebec families to date.
On the other hand, one should consider the intriguing perspective that the FLNC mutation is responsible for both muscle and cerebellar diseases. Mutations in FLNA, another filamin gene, are associated with periventricular heterotopia, a disorder of cerebral cortex neuron migration25, and the possibility that C-filaminopathy represents a multisystem disorder involving tissues other than skeletal and cardiac muscle is also suggested by the presence of chronic diarrhea in the Chinese patients9. However, although FLNC transcript has been demonstrated in the fetal brain in one study26, this remains a very controversial finding27, 28, and it is generally accepted that filamin C expression is limited to striated muscle6.
In conclusion, we report a novel FLNC mutation causing proximal MFM, the fourth described to date and the first in a non-German European patient. Our report confirms previous findings that the muscle phenotype associated with FLNC mutations is consistent, and we describe the coexistence of filaminopathy-associated muscle disease with adult-onset cerebellar ataxia of unknown etiology. The possible association between these two diseases deserves further study.
Acknowledgements
This work was supported in part by the Intramural Research Program of the National Institute of Neurological Disorders and Stroke, National Institutes of Health. We want to thank the patient and his family for participation to the study and Manuela Papacci for technical assistance. We thank Dr Nigel Laing, University of Western Australia, for analysis of exons 30–40 of MYH7.
ABBREVIATIONS
- EM
electron microscopy
- MFM
myofibrillar myopathy
REFERENCES
- 1.Selcen D. Myofibrillar myopathies. Neuromuscul Disord. 2011;21:161–171. doi: 10.1016/j.nmd.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Selcen D, Engel AG. Myofibrillar Myopathy. In: Pagon RA, Bird TD, Dolan CR, Stephens K, editors. GeneReviews. University of Washigton; Seattle: 1993–2010. Internet. [PubMed] [Google Scholar]
- 3.Kley RA, Hellenbroich Y, van der Ven PF, Furst DO, Huebner A, Bruchertseifer V, et al. Clinical and morphological phenotype of the filamin myopathy: a study of 31 German patients. Brain. 2007;130:3250–3264. doi: 10.1093/brain/awm271. [DOI] [PubMed] [Google Scholar]
- 4.Schroder R, Schoser B. Myofibrillar myopathies: a clinical and myopathological guide. Brain Pathol. 2009;19:483–492. doi: 10.1111/j.1750-3639.2009.00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shatunov A, Olive M, Odgerel Z, Stadelmann-Nessler C, Irlbacher K, van Landeghem F, et al. In-frame deletion in the seventh immunoglobulin-like repeat of filamin C in a family with myofibrillar myopathy. Eur J Hum Genet. 2009;17:656–663. doi: 10.1038/ejhg.2008.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van der Flier A, Sonnenberg A. Structural and functional aspects of filamins. Biochim Biophys Acta. 2001;1538:99–117. doi: 10.1016/s0167-4889(01)00072-6. [DOI] [PubMed] [Google Scholar]
- 7.Vorgerd M, van der Ven PF, Bruchertseifer V, Lowe T, Kley RA, Schroder R, et al. A mutation in the dimerization domain of filamin c causes a novel type of autosomal dominant myofibrillar myopathy. Am J Hum Genet. 2005;77:297–304. doi: 10.1086/431959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Selcen D, Carpen O. The Z-disk diseases. Adv Exp Med Biol. 2008;642:116–130. doi: 10.1007/978-0-387-84847-1_10. [DOI] [PubMed] [Google Scholar]
- 9.Luan X, Hong D, Zhang W, Wang Z, Yuan Y. A novel heterozygous deletion-insertion mutation (2695–2712 del/GTTTGT ins) in exon 18 of the filamin C gene causes filaminopathy in a large Chinese family. Neuromuscul Disord. 2010;20:390–396. doi: 10.1016/j.nmd.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 10.Duff RM, Tay V, Hackman P, Ravenscroft G, McLean C, Kennedy P, et al. Mutations in the N-terminal actin-binding domain of filamin C cause a distal myopathy. Am J Hum Genet. 2011;88:729–740. doi: 10.1016/j.ajhg.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guergueltcheva V, Peeters K, Baets J, Ceuterick-de Groote C, Martin JJ, Suls A, et al. Distal myopathy with upper limb predominance caused by filamin C haploinsufficiency. Neurology. 2011;77:2105–2114. doi: 10.1212/WNL.0b013e31823dc51e. [DOI] [PubMed] [Google Scholar]
- 12.Altshuler D, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Odgerel Z, van der Ven PF, Furst DO, Goldfarb LG. DNA sequencing errors in molecular diagnostics of filamin myopathy. Clin Chem Lab Med. 2010;48:1409–1414. doi: 10.1515/CCLM.2010.272. [DOI] [PubMed] [Google Scholar]
- 14.Lehtokari VL, Ceuterick-de Groote C, de Jonghe P, Marttila M, Laing NG, Pelin K, et al. Cap disease caused by heterozygous deletion of the beta-tropomyosin gene TPM2. Neuromuscul Disord. 2007;17:433–442. doi: 10.1016/j.nmd.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 15.Ohlsson M, Fidzianska A, Tajsharghi H, Oldfors A. TPM3 mutation in one of the original cases of cap disease. Neurology. 2009;72:1961–1963. doi: 10.1212/WNL.0b013e3181a82659. [DOI] [PubMed] [Google Scholar]
- 16.Hung RM, Yoon G, Hawkins CE, Halliday W, Biggar D, Vajsar J. Cap myopathy caused by a mutation of the skeletal alpha-actin gene ACTA1. Neuromuscul Disord. 2010;20:238–240. doi: 10.1016/j.nmd.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 17.Tajsharghi H, Thornell LE, Lindberg C, Lindvall B, Henriksson KG, Oldfors A. Myosin storage myopathy associated with a heterozygous missense mutation in MYH7. Ann Neurol. 2003;54:494–500. doi: 10.1002/ana.10693. [DOI] [PubMed] [Google Scholar]
- 18.Engel AG, Franzini-Armstrong C. Myology. third edition Vol. 2. McGraw-Hill; 2004. p. 1151.p. 1373. [Google Scholar]
- 19.Claeys KG, Fardeau M, Schroder R, Suominen T, Tolksdorf K, Behin A, et al. Electron microscopy in myofibrillar myopathies reveals clues to the mutated gene. Neuromuscul Disord. 2008;18:656–666. doi: 10.1016/j.nmd.2008.06.367. [DOI] [PubMed] [Google Scholar]
- 20.Domazetovska A, Ilkovski B, Cooper ST, Ghoddusi M, Hardeman EC, Minamide LS, et al. Mechanisms underlying intranuclear rod formation. Brain. 2007;130:3275–3284. doi: 10.1093/brain/awm247. [DOI] [PubMed] [Google Scholar]
- 21.Fischer D, Kley RA, Strach K, Meyer C, Sommer T, Eger K, et al. Distinct muscle imaging patterns in myofibrillar myopathies. Neurology. 2008;71:758–765. doi: 10.1212/01.wnl.0000324927.28817.9b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wattjes MP, Kley RA, Fischer D. Neuromuscular imaging in inherited muscle diseases. Eur Radiol. 2010;20:2447–2460. doi: 10.1007/s00330-010-1799-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manto M, Marmolino D. Cerebellar ataxias. Curr Opin Neurol. 2009;22:419–429. doi: 10.1097/WCO.0b013e32832b9897. [DOI] [PubMed] [Google Scholar]
- 24.Gros-Louis F, Dupre N, Dion P, Fox MA, Laurent S, Verreault S, et al. Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat Genet. 2007;39:80–85. doi: 10.1038/ng1927. [DOI] [PubMed] [Google Scholar]
- 25.Fox JW, Lamperti ED, Eksioglu YZ, Hong SE, Feng Y, Graham DA, et al. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron. 1998;21:1315–1325. doi: 10.1016/s0896-6273(00)80651-0. [DOI] [PubMed] [Google Scholar]
- 26.Xie Z, Xu W, Davie EW, Chung DW. Molecular cloning of human ABPL, an actin-binding protein homologue. Biochem Biophys Res Commun. 1998;251:914–919. doi: 10.1006/bbrc.1998.9506. [DOI] [PubMed] [Google Scholar]
- 27.Maestrini E, Patrosso C, Mancini M, Rivella S, Rocchi M, Repetto M, et al. Mapping of two genes encoding isoforms of the actin binding protein ABP-280, a dystrophin like protein, to Xq28 and to chromosome 7. Hum Mol Genet. 1993;2:761–766. doi: 10.1093/hmg/2.6.761. [DOI] [PubMed] [Google Scholar]
- 28.Chiang W, Greaser ML, Lyons GE. Filamin isogene expression during mouse myogenesis. Dev Dyn. 2000;217:99–108. doi: 10.1002/(SICI)1097-0177(200001)217:1<99::AID-DVDY9>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]