

Abstract
The availability of suitable animal models and of the opportunity to record electrophysiologic data in movement disorder patients undergoing neurosurgical procedures has allowed researchers to investigate parkinsonism-related changes in neuronal firing patterns in the basal ganglia and associated areas of thalamus and cortex. These studies have shown that parkinsonism is associated with increased activity in the basal ganglia output nuclei, along with an increase in burst discharges, oscillatory firing, and synchronous firing patterns throughout the basal ganglia. Computational approaches have the potential to play an important role in the interpretation of these data. Such efforts can provide a formalized view of neuronal interactions in the network of connections between basal ganglia, thalamus and cortex, allow for the exploration of possible contributions of particular network components to parkinsonism, and potentially result in new conceptual frameworks and hypotheses that can be subjected to biological testing. It has proven very difficult, however, to integrate the wealth of the experimental findings into coherent models of the disease. In this review, we provide an overview of the abnormalities in neuronal activity that have been associated with parkinsonism. Subsequently, we discuss some particular efforts to model the pathophysiologic mechanisms that may link abnormal basal ganglia activity to the cardinal parkinsonian motor signs and may help explain the mechanisms underlying the therapeutic efficacy of deep brain stimulation for Parkinson’s disease. We emphasize the logical structure of these computational studies, making clear the assumptions from which they proceed and the consequences and predictions that follow from these assumptions.
Keywords: Parkinson’s disease, dopamine, striatum, globus pallidus, subthalamic nucleus, deep brain stimulation, burst, oscillation, synchrony
1. Motivation
There is little doubt that degeneration of the dopaminergic innervation of the basal ganglia (BG) is the essential pathologic defect that results in the motor signs of Parkinson’s disease (PD), which include akinesia, bradykinesia, rigidity and tremor. A major advance toward understanding how this local loss of dopamine leads to the genesis of parkinsonian signs came with the discovery that abnormalities in the discharge of BG output neurons constitute a critical intermediate step in the pathophysiology of PD. However, wide gaps remain in our understanding. One serious impediment to further progress is the fundamental challenge of understanding how the firing patterns of large populations of neurons influence neuronal network function.
Computational models provide a way to formalize and quantify otherwise vague concepts of neuronal network function and how abnormalities in neuronal firing, like those observed in PD, may disrupt network function. One might argue that the only way to truly “understand” the pathophysiology of PD is to model it computationally.
In this review, we provide an overview of the abnormalities in neuronal activity that have been associated with PD and then discuss efforts to model some particular pathophysiologic mechanisms that may translate abnormal patterns of neuronal activity in the BG into the cardinal signs associated with that disorder, as well as how this process may be affected by deep brain stimulation (DBS).
2. Anatomy and pathology in parkinsonism
The BG are a group of heavily interconnected subcortical nuclei (Alexander et al., 1990) (Fig. 1a). The striatum, the primary receptive nucleus of the BG, receives afferent projections from almost all areas of the neocortex, from specific nuclei of the thalamus, and from dopaminergic neurons of the substantia nigra compacta (SNc). The neocortical projections are organized into parallel anatomically-segregated pathways for skeletomotor, oculomotor, associative, and limbic regions of cortex and the striatum (Alexander et al., 1990). A second important input pathway into the BG arises from pre-central cortical areas and terminates topographically in the subthalamic nucleus (STN).
Figure 1.

Circuit diagram of the BG and changes in discharge rate predicted by the standard ‘rate model’ of PD. (a) The basic loop circuit includes an excitatory glutamatergic (black arrow) projection from the neocortex to the striatum (putamen) and then inhibitory (γ-amino butyric acid-containing; GABAergic, gray lines) striatal projection (the ‘direct’ pathway) to the GPi. GABAergic neurons in GPi project to targets in the thalamus [VLa and centromedian nucleus (CM), a posterior intralaminar nucleus] and the brainstem (PPN). The VLa thalamus projects to the frontal cortex including parts of the premotor and primary motor cortex. Only principal pathways are shown for the internal connectivity of the BG. Direct and indirect pathways start in projection neurons of the putamen that express D1- and D2-type dopamine receptors, respectively. D2-type neurons project to GPe. GPe projects to the STN and GPi. STN also receives monosynaptic glutamatergic input from the motor cortices and projects to GPi and GPe. Dopaminergic neurons of the SNc innervate the striatum and, less densely, the GP and STN (not shown). (b) Changes in mean discharge rate that the ‘rate model’ predicts will result from degeneration of dopamine neurons of the SNc and their terminals in the putamen. The thickness of lines indicates the predicted changes in discharge rate. The diagram does not show parkinsonism-related changes for anatomical connections that are not part of the standard rate model (e.g., corticostriatal and PPN projections). See text for abbreviations.
The primary output projections from the BG are GABAergic efferents arising from neurons in the internal segment of the globus pallidus (GPi) and the substantia nigra pars reticulata (SNr). These efferents terminate in specific nuclei of the thalamus [the anterior portion of the VL nucleus (VLa), the ventral anterior nucleus of the thalamus (VA) and intralaminar nuclei] (Yoshida et al., 1972; DeVito & Anderson, 1982) and in midbrain nuclei such as the pedunculopontine nucleus (PPN) and superior colliculus. BG efferent neurons have high spontaneous discharge rates in neurologically-normal animals at rest [mean firing rates of 40–80 Hz (DeLong, 1971; Wichmann et al., 1999; Starr et al., 2005)], which are thought to produce a tonic inhibition of their targets in the thalamus and midbrain.
Similar to the organization of BG input, efferent projections from the BG show an anatomically-segregated functional organization such that distinct regions of the GPi and SNr project to skeletomotor-, oculomotor-, associative-, and limbic-related regions of the thalamus (Hoover & Strick, 1993; Middleton & Strick, 2000). Neurons that project to motor- and premotor-related regions of thalamus are located in the posterior GPi, whereas those projecting to prefrontal-related thalamic nuclei are located in the anterior dorsal GPi and the SNr.
Much of the intrinsic connectivity of the BG can be captured by the classic model that identifies “direct” and “indirect” pathways that connect the striatum to the BG output nuclei [GPi and SNr; Fig. 1a (Albin et al., 1989; DeLong, 1990)]. The striatum contains two distinct populations of GABAergic projection neurons (termed “medium spiny neurons,” MSNs), those that project directly to the BG output nuclei and those that project only indirectly (Albin et al., 1989; Gerfen et al., 1990). Indirect-type MSNs project to an intermediate nucleus, the external globus pallidus (GPe), which in turn sends GABAergic projections to the output nuclei and to STN. The STN sends glutamatergic efferents to the GPi and the GPe. As will be seen below, the direct/indirect pathway model provides a useful framework for understanding initial stages of the pathophysiology of PD.
Some BG connectivity is not included in the standard direct/indirect pathway model. For example, the cortico-subthalamic pathway provides what has been described as a “hyper-direct” pathway by which excitatory cortical input can influence the activity of both segments of the globus pallidus (Nambu et al., 2004). Also, a sub-population of GPe neurons project “back” to the striatum where they differentially innervate a specific type of striatal interneuron (Parent & Parent, 2002). Recent work has also reported disynaptic interactions between the BG and the cerebellum. The deep cerebellar nuclei project to the striatum via a cerebello-thalamo-striatal pathway (Hoshi et al., 2005) and the STN projects to cerebellar cortex via pre-cerebellar nuclei of the brainstem (Bostan et al., 2010). The functional significance of these newly-discovered pathways remains unclear.
The cardinal motor signs of PD arise from degeneration of the dopaminergic neurons in the SNc and of their extensive axonal arborizations in the striatum and other BG nuclei (Fig. 1b). It is important to recognize, however, that PD is a complex disease associated with progressive degeneration of neurons from many sites in the central and peripheral nervous systems (Ruberg et al., 1986; Scatton et al., 1986; Zweig et al., 1993; Braak & Braak, 2000; Henderson et al., 2000a; Bohnen & Albin, 2011). Degeneration of the dopaminergic neurons of the SNc is one feature of that complex pathology. Some of the other, non-dopaminergic, features of the disease will be mentioned below. Despite the complexity of the disease, the clinical importance of the association between the cardinal motor signs of PD and the loss of dopamine is indicated by the spectacular therapeutic efficacy of dopamine replacement therapies (Hornykiewicz & Kish, 1987).
3. Activity patterns associated with PD
Loss of dopaminergic innervation is known to induce a variety of abnormalities in cellular excitability, synaptic plasticity, and even cell morphology in the striatum (Ingham et al., 1989; Day et al., 2006; Shen et al., 2008; Gerfen & Surmeier, 2011) and, though investigated to a lesser extent, in other BG nuclei as well (Rommelfanger & Wichmann, 2010). One (potentially indirect network-mediated) consequence of these diverse cellular changes is the appearance of abnormalities in neuronal discharge in BG nuclei and in connected regions of the thalamus and cortex (figure 2). Abnormal discharge exiting the BG via projections from the GPi constitutes an essential intermediate step in the genesis of parkinsonian motor signs. There is little doubt that this is the case given the remarkable antiparkinsonian effects of lesions of the GPi (pallidotomy) in parkinsonian patients (Laitinen, 1995; Lozano et al., 1995; Vitek et al., 2003) and GPi inactivation in animal models of parkinsonism (Brotchie et al., 1991; Lieberman et al., 1999; Baron et al., 2002).
Figure 2.

Changes in the activity of single cells in GPe, STN or GPi of parkinsonian monkeys. Shown are examples of separate neurons, recorded with standard extracellular electrophysiologic recording methods in normal and parkinsonian animals. Each data segment is 5 seconds in duration. Figure from Galvan and Wichmann (Galvan & Wichmann, 2008), used with permission.
a. Rate changes
An inherent component of the direct/indirect pathway model, as originally stated, was the prediction that abnormalities in mean discharge rate play an essential role in the pathophysiology of PD. According to the model, loss of striatal dopamine causes reduced discharge rates in direct pathway MSNs and increased discharge in indirect pathway MSNs. Both of these changes promote increased spontaneous discharge in GPi (Figure 1b and Albin et al., 1989; DeLong, 1990). Abnormally-elevated discharge of inhibitory GPi neurons was proposed to interfere with the normal movement-related activation of GPi-recipient thalamus and thereby generate the hypokinetic features of PD. The rate model predicts that resting discharge rates should be elevated in STN and GPi and depressed in GPe, GPi-recipient thalamus and in connected regions of the motor cortices.
Predictions of the rate model have been supported by single unit recording studies in monkeys rendered parkinsonian by treatment with the dopaminergic neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). These studies found elevated firing rates in the GPi and STN and depressed rates in GPe, thalamus, and motor cortex (Miller & DeLong, 1988; Filion & Tremblay, 1991; Bergman et al., 1994; Schneider & Rothblat, 1996; Elder & Vitek, 2001; Pasquereau & Turner, 2010). Studies using indirect markers of neuronal activity (levels of 2-deoxyglucose uptake, cytochrome oxidase or immediate-early gene expression) have also reported changes in the BG, thalamus and motor cortices consistent with the rate model (Crossman et al., 1985; Vila et al., 1997; Steiner & Kitai, 2000; 2001; Orieux et al., 2002; Emborg et al., 2007; Rolland et al., 2007). Moreover, single unit recording studies in patients undergoing surgical therapies have yielded increased firing rates in the parkinsonian GPi and reduced rates in thalamus as compared with rates in those structures in other neurologic disorders (Hutchison et al., 1994; Molnar et al., 2005; Chen et al., 2010) or in neurologically normal monkeys (Starr et al., 2008). Further support for the rate model came from the observation that parkinsonian signs are alleviated by manipulations that reduce STN discharge rates [i.e., permanent lesion (Bergman et al., 1990; Gill & Heywood, 1997; Alvarez et al., 2005), transient inactivation (Wichmann et al., 1994; Levy et al., 2001b; Baron et al., 2002), or genetic manipulation (Luo et al., 2002; Emborg et al., 2007; Lewitt et al., 2011)].
Despite those results, it is now recognized that aspects of the rate model are not tenable. Many studies have reported that firing rates in the pallidum, STN, thalamus, or cortex do not change in the way the rate model would predict with the induction of parkinsonism. Individual animals may be severely parkinsonian, but show no significant increase in STN or GPi discharge rates (Wichmann et al., 1999; Raz et al., 2001; Rivlin-Etzion et al., 2008) or decrease in thalamic or motor cortical activity (Doudet et al., 1990; Watts & Mandir, 1992; Goldberg et al., 2002; Pessiglione et al., 2005; Rivlin-Etzion et al., 2008). In addition, interventions such as DBS that yield significant therapeutic benefit can be associated with no net change in GPi firing rates (McCairn & Turner, 2009), or even increased rates (Anderson et al., 2003; Hashimoto et al., 2003; Hahn et al., 2008). Conversely, manipulations that the rate model predicts should induce parkinsonism (e.g., lesions of GPe, see Soares et al., 2004) do not result in the predicted effect.
Considering the mass of evidence in conflict with the classic rate model, it now seems clear that abnormalities in neuronal discharge pattern, beyond firing rate alone, play central roles in the genesis of parkinsonian signs. Taking a more nuanced perspective, however, it is still possible that abnormalities in discharge rate are important at specific points in the pathophysiologic process. Consistent with that idea, a recent study confirmed the predictions of the rate model for discharge rates of direct and indirect pathway MSNs (Kravitz et al., 2010). Using optogenetic techniques, Kravitz et al. demonstrated that inhibition of direct pathway MSNs or increased activation of indirect pathway MSNs can induce parkinsonian signs. These results suggest that abnormalities in neuronal discharge rate and discharge pattern interact and possibly reinforce each other at different steps in the pathophysiologic chain. Such a mechanism could explain the continued accumulation of evidence for abnormal discharge rates at particular stages of the BG-thalamocortical (TC) loop circuit (Orieux et al., 2002; Molnar et al., 2005; Rolland et al., 2007; Chen et al., 2010; Pasquereau & Turner, 2010) despite the equally compelling evidence in conflict with a classic “monolithic” version of the rate model.
b. Burst discharges
Neurons in the GP and STN of parkinsonian animals frequently emit action potentials in bursts (short epochs of markedly elevated firing rate). Significant increases in the prevalence of burst firing have been reported for neurons in the GPi, GPe, and STN in neurotoxin models of PD (Miller & DeLong, 1988; Filion & Tremblay, 1991; Bergman et al., 1994; Wichmann & Soares, 2006). Burst discharges are also a common feature of neuronal activity sampled from the GPi and STN of PD patients undergoing surgical therapies (Hutchison et al., 1994; Magnin et al., 2000). The increase in burst discharge appears early during the induction of experimental parkinsonism, roughly paralleling the time course of changes in discharge rate and metabolic activity (Ni et al., 2000; Vila et al., 2000; Breit et al., 2007).
The burst discharges of the parkinsonian BG are often described as “oscillatory-” or “rhythmic-bursting,” thereby implying that bursts and oscillatory modulations in firing rate (see below) are essentially two facets of one underlying phenomenon (Raz et al., 2001; Rivlin-Etzion et al., 2008). Several observations suggest, however, that bursts and rhythmic modulations in firing rate may be independent phenomena (Kaneoke & Vitek, 1996). The prevalence of bursts and oscillatory firing have been found to vary independently in GPi neurons sampled from PD patients undergoing DBS implantation surgery (Wichmann & Soares, 2006; Chan et al., 2011). Furthermore, therapeutic interventions may affect one measure (e.g., rhythmic firing) without changing the other (e.g., burst firing) (Levy et al., 2001a; Garcia et al., 2003; Heimer et al., 2006; Hahn et al., 2008; McCairn & Turner, 2009; Rosin et al., 2011). Based on these observations, we consider bursts and oscillations separately here.
Increased burst firing has also been described for neurons in GPi-recipient regions of thalamus (Raeva et al., 1999; Elder & Vitek, 2001; Pessiglione et al., 2005), and in the motor cortices (Goldberg et al., 2002; Pasquereau & Turner, 2011; Rosin et al., 2011) in parkinsonism. It is important to note, however, that the bursts reported for thalamic and cortical activity have markedly different durations and timing than the bursts reported for neurons in the parkinsonian GP or STN. The difference in burst metrics may be explained by the fact that GPi-to-thalamic communication is mediated via GABAergic inhibitory synapses; bursts in GPi activity cannot generate bursts directly in thalamic and cortical activity. This nonlinearity at the GPi-to-thalamic synapse is a central feature in computational models discussed below. Whether thalamic and cortical bursts have metrics consistent with a post-inhibitory rebound mechanism is unknown at present.
A potentially confounding factor to be addressed in future studies is the possibility that previous observations of increased burst discharge in parkinsonism, particularly in thalamus and cortex, may be attributed to reduced levels of arousal and attentiveness in parkinsonian animals. A marked increase in burst discharges throughout thalamus and cortex is a well-established feature of reduced arousal and wakefulness (Steriade & Llinas, 1988) and reduced arousal and attentiveness are common in parkinsonian subjects (Rye et al., 2000; Gatev & Wichmann, 2003; Barraud et al., 2009).
c. Oscillatory firing patterns
Regularly recurring fluctuations in firing have been documented in the STN, GPi, GPe, and tonically active striatal interneurons (corresponding to cholinergic interneurons) in MPTP-treated monkeys (Bergman et al., 1994; Nini et al., 1995; Raz et al., 1996) and in the STN and GPi of patients with PD undergoing electrophysiological recordings as part of neurosurgical procedures (Levy et al., 2000; Levy et al., 2002a; Levy et al., 2002b). Most likely, oscillatory activity patterns arise as network phenomena, an aspect of BG activity that has been extensively studied with network simulations (see below). For instance, there is experimental support that oscillations can arise in the GPe-STN network, through interactions by which excitatory output from the STN leads to a burst of GPe spiking, which, in turn, leads first to hyperpolarization and then rebound bursting in the STN, resulting in renewed GPe bursting activity (Plenz & Kitai, 1999; Holgado et al., 2010). Other mechanisms, for instance, STN driving by oscillatory cortico-subthalamic inputs, may also lead to oscillatory bursting in the STN and related nuclei (Magill et al., 2001; Magill et al., 2004a; Magill et al., 2004b).
Oscillatory activities in the BG-TC network of connections are also frequently studied in local field potential signals (LFPs). LFPs reflect synchronous membrane potential fluctuations of groups of neurons. The amplitude of these potentials is strongly dependent on the spatial arrangement of the electrically-excitable tissue elements in the recorded area. The study of LFP recordings became very popular after the discovery that electrodes implanted in the BG of movement disorder patients for DBS therapy can be used as LFP recording devices. Analysis of oscillations in LFP records from such electrodes has revealed the occurrence of oscillatory activity in the beta frequency range (approximately 10–35Hz) throughout the extrastriatal BG (specifically in the STN), which can be suppressed by dopaminergic replacement therapies (Brown et al., 2001; Levy et al., 2002a; Williams et al., 2002; Priori et al., 2004; Kuhn et al., 2009) or DBS therapy (Kuhn et al., 2008; Bronte-Stewart et al., 2009). At least in the STN of parkinsonian patients, single cell oscillations and beta-band LFP oscillations are related to one another (Kuhn et al., 2005; Weinberger et al., 2006). It is thought that LFP oscillations reflect dopamine-dependent oscillatory phenomena involving the entire BG–TC network of connections (Brown & Williams, 2005; Silberstein et al., 2005; Hammond et al., 2007). This conjecture is supported by evidence that beta oscillations in the LFP signals recorded in STN and GPi are coherent with cortical oscillatory electroencephalographic activity (Brown et al., 2001; Marsden et al., 2001; Cassidy et al., 2002; Williams et al., 2002; Fogelson et al., 2005). In parallel with the presence of beta-band oscillatory activities, gamma-band oscillatory activities (frequencies > 35Hz) are found to be less prominent in the BG and cortex of parkinsonian patients and in animal models of the disease (Wang et al., 1999; Brown, 2003; Lalo et al., 2008).
Interestingly, while oscillatory activities are easily identified in the BG of parkinsonian patients and animal models, they are less clear in single-cell recordings from VLa and VA thalamic nuclei (but see Guehl et al., 2003; Pessiglione et al., 2005), although reduced gamma-band activation has been shown for thalamic LFP recordings from parkinsonian patients (Kempf et al., 2009). Oscillatory activity is also less prominent in single cell recordings in primary motor cortex (Goldberg et al., 2002; Pasquereau & Turner, 2011). A nonlinear transformation of activity in TC circuits may prevent BG oscillatory activity from directly inducing similar oscillatory activities at the single-cell level in cortex. However, the importance of cortical oscillatory activity continues to be a matter of debate. Interestingly, a recent study has shown that electrical stimulation of GPi with short trains of stimuli that were triggered by oscillatory single-cell activity in the primary motor cortex had strong antiparkinsonian effects (Rosin et al., 2011).
d. Synchronization
There is rarely synchrony between the spontaneous discharges of different neurons in the BG of neurologically-normal subjects, supporting the general concept that the BG function as a series of parallel, largely independent modules (see above). This independence changes significantly in PD: neurons that are close to each other within areas located throughout the BG, the BG-receiving areas of the thalamus, and the cortex start to fire in synchrony (Bergman et al., 1994; Goldberg et al., 2002; Heimer et al., 2002; Goldberg et al., 2004; Rivlin-Etzion et al., 2006; Hammond et al., 2007). Studies of correlation patterns in human patients with PD and in non-human primates with parkinsonism have demonstrated that systemic treatment with dopamine receptor agonists acts to lower the level of abnormal synchronization in the firing of BG neurons (Levy et al., 2001a; Heimer et al., 2006), suggesting that the segregation of neuronal activity in the BG is, at least in part, actively maintained through the presence of dopamine.
Synchronous firing is often associated with oscillatory discharges. Such oscillatory synchrony is not only found within but also across the BG nuclei. For instance, oscillatory activity is synchronized across the STN, GPi and cortex, and this synchrony is suppressed by the administration of levodopa (Brown, 2003; Gatev et al., 2006; Hammond et al., 2007). As is the case for oscillatory activities at the single cell level (see above), the synchronous firing of single neurons in the STN is coherent with concomitantly recorded beta-band LFP oscillations (Kuhn et al., 2005; Weinberger et al., 2006).
The loss of independence between neighboring trans-BG channels is also apparent in the increased tendency of BG neurons to widen their receptive fields under parkinsonian conditions (Bronfeld & Bar-Gad, 2011). Under normal circumstances, BG neurons are usually highly specific in terms of their responses to sensory inputs, such as proprioceptive inputs during joint rotation. A widening of these receptive fields was found in recordings of neuronal responses in the STN, GP and thalamus of MPTP-treated monkeys (e.g., Filion et al., 1988; Pessiglione et al., 2005). The altered sensory field size may, in part, be a functional correlate of the greater degree of synchronized activities within the BG, but it may also reflect altered convergence patterns of sensory processing in these structures. It is not known whether the size of receptive fields is modulated specifically by the level of dopamine in the BG.
e. Generation of abnormal firing patterns
For many years, the absence of striatal dopamine has been thought to be solely responsible for the abnormal BG discharge patterns in parkinsonian conditions. Clearly, dopamine loss is the most prominent parkinsonism-related biochemical change in the BG, and the loss of dopamine at striatal synapses is likely to strongly influence corticostriatal transmission, and, thus, to affect activity patterns along the direct and indirect pathways (see above). Detailed computational models of the resulting activity changes have been developed, as is discussed in more detail below.
However, the dogma that BG firing abnormalities are entirely due to striatal dopamine loss has been challenged by recent studies documenting a widespread pattern of additional changes in the brains of parkinsonian subjects, which may also influence activity patterns in the BG in parkinsonism. One of these recently recognized parkinsonism-associated changes is that dopamine is not only lost in the striatum, but throughout the extrastriatal BG, thalamus and frontal cortex, which, based on electrophysiological studies, may strongly (and directly) affect the activity of neurons in these areas (reviewed in Rommelfanger & Wichmann, 2010). Furthermore, PD in humans and experimental parkinsonism in animals is associated with the early loss of norepinephrinergic cells in the locus coeruleus and other catecholaminergic brain stem regions (Braak et al., 2004; Masilamoni et al., 2011). As mentioned above, PD and toxin-induced parkinsonism in animals is also associated with a variety of structural changes in the BG and associated areas, which may further affect firing patterns in the BG. For instance, it is known that synapses of corticostriatal projections and dendritic spines of striatal medium spiny neurons degenerate in PD (reviewed in Villalba & Smith, 2010; 2011), and similar changes may also occur at glutamatergic terminals of the cortico-subthalamic projection (Mathai et al., 2011). Another recently documented change is the finding that the thalamic source neurons of the massive thalamostriatal projection system (located in the caudal intralaminar nuclei of the thalamus) degenerate in patients with advanced PD and in animal models of the disease (Freyaldenhoven et al., 1997; Henderson et al., 2000a; b; Ghorayeb et al., 2002; Henderson et al., 2005; Aymerich et al., 2006; Villalba et al., 2011).
f. The role of abnormal BG discharge in the expression of parkinsonism
The link(s) between specific changes in the discharge patterns of BG neurons and the behavioral manifestations of PD remain(s) tenuous. One approach to investigating this issue is to examine the temporal relationship between the development of parkinsonism and the occurrence of abnormal discharge patterns in the BG. Such studies have confirmed that the neuronal activity in STN and GPi is increased prior to the onset of motor symptoms (Bezard et al., 1999). Furthermore, BG interventions such as lesions or DBS of GPi or STN dramatically and immediately improve parkinsonian signs, supporting a role of BG discharge abnormalities in the development of parkinsonism (Wichmann & Delong, 2006).
A very large body of literature is devoted to an exploration of the role of oscillatory activity in movement, and the possible disturbing effects of enhanced beta-band oscillations in the BG-TC network of connections. In normal individuals, beta band oscillations are reduced immediately prior to and during voluntary movements in cortex (Pfurtscheller & Neuper, 1992; Toro et al., 1994; Leocani et al., 1997; Ohara et al., 2000; Alegre et al., 2002; Doyle et al., 2005b) and putamen (Courtemanche et al., 2003; Sochurkova & Rektor, 2003), concomitant with an increase in gamma-band activities. Movements are followed by a resurgence of beta-band oscillations, suggesting that beta-band activity may have movement-terminating or suppressing effects. In patients with implanted DBS electrodes, a similar general relationship between beta-band activity and movement was demonstrated for LFPs in the STN (Cassidy et al., 2002; Williams et al., 2003; Kuhn et al., 2004; Doyle et al., 2005a; Williams et al., 2005; Kuhn et al., 2006; Kempf et al., 2007). It is thought that the increased ‘anti-kinetic’ oscillatory activity in the beta-band in the BG may interfere with movement initiation in parkinsonian patients (akinesia).
Evidence for a direct role of abnormal BG activities in parkinsonism also comes from studies involving electrical stimulation of the STN in monkeys. These experiments have shown that motoric impairments can be induced with stimulation patterns fashioned after those recorded in parkinsonian animals (Ma & Wichmann, 2004), that movement is slowed by stimulation at beta-band frequencies (Timmermann et al., 2004; Chen et al., 2007; Eusebio et al., 2009), and that parkinsonism can be improved with oscillatory trains of stimuli timed to eliminate the beta-band activities in the BG (Rosin et al., 2011).
In contrast to these studies, several animal studies have found that the neuronal activity changes (particularly oscillatory activities) appear only after the emergence of parkinsonism and, therefore, cannot be fully responsible for it (Leblois et al., 2007). Likewise, in studies in rodents in which nigrostriatal dopaminergic transmission was blocked acutely with dopamine antagonists at doses that induce parkinsonism, oscillatory neuronal activities were not seen in the BG and cortex (Mallet et al., 2008; Degos et al., 2009), contrasting with the outcome of more chronic dopamine depletion strategies. Furthermore, despite the consistent finding of increased burst firing in the BG in parkinsonism, treatments with dopaminergic agents do not always reduce burst firing in the BG of parkinsonian animals or patients (compare Tseng et al., 2000; Lee et al., 2001; Levy et al., 2001a). Local injections of dopamine D1-like receptor agonists into the primate GPi or SNr, or D5 receptor activation in the rodent STN, were also found to increase rather than decrease burst firing in these nuclei (Baufreton et al., 2003; Kliem et al., 2007). Additionally, therapeutic DBS of the GPi (McCairn & Turner, 2009; Rosin et al., 2011) or STN (Hahn et al., 2008) is not accompanied consistently by reductions in the prevalence of GPi burst discharges.
4. A perspective on computational modeling
Given the complexity of the brain, there is little hope of building a computational representation of even a limited brain area, much less of something like the BG, that is both a complete model and of practical utility. Nonetheless, we claim that computational and mathematical methods (Dayan & Abbott, 2001; Izhikevich, 2007; Ermentrout & Terman, 2010) offer a means to explore and generate hypotheses and experimentally testable predictions about the BG in parkinsonism. As we have described in the earlier sections of this article, there are a variety of alterations that have been experimentally and clinically observed to occur in the BG in the parkinsonian condition. Many of these are changes in activity in various nuclei, including modulations of firing rates, of temporal patterns of firing, and of correlation patterns and response specificities. One natural direction for computational efforts is to explore the mechanisms underlying the emergence and properties of parkinsonian activity within model BG circuits.
It is possible that some or all of these changes actively contribute to motor impairment, or it may be that they are consequences of some other factors that cause the dysfunctions. To address this paradigmatic dichotomy, one alternative but reasonable approach is to pick a particular change in activity and ask, given that this change occurs, how could it lead to motor effects, and what would these effects be? While this is a reductionist step, it is potentially powerful in allowing for the examination of the logical consequences of a small set of initial assumptions (Silva, 2011). Indeed, we claim that this approach is essential to developing a complete understanding of parkinsonian pathophysiology, bridging from the loss of dopamine and other aspects of the onset of parkinsonism to the emergence of its motor (our focus here) and other signs, and to explaining the impact and efficacy of treatments for parkinsonian patients.
A variety of models and modeling frameworks that have been employed in the context of parkinsonism and DBS therapy have been reviewed elsewhere (Titcombe et al., 2004; Modolo et al., 2011). To constrain the scope of this article and match the emphasis in the literature, we will focus on modeling the link from alterations in BG activity to their possible downstream effects, along with the possible impact of STN DBS on this pathway. We first discuss a specific reduced model, with an emphasis on the underlying logic (assumptions, consequences, and predictions). Beyond this particular line of investigation, we subsequently review some complementary and alternative computational approaches to understanding the therapeutic effects of STN DBS, which also build from starting points based on certain aspects of parkinsonian activity.
5. A computational framework based on thalamic relay
a. Foundations for the framework
Taking this approach, consider the excessive bursting found in the parkinsonian BG. The dynamic mechanisms underlying the bursting are critical in terms of evaluating how possible therapeutic interventions might impact or target this phenomenon, and various mechanisms have been proposed and studied computationally (Terman et al., 2002; Kubota & Rubin, 2011). First, however, to determine the possible pathological motor implications of excessive bursting, a natural starting point is to consider where it occurs and, crucially, what other brain areas receive inputs from the sites exhibiting excessive bursting. Two sites with prominent increases in bursting that project out of the BG are the STN and the GPi. The STN projects to the PPN, so one possibility is that the effects of STN bursting on PPN are critical to motor complications. However, there is significant loss of PPN neurons in parkinsonian conditions (Pahapill & Lozano, 2000). Thus, we again make a reductionist choice and ignore this pathway to focus on outputs of GPi. That is, we ask the specific question, how could excessive bursting in GPi translate into altered motor behavior? As reviewed above, GABAergic neurons of the “motor” GPi project to the anterior ventrolateral nucleus of thalamus (VLa) (Yoshida et al., 1972; DeVito & Anderson, 1982; Jones, 2007). Therefore, to pursue this line of reasoning, the key issue is how excessive bursting in GPi impacts VLa thalamus.
In fact, how GPi output affects the spiking activity in VLa is poorly understood. In the normal brain, BG projections to the thalamus are not principal drivers of thalamic activity. Sensory-evoked responses typically begin earlier in VLa than in GPi (DeLong et al., 1985; Vitek et al., 1994), suggesting that thalamic responses are driven by an earlier-firing non-BG source. Also, transient inactivation of the GPi does not alter task-related activity in VLa, even when it increases resting firing rates (Inase et al., 1996). Thus, in the normal brain, BG afferents appear to modulate thalamic activity that is driven by some other component (e.g., excitatory inputs from cortex) (Deniau & Chevalier, 1985; Inase et al., 1996). Rubin and Terman introduced the idea of using computational models to study how bursty inhibition from GPi might affect the response of TC relay cells to this excitatory drive (Rubin & Terman, 2004). Reduced TC cell models had been developed previously, for example, for the study of sleep spindles and absence epilepsy (Golomb et al., 1994; Destexhe et al., 1998; Sohal & Huguenard, 2002). These models feature various currents, but their dynamics are dominated by standard sodium and potassium spiking currents as well as a low-threshold or T-type calcium current. This T-current can give rise to post-inhibitory rebound or anodal break bursting, in which a sustained hyperpolarizing input slowly de-inactivates the current, opening one set of ion gates, and the subsequent abrupt removal of hyperpolarization activates the current, opening another set of gates and yielding a burst of rapid spikes (Jahnsen & Llinas, 1984a; b). In fact, a standard sodium current can also give rise to a similar but less pronounced rebound effect, due to its own slow inactivation component. While the T-type current tends to be more inactivated in awake than sleep states, evidence suggests that TC cell bursting is a component of awake dynamics, such as responses to novel stimuli, in sensory-driven thalamic areas (Sherman, 2001; Sherman & Guillery, 2002) and therefore the T-current should be included in models for awake states as well.
b. The Rubin/Terman (RT) model
For the study of what happens when a TC cell receives a burst of inhibition from GPi, Rubin and Terman considered several different sets of computational components (Rubin & Terman, 2004). In their most complete simulations and analysis, they synaptically connected model STN and GPe cells developed previously (Terman et al., 2002) together with GPi and TC cells to form a small network featuring indirect and direct pathway components, with GPi cells inhibiting TC cells. Guided by earlier work (Terman et al., 2002), they developed an architecture for the BG components of the network that allowed it to be tuned to generate irregular, asynchronous activity or to be retuned to yield bursty, synchronous activity in the 3–8 Hz range. The retuning consisted of changes in just two parameters, the strengths of inhibition from striatum to GPe and within GPe, which have been observed to occur experimentally in parkinsonian conditions (Albin et al., 1989; Stanford & Cooper, 1999; Ogura & Kita, 2000). The model TC cells received inhibitory inputs from GPi as well as a computationally generated excitatory input train of spikes that were either periodic or Poisson. The authors calculated an error index to quantify the fidelity of the TC cell relay of its excitatory input. A successful relay consisted of a single TC spike within a small time window after an excitatory signal. Any other response to an excitatory input was considered an error. Specifically, unsuccessful events could be misses, in which no TC spike occurred during that window, or bad responses, consisting of multiple spikes after a single stimulus that did not reflect the excitatory input characteristics. The error index was given by the ratio of errors to the total number of excitatory input spikes.
The authors found that the switch from irregular to bursty activity within the BG significantly compromised the fidelity of TC relay, as quantified by the error index. Importantly, during a burst of GPi activity, the resulting TC cell inhibitory synaptic conductance grows and then saturates at a high level, since the GPi spike rate is high relative to the time constant of inhibitory conductance decay. At the end of the burst, the conductance decays, and it subsequently remains low, until the next burst begins. Based on this observation, it is possible to analyze the loss of relay fidelity in terms of bifurcation diagrams or in terms of a simplified model and its nullclines (Rubin & Terman, 2004). This analysis reveals that under parkinsonian conditions, the TC neuron passes through four phases of distinct relay capacity: baseline, corresponding to low inhibition and low T-current availability; compromised, corresponding to high inhibition and low T-current availability shortly after GPi burst onset; recovered, corresponding to high T-current availability during sustained inhibition; and excessive, corresponding to low inhibition yet high T-current availability shortly after GPi burst offset (see Figure 3 for schematic illustration).
Figure 3.
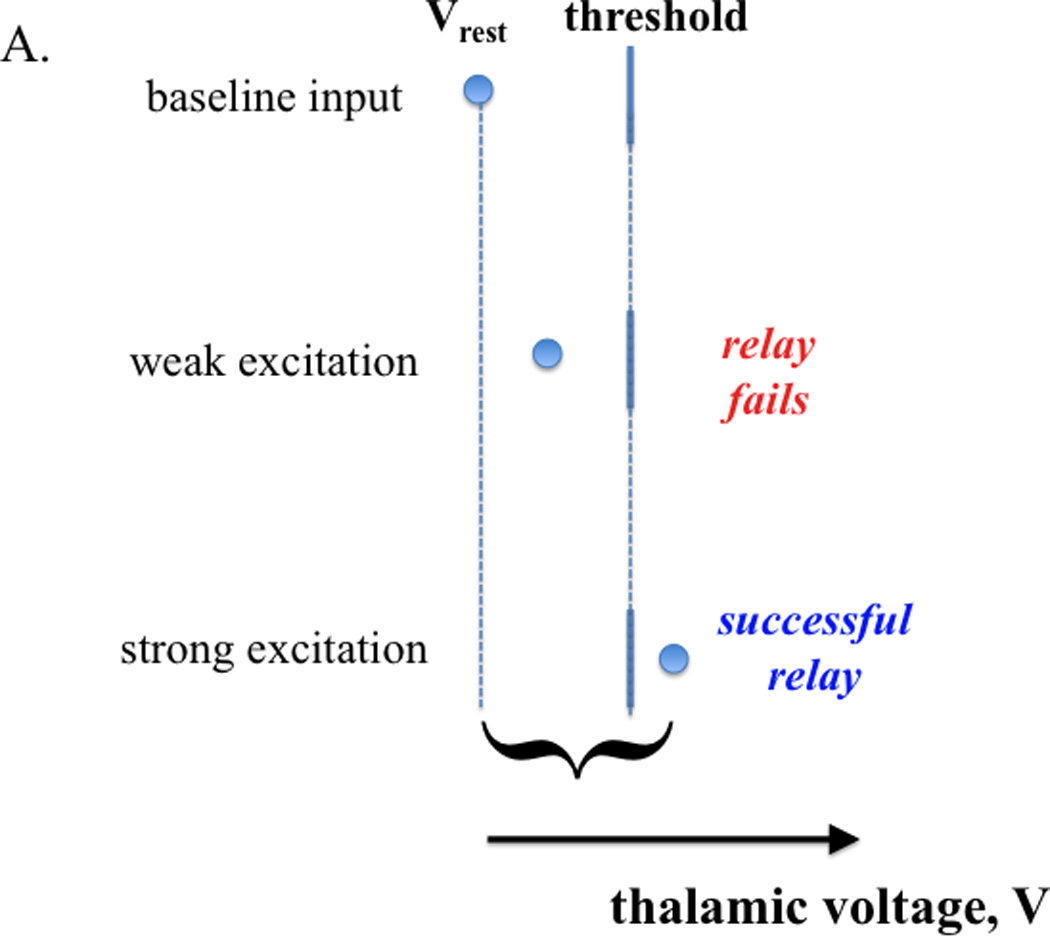
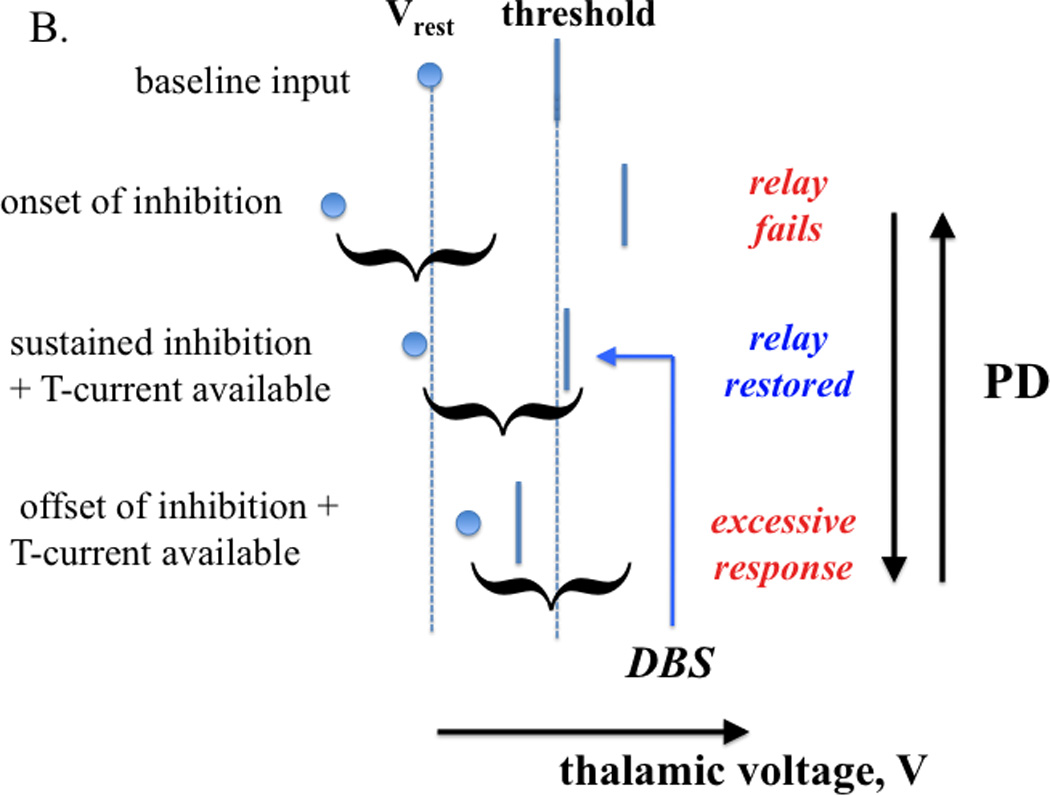
Inhibition influences thalamic relay capability (schematic illustration). A. Baseline input conditions establish a rest potential and a threshold for action potential generation (top). If an excitatory input arrives, a successful relay response (i.e., spike generation) is determined by the strength of that input relative to the separation between rest potential and threshold (middle, bottom); a sufficiently input strength to yield relay is represented by a curly bracket. B. Parkinsonian conditions are characterized by oscillations in the inhibitory input to thalamus (from GPi). At the onset of strong inhibition, a formerly relay-inducing excitatory input fails to yield relay (top). If inhibition is sustained, T-current deinactivation can restore relay by raising rest potential and lowering threshold (middle). Since T- current inactivation is slow, the arrival of the same input after a relatively abrupt withdrawal of inhibition can yield an excessive response (bottom). One possibility is that DBS of STN pins the inhibitory input from GPi to thalamus at a high level, where relay is restored by T-current availability.
Part of the power of this framework is that it also offers an explanation for how STN DBS could achieve therapeutic efficacy. Experiments have shown that high-frequency stimulation can boost activity in its targets or can at least elicit effects consistent with augmented synaptic outputs from stimulated sites (Paul et al., 2000; Windels et al., 2000; Jech et al., 2001; Anderson et al., 2003; Hashimoto et al., 2003; Hershey et al., 2003; Windels et al., 2003; Garcia et al., 2005). If STN DBS drives GPi neurons in a way that leads to high amplitude but effectively sustained inhibition from GPi to its thalamic targets, then the computational model shows a significant restoration of thalamic relay fidelity. This effect arises even if the net TC cell inhibitory conductance oscillates, as long as the oscillation is of sufficiently high frequency and small amplitude. Mechanistically, the sustained inhibition leads to a sustained T-current deinactivation. The resulting current availability provides an extra boost that allows the model TC neuron to respond reliably to excitatory input spikes through its standard sodium and potassium dynamics. A similar effect would be expected with GPi DBS, again assuming that it results in a regularization of inhibition to pallidal targets in thalamus.
These results offer the first mechanistic theory for the proposed conceptual idea that pathological temporal variations in spike timing in the parkinsonian BG, such as burstiness or rhythmicity, could be disruptive to brain function and that elimination of these firing patterns could be correspondingly beneficial (Montgomery & Baker, 2000; Vitek, 2002; Foffani et al., 2003; Grill et al., 2004; Garcia et al., 2005; Meissner et al., 2005; Foffani & Priori, 2006). It is important to recognize the assumptions underlying this theory: the theory assumes that net inhibitory inputs to neurons within VLa thalamus are bursty in parkinsonism and that faithful relay of excitatory inputs by VLa thalamus is important for some aspects of normal motor activation. The theory also makes strong predictions, namely that at least some parkinsonian states should feature significant bursting in VLa thalamus, that elimination of prominent bursting or rhythmicity in the total GPi inputs to most cells in VLa thalamus should improve some motor features, and that changes in time constants of inhibition, T-current inactivation, or in sodium current inactivation within VLa thalamus should alter parkinsonism. More subtly, the theory implicitly predicts that for non-parkinsonian animals in states under which at least some symptoms occur in parkinsonism (e.g., awake resting state for rest tremor, movement initiation for bradykinesia), GPi outputs should be sufficiently irregular and asynchronous that TC cells can respond faithfully to excitatory inputs. Finally, the explanation offered for the mechanism of therapeutic efficacy of STN or GPi DBS relies on the idea that DBS yields a regularization of the inhibition from GPi to VLa thalamus, although the actual level of inhibition that results is not important. A prediction that follows is that any form of DBS, or other therapeutic modalities that eliminate GPi output patterning without compromising other aspects of BG output should also yield therapeutic benefit.
c. A data-driven version of the RT model and some limitations
Testing the importance of VLa relay fidelity for motor outcomes has thus far been beyond the reach of experiments. There is experimental data, however, on parkinsonian activity patterns in GPi and VLa thalamus that is relevant to this theoretical framework. Most directly, experimentally recorded spike trains from single primate GPi neurons were collected under several conditions: normal; MPTP-induced parkinsonism; MPTP plus STN DBS without therapeutic benefit (sub-DBS); and MPTP plus therapeutically effective STN DBS (Hashimoto et al., 2003). In work by Guo et al. (Guo et al., 2008), a collection of these trains were used to computationally generate continuous conductance signals. Each signal was used as an inhibitory synaptic conductance in a computational TC cell model, subject to the same forms of excitatory input used in previous work (Rubin & Terman, 2004), and again, a relay error index was computed. The GPi signals from the normal condition led to low error index scores, while, following MPTP, GPi signals yielded a very significant increase in error index. Crucially, the error index scores during sub-therapeutic DBS were also significantly elevated relative to normal, while scores were returned to normal levels with therapeutic DBS. Thus, regardless of the dynamic mechanisms conspiring to generate GPi activity patterns in various conditions, and independent of whether DBS activates, shuts down, or otherwise alters firing (where it is applied or upstream or downstream from there), we can conclude that the GPi signals that are present in parkinsonian conditions can compromise TC relay fidelity, while the application of therapeutic, but not sub-therapeutic, DBS leads to signals that support highly reliable relay.
While these results are exciting, there are reasons for caution: First, GPi spike trains had to be converted to continuous synaptic conductances, and this step required assumptions about synaptic parameters and dynamics. Second, a single-compartment computational TC cell model was used, necessitating an additional set of choices about currents to include and parameter values to use. Third, data was only available from one GPi neuron at a time, so no information about the patterns of activity across GPi within an experimental condition was available. Fourth, the data used was limited to 38 5-second blocks of spikes from 11 cells. Fifth, the data was obtained from non-human primates, rather than human patients. A subsequent study, however, did include data from GPi recordings in human PD patients; when this data was used in TC simulations, relay was compromised, while a phenomenological representation of DBS restored relay when DBS frequency was sufficiently high and DBS amplitude was within a particular band (Meijer et al., 2011). Sixth, the simulations did not incorporate any specific features of the architecture of synaptic connections, such as divergence or convergence, from GPi to thalamus. Finally, results from a number of empirical studies have brought into question the pathophysiologic importance of burst discharges in the GPi (see sections 3b and 3f, above) Follow-up studies that link additional experiments and simulations to move beyond these limitations could make an important contribution to our understanding of these effects.
Interestingly, several aspects of the data-driven computational results (Guo et al., 2008) point to the importance of GPi bursting in determining relay outcomes. The fraction of time over which the data-driven GPi inhibitory input signals were elevated rose steadily from normal to MPTP to sub-DBS to DBS conditions, corresponding to a progression from irregular to bursty to high frequency, relatively sustained spiking. Supplying the same GPi signal to a heterogeneous population of model TC cells caused independent TC relay failures in normal and DBS cases, but the failures were synchronized across the TC population in MPTP and sub-DBS cases, pointing to the robustness of the effects of inhibitory bursts on the model TC neurons. Finally, averaging GPi signals that gave identical TC responses to an excitatory input (successful relay, failure to spike, or excessive response), aligned relative to each excitatory input (i.e., using the 20 ms of signal before each input and the 5 ms of signal after it), revealed significant differences across the three response types. Average inhibitory conductance was relatively constant before successful responses but showed a pronounced rise before spike failures and decline before excessive responses (Figure 4). These results reflect the power of relatively rapid changes in inhibitory conductance, as would be associated with synchronized GPi bursting, to interfere with the straightforward relay of an excitatory input.
Figure 4.
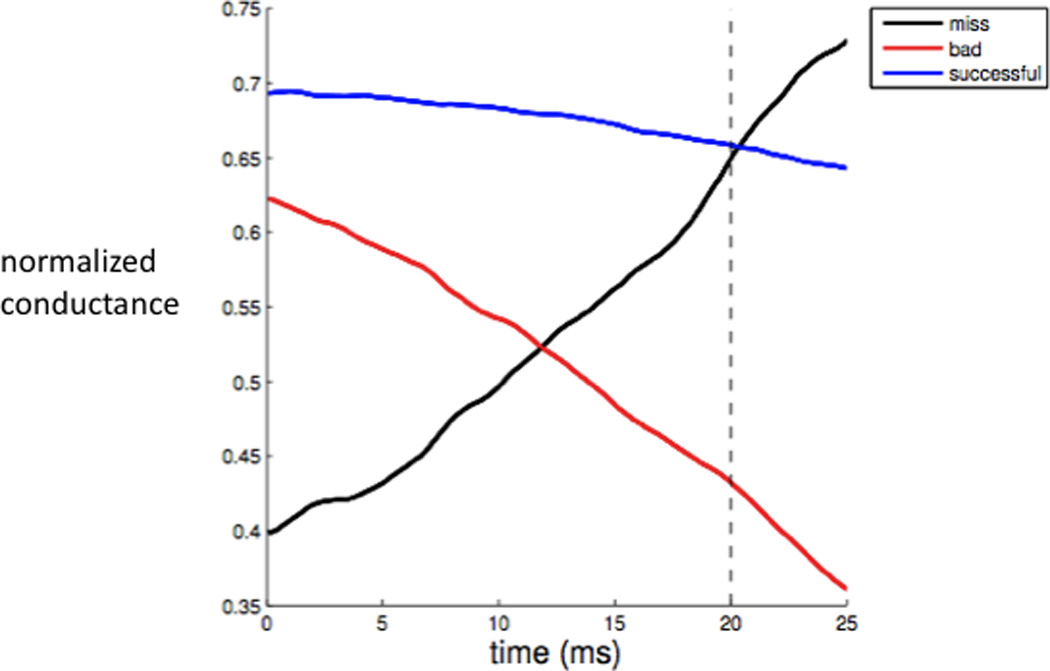
Response-triggered average GPi input signals to computational model TC relay neuron. Input signal strength is measured as normalized conductance, given by synaptic conductance divided by maximal synaptic conductance. The GPi inhibitory output patterns preceding different types of TC responses to excitatory inputs are qualitatively different, consistent with the impact of relatively abrupt changes in GPi signaling on TC response capabilities (see text for details). Modified from Guo et al. (Guo et al., 2008).
Thus, while these data-driven computational results circumvent the issue of burst generation mechanisms, they bring us back to the question of bursting in VLa thalamus. A small number of monkey studies have suggested that some bursting is present and may be reduced by DBS. Specifically, following MPTP injections, effective but not ineffective STN DBS regularized firing and reduced bursts in the pallidal receiving areas of motor thalamus (Xu et al., 2008). Moreover, in monkeys in which bursting was observed in a small subset of downstream thalamic neurons, GPi DBS reduced the prevalence of this activity pattern and the number of spikes per burst when bursting remained (Anderson et al., 2003). Without DBS, the prevalence of VLa thalamic bursting has not been thoroughly explored. In MPTP-treated monkeys, bursting was observed in only about 10% of motor thalamic neurons recorded extracellularly (Guehl et al., 2003). However, the great majority of these neurons exhibited rhythmic activity, which did not feature the long pauses associated with bursting but did include shorter pauses, leading to a bimodal ISI distribution; the impact of such ISIs on thalamocortical relay has not been studied. Interestingly, in these experiments, the bursting fraction did not change significantly between tremor and non-tremor periods, while rhythmic activity became more prominent with tremor. Moreover, motor thalamic oscillations predominantly occurred in the 5–7 Hz range that has been correlated with EMG activity during tremor in parkinsonian patients (Lenz et al., 1988), not in the beta-band (Guehl et al., 2003). In light of this result, and the recent accumulation of evidence about the prominence of enhanced oscillations in parkinsonian conditions (see above), an important direction for future investigations within this framework will be to explore the impact of oscillatory activity in GPi on thalamic activity patterns. Finally, a subsequent analysis of the same thalamic data emphasized changes in correlation structure and response specificity, rather than activity patterns, although this work excluded data from tremor episodes (Pessiglione et al., 2005). It has been shown computationally that altered GPi activity patterns could impact the transfer of correlated activity from BG to thalamus across normal, parkinsonian, and DBS conditions (Reitsma et al., 2011), but additional work on this topic is also needed. Clearly, a thorough investigation of bursting and oscillation properties of VLa thalamus under normal, parkinsonian, and DBS conditions, including establishing relationships between these properties, GPi activity patterns, and motor impairments, remains an important target for future work.
d. Additional computational studies
In the meantime, it is important to note that additional, purely computational studies have been performed that clarify the generality of this relay framework. These studies have explored an alternative, multi-compartmental TC cell model (Cagnan et al., 2009) and a broader range of GPi burst frequencies and DBS parameters and sites (Cagnan et al., 2009; Pirini et al., 2009). Results obtained have been consistent with the idea that overly rhythmic inhibitory signals from GPi compromise TC relay, whereas effectively constant inhibitory signals from GPi to thalamus, which can be achieved via certain forms of BG DBS, promote effective relay.
The RT model and these subsequent studies established the theoretical importance of modifying bursting activity in the BG with DBS to achieve a more regularized input to thalamus. However, the RT model was criticized as a relatively simplified representation of the BG and, in its original formulation, it did use the controversial assumption that burst activity was generated by STN-GPe interactions. Hahn & McIntyre (Hahn & McIntyre, 2010) attempted to address these issues with the creation of a more detailed subthalamopallidal microcircuit model. Synaptic weights in the network were trained to fit in vivo neural firing patterns from parkinsonian monkeys, and the model system was driven by stochastically defined cortical beta rhythms. STN DBS applied to the model in the parkinsonian condition reproduced the reduction in GPi bursting found in experimental data. The Hahn and McIntyre model also predicted that regularization of GPi firing was dependent on the volume of STN tissue affected and a threshold level of burst reduction may be necessary for therapeutic effect, supporting the general hypotheses of the RT model.
6. A complementary approach: the information lesion
Around the same time that Rubin and Terman were developing their network model of DBS, McIntyre and Grill were developing models of the response of individual neurons to DBS (McIntyre et al., 2004). These individual neuron models predicted that DBS generates efferent axonal activation at or near the stimulation frequency. These results led to the hypothesis that effective DBS overrides oscillatory pathological activity and replaces it with more regularized neuronal firing patterns. This concept was further expanded by Grill et al. (Grill et al., 2004) with the introduction of the term “Informational Lesion”. Using a TC neuron model, they showed that DBS produced a frequency-dependent modulation of the variability of neuronal output, and above a critical frequency, stimulation resulted in regular output with zero variance. They then hypothesized that zero output variance is analogous to a lesion in terms of network processing of information. In other words, the logic here is that DBS replaces a pathological signal with an innocuous one; however, the question of what downstream effects were induced that made one signal problematic and the other harmless was not answered.
Excitingly, the concepts of stimulator pulse variance and the corresponding neuronal output variance were further evaluated by the Grill group with human experiments, as well as with computer models (Birdno et al., 2007). In the human experiments, thalamic DBS with an average stimulus pulse rate of 130 Hz was more effective at reducing tremor when the pulses were evenly spaced than when there were large variances in the inter-pulse interval. These experiments do not reveal the mechanism underlying tremor reduction, however. In the thalamic neuron model, increasing the difference between the intra-pair and inter-pair pulse intervals rendered model neurons more likely to fire synchronous bursts, more likely to fire irregularly, and less likely to entrain to the stimulus. This observation could be consistent with an information lesion viewpoint, that the failure to entrain represents a failure to eliminate pathological “information”, or with the relay framework, that the presence of synchronous thalamic bursts compromises responses to cortical signals. Indeed, it is possible that these viewpoints are in fact the same, and that what makes signals pathological or not is determined by their impact on relay. An indication of this convergence of theories is provided by the work of Dorval et al. (Dorval et al., 2010). These authors expanded the analysis of DBS regularity to the PD symptom of bradykinesia, showing that DBS delivered with low inter-pulse variability was more effective than irregular DBS at reducing symptoms. They then used the RT model to show that high-frequency stimulation alone is insufficient to improve relay. In summary, DBS pulses must be highly regular to be effective.
7. An alternative framework based on (de)synchronization
Various computational models relating to parkinsonian activity and/or DBS have been developed and reviewed elsewhere (Titcombe et al., 2004; Modolo et al., 2011). This paper does not provide a comprehensive review but instead considers a particular mechanistic theory for how parkinsonian activity could induce specific downstream effects and how DBS could eliminate these effects. We have spelled out the assumptions underlying this theory, the consequences of these assumptions, and a set of predictions that follow. Such an elaboration of the logical structure of a theory is critical for the objective evaluation of its biological and clinical relevance. For comparison, we will now consider some of the logic underlying a alternative computational approach to parkinsonism and DBS in the extensive work of Tass and collaborators (see, e.g., Tass, 2006).
Many of the works from this group build off of the experimental observation that excessive synchronization of BG activity is a central feature of parkinsonism. The central assumption made, in light of this observation, is that the efficacy of DBS depends critically on its ability to desynchronize neurons in its target site. In other words, a key prediction on which this theory depends is that a clinical intervention that achieves desynchronization will be effective, while failure to desynchronize will correspond to persistence of parkinsonian symptoms. Note, however, that the precise meaning of these statements depends on the definition of synchrony. The models used in some works in this vein consist of phase equations. Such equations treat each neuron as an intrinsic oscillator, characterized by a time-dependent phase θ ∈[0, 2π]. Alternatively, partial or integrodifferential equations can be used to track the time evolution of the density of phases within a neuronal population treated as a continuum, or discrete equations in higher dimensions can be used to represent each neuron and a corresponding phase for each neuron can be derived, as long as each neuron exhibits regular intrinsic oscillations. Synchrony of a discrete population of phase oscillators is measured using an order parameter, R(t). We note that R(t)=0 occurs whenever neuronal phases are equally distributed on [0, 2π]. Thus, the synchrony measure gives the same result whether neurons are fully desynchronized or they form perfectly synchronized clusters that are uniformly spaced in phase. So, activity that is still very structured can be characterized as desynchronized, and this issue must be borne in mind in considering results based on an order parameter definition of synchrony.
Starting from the assumptions that the parkinsonian BG is characterized by a stable regime of synchronized oscillations of a collection of intrinsic oscillators and that DBS works through desynchronization, Tass et al. have shown that several consequences emerge. One major consequence is that non-standard DBS paradigms may be more effective than standard high frequency DBS at achieving a non-pathological state. Indeed, Tass and collaborators have been creative in proposing a variety of DBS techniques, including two-pulse (or soft-resetting) stimulation, multisite stimulation, and delayed feedback stimulation based on LFP signals (see also, Rosenblum & Pikovsky, 2004a; Rosenblum & Pikovsky, 2004b; Tukhlina et al., 2007), that work well at achieving desynchronization in their computational models. Preliminary testing in patients with some of these ideas has been promising (Tass, 2011).
Suppose, within this framework, that we make the additional assumption that there is some form of plasticity that can occur in the BG network. The parkinsonian state would have to be stable despite the presence of this plasticity. DBS, however, represents an external forcing to the system. So as long as DBS is present, the stable states of the system would likely differ from what they were without DBS. Thus, whatever quantities in the system were involved in its plasticity mechanisms could behave differently in the presence of DBS than in its absence. If DBS were subsequently removed, then the system would be in a different state than it was in before the application of DBS, such that it could possibly converge to a different state than it was in previously. This insight opens the door to the possibility that the right form of DBS, applied for a sufficient amount of time, could alter the previously parkinsonian BG in such a way that DBS would be no longer needed to avoid pathological activity.
As a specific example of this principle, Tass and collaborators consider synaptic plasticity within the BG (Tass & Majtanik, 2006; Hauptmann & Tass, 2010). Their results show that if excitatory connections are assumed to exist between pairs of neurons within the STN, and the strengths of these connections evolve via certain spike timing-dependent plasticity rules, then multisite coordinated reset stimulation (MCRS) of STN will cause these to change. Once DBS is removed, the model network is in the basin of attraction of a different stable state than it had been before DBS was applied. Thus, from its new state, the system will evolve toward a different activity regime lacking parkinsonian synchronization. Effectively, the DBS pushes the BG out of its pathological state, such that DBS is no longer needed. Alternatively, the new state that is achieved by the time that DBS is terminated may fail to be stable, but it may be sufficiently far removed from the pathological state that the return to pathology is very gradual. In this case, while DBS could not be eliminated indefinitely, its delivery could be interrupted, avoiding buildup of tissue damage, use of battery power, and induction of DBS-related side effects for some period until the pathological state is recovered to an extent that DBS is needed again.
While very appealing for its elegance and its potential to improve the how DBS is applied, this theory in its current formulation does depend critically on STDP in connections among excitatory neurons within the STN, the presence of which currently lacks experimental confirmation. It is possible that the general theory is correct but that the site of plasticity is elsewhere in BG, an idea that remains to be explored. The logic of these ideas leads to the prediction that if MCRS is applied for a period of time that effectively harnesses plasticity, then it should be possible to turn it off after this time and observe sustained elimination of symptoms. While the experimental search for the presence of long-term synaptic plasticity somewhere in the BG and for the relevance between such plasticity and the efficacy of DBS could prove arduous, the testing of this prediction about the removal of MCRS should be more immediately accessible.
10. Other effects of DBS treatment
There is a growing body of evidence suggesting that stimulation effects in the STN may not be entirely explained by effects of the stimulation on basal ganglia output. In particular, optogenetic and electrophysiologic studies in experimental animals have suggested that antidromic activation of motor cortical areas occurs and may be relevant for the clinical effects of STN DBS (Li et al., 2007; Dejean et al., 2009; Gradinaru et al., 2009), explained by the proximity of the stimulation site in the STN to the internal capsule and the existence of the corticosubthalamic pathway. Experiments in human patients in whom cortical evoked potentials were recorded have also arrived at the conclusion that antidromic stimulation effects occur in STN DBS (Ashby et al., 2001; Hanajima et al., 2004; MacKinnon et al., 2005; Kuriakose et al., 2010; Devergnas & Wichmann, 2011). Such effects are much less likely to occur in GPi DBS, because the electrodes are positioned away from the internal capsule, and GPi does not receive direct cortical input (Devergnas & Wichmann, 2011). These antidromic effects of DBS, as well as possible downstream effects of the stimulation of fibers of passage that reach brain stem and spinal cord, have not yet been incorporated into the existing models of DBS effects, representing a significant and exciting challenge for this this field. We also note that theories focusing on thalamocortical relay implicitly predict that cortical alterations will arise in parkinsonism due to compromised relay, and that STN DBS will modify these cortical effects.
11. Conclusions
We have reviewed many of the changes in activity observed in parkinsonism in animal models and human PD patients. Our review includes a discussion of the connection between these changes and the expression of motor aspects of parkinsonism, but these relationships are currently not well understood, and much new work is needed to firmly establish the pathways through which modulations of BG activity lead to motor and other effects. A variety of computational models exist that are relevant to BG activity patterns under various conditions (e.g., tremor-band oscillations in the STN-GPe network (Terman et al., 2002) and beta or other oscillations (Leblois et al., 2006; Holgado et al., 2010; McCarthy et al., 2011), the emergence of particular clinical conditions (e.g., bradykinesia, see Cutsuridis & Perantonis, 2006; Moroney et al., 2008), and the application of DBS to the parkinsonian BG (e.g., Wilson et al., 2011). Some of these are reviewed elsewhere, with an emphasis on parkinsonian activity and DBS (Titcombe et al., 2004; Modolo et al., 2011). Rather than providing a direct review or overview of the galaxy of computational models of BG, we have provided a new perspective on the logic underlying a small number of modeling frameworks that focus on downstream effects of parkinsonian activity and possible mechanisms for the therapeutic efficacy of DBS. In particular, some of these models provide mechanistic hypotheses for how parkinsonian activity may compromise downstream thalamic function and suggest that the effectiveness of certain therapies may relate to their impact on this processing, and these ideas merit future experimental exploration. Our discussion emphasizes the key assumptions and predictions of the computational modeling approaches that we consider. A careful detailing of the logical underpinnings of computational models is crucial for their utility, since this step makes clear where experiments should be directed to attempt to falsify the hypotheses that they embody and thereby refine our understanding of the biological components that they represent.
Acknowledgements
The writing of this review was supported by the following grants: NIH R01 NS070865 (JR, RT), NSF DMS1021701 (JR), NIH R01 NS047388 (CM), NIH P50 NS071669 (TW), NIH R01 NS062876 (TW), NIH R01 NS054976 (TW), NIH P51 RR-000165 (Yerkes National Primate Research Center).
Abbreviations
- BG
basal ganglia
- CM
centromedian nucleus of the thalamus
- DBS
deep brain stimulation
- EMG
electromyogram
- GABA
gamma-amino butyric acid
- GPe
external segment of the globus pallidus
- GPi
internal segment of the globus pallidus
- LFP
local field potential
- MCRS
multisite coordinated reset stimulation
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MSN
medium spiny neuron
- PD
Parkinson’s disease
- PPN
pedunculopontine nucleus
- RT model
Rubin/Terman model
- SNc
substantia nigra, pars compacta
- SNr
substantia nigra, pars reticulata
- STN
subthalamic nucleus
- TC
thalamocortical
- VA
ventral anterior nucleus of the thalamus
- VLa
anterior portion of the ventrolateral nucleus of the thalamus
Footnotes
The authors have no conflict of interest to declare.
References
- Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–375. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- Alegre M, Labarga A, Gurtubay IG, Iriarte J, Malanda A, Artieda J. Beta electroencephalograph changes during passive movements: sensory afferences contribute to beta event-related desynchronization in humans. Neurosci Lett. 2002;331:29–32. doi: 10.1016/s0304-3940(02)00825-x. [DOI] [PubMed] [Google Scholar]
- Alexander GE, Crutcher MD, DeLong MR. Basal ganglia-thalamocortical circuits: parallel substrates for motor, oculomotor, `prefrontal' and `limbic' functions. Prog. Brain Res. 1990;85:119–146. [PubMed] [Google Scholar]
- Alvarez L, Macias R, Lopez G, Alvarez E, Pavon N, Rodriguez-Oroz MC, Juncos JL, Maragoto C, Guridi J, Litvan I, Tolosa ES, Koller W, Vitek J, DeLong MR, Obeso JA. Bilateral subthalamotomy in Parkinson's disease: initial and long-term response. Brain. 2005;128:570–583. doi: 10.1093/brain/awh397. [DOI] [PubMed] [Google Scholar]
- Anderson ME, Postupna N, Ruffo M. Effects of high-frequency stimulation in the internal globus pallidus on the activity of thalamic neurons in the awake monkey. J. Neurophys. 2003;89:1150–1160. doi: 10.1152/jn.00475.2002. [DOI] [PubMed] [Google Scholar]
- Ashby P, Paradiso G, Saint-Cyr JA, Chen R, Lang AE, Lozano AM. Potentials recorded at the scalp by stimulation near the human subthalamic nucleus. Clin Neurophysiol. 2001;112:431–437. doi: 10.1016/s1388-2457(00)00532-0. [DOI] [PubMed] [Google Scholar]
- Aymerich MS, Barroso-Chinea P, Perez-Manso M, Munoz-Patino AM, Moreno-Igoa M, Gonzalez-Hernandez T, Lanciego JL. Consequences of unilateral nigrostriatal denervation on the thalamostriatal pathway in rats. Eur. J. Neurosci. 2006;23:2099–2108. doi: 10.1111/j.1460-9568.2006.04741.x. [DOI] [PubMed] [Google Scholar]
- Baron MS, Wichmann T, Ma D, DeLong MR. Effects of transient focal inactivation of the basal ganglia in parkinsonian primates. J. Neurosci. 2002;22:592–599. doi: 10.1523/JNEUROSCI.22-02-00592.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barraud Q, Lambrecq V, Forni C, McGuire S, Hill M, Bioulac B, Balzamo E, Bezard E, Tison F, Ghorayeb I. Sleep disorders in Parkinson's disease: the contribution of the MPTP non-human primate model. Exp Neurol. 2009;219:574–582. doi: 10.1016/j.expneurol.2009.07.019. [DOI] [PubMed] [Google Scholar]
- Baufreton J, Garret M, Rivera A, de la Calle A, Gonon F, Dufy B, Bioulac B, Taupignon A. D5 (not D1) dopamine receptors potentiate burst-firing in neurons of the subthalamic nucleus by modulating an L-type calcium conductance. J. Neurosci. 2003;23:816–825. doi: 10.1523/JNEUROSCI.23-03-00816.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman H, Wichmann T, DeLong MR. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science. 1990;249:1436–1438. doi: 10.1126/science.2402638. [DOI] [PubMed] [Google Scholar]
- Bergman H, Wichmann T, Karmon B, DeLong MR. The primate subthalamic nucleus. II. Neuronal activity in the MPTP model of parkinsonism. J. Neurophys. 1994;72:507–520. doi: 10.1152/jn.1994.72.2.507. [DOI] [PubMed] [Google Scholar]
- Bezard E, Boraud T, Bioulac B, Gross CE. Involvement of the subthalamic nucleus in glutamatergic compensatory mechanisms. Eur. J. Neurosci. 1999;11:2167–2170. doi: 10.1046/j.1460-9568.1999.00627.x. [DOI] [PubMed] [Google Scholar]
- Birdno MJ, Cooper SE, Rezai AR, Grill WM. Pulse-to-Pulse Changes in the Frequency of Deep Brain Stimulation Affect Tremor and Modeled Neuronal Activity. J. Neurophys. 2007;98:1675–1684. doi: 10.1152/jn.00547.2007. [DOI] [PubMed] [Google Scholar]
- Bohnen NI, Albin RL. White matter lesions in Parkinson disease. Nature reviews. Neurology. 2011;7:229–236. doi: 10.1038/nrneurol.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostan AC, Dum RP, Strick PL. The basal ganglia communicate with the cerebellum. Proc Natl Acad Sci U S A. 2010;107:8452–8456. doi: 10.1073/pnas.1000496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Pathoanatomy of Parkinson's disease. J Neurol. 2000;247(Suppl 2):II3–II10. doi: 10.1007/PL00007758. [DOI] [PubMed] [Google Scholar]
- Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K. Stages in the development of Parkinson's disease-related pathology. Cell Tissue Res. 2004;318:121–134. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- Breit S, Bouali-Benazzouz R, Popa RC, Gasser T, Benabid AL, Benazzouz A. Effects of 6-hydroxydopamine-induced severe or partial lesion of the nigrostriatal pathway on the neuronal activity of pallido-subthalamic network in the rat. Exp. Neurol. 2007;205:36–47. doi: 10.1016/j.expneurol.2006.12.016. [DOI] [PubMed] [Google Scholar]
- Bronfeld M, Bar-Gad I. Loss of specificity in Basal Ganglia related movement disorders. Front Syst Neurosci. 2011;5:38. doi: 10.3389/fnsys.2011.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronte-Stewart H, Barberini C, Koop MM, Hill BC, Henderson JM, Wingeier B. The STN beta-band profile in Parkinson's disease is stationary and shows prolonged attenuation after deep brain stimulation. Exp Neurol. 2009;215:20–28. doi: 10.1016/j.expneurol.2008.09.008. [DOI] [PubMed] [Google Scholar]
- Brotchie JM, Mitchell IJ, Sambrook MA, Crossman AR. Alleviation of parkinsonism by antagonism of excitatory amino acid transmission in the medial segment of the globus pallidus in rat and primate. Mov Disord. 1991;6:133–138. doi: 10.1002/mds.870060208. [DOI] [PubMed] [Google Scholar]
- Brown P. Oscillatory nature of human basal ganglia activity: relationship to the pathophysiology of Parkinson's disease. Mov Disord. 2003;18:357–363. doi: 10.1002/mds.10358. [DOI] [PubMed] [Google Scholar]
- Brown P, Oliviero A, Mazzone P, Insola A, Tonali P, Di Lazzaro V. Dopamine dependency of oscillations between subthalamic nucleus and pallidum in Parkinson's disease. J. Neurosci. 2001;21:1033–1038. doi: 10.1523/JNEUROSCI.21-03-01033.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown P, Williams D. Basal ganglia local field potential activity: character and functional significance in the human. Clin Neurophysiol. 2005;116:2510–2519. doi: 10.1016/j.clinph.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Cagnan H, Meijer HG, van Gils SA, Krupa M, Heida T, Rudolph M, Wadman WJ, Martens HC. Frequency-selectivity of a thalamocortical relay neuron during Parkinson's disease and deep brain stimulation: a computational study. Eur J Neurosci. 2009;30:1306–1317. doi: 10.1111/j.1460-9568.2009.06922.x. [DOI] [PubMed] [Google Scholar]
- Cassidy M, Mazzone P, Oliviero A, Insola A, Tonali P, Di Lazzaro V, Brown P. Movement-related changes in synchronization in the human basal ganglia. Brain. 2002;125:1235–1246. doi: 10.1093/brain/awf135. [DOI] [PubMed] [Google Scholar]
- Chan V, Starr PA, Turner RS. Bursts and oscillations as independent properties of neural activity in the parkinsonian globus pallidus internus. Neurobiol Dis. 2011;41:2–10. doi: 10.1016/j.nbd.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Litvak V, Gilbertson T, Kuhn A, Lu CS, Lee ST, Tsai CH, Tisch S, Limousin P, Hariz M, Brown P. Excessive synchronization of basal ganglia neurons at 20 Hz slows movement in Parkinson's disease. Exp. Neurol. 2007;205:214–221. doi: 10.1016/j.expneurol.2007.01.027. [DOI] [PubMed] [Google Scholar]
- Chen H, Zhuang P, Miao SH, Yuan G, Zhang YQ, Li JY, Li YJ. Neuronal firing in the ventrolateral thalamus of patients with Parkinson's disease differs from that with essential tremor. Chin Med J (Engl) 2010;123:695–701. [PubMed] [Google Scholar]
- Courtemanche R, Fujii N, Graybiel AM. Synchronous, focally modulated beta-band oscillations characterize local field potential activity in the striatum of awake behaving monkeys. J. Neurosci. 2003;23:11741–11752. doi: 10.1523/JNEUROSCI.23-37-11741.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossman AR, Mitchell IJ, Sambrook MA. Regional brain uptake of 2-deoxyglucose in N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism in the macaque monkey. Neuropharmacology. 1985;24:587–591. doi: 10.1016/0028-3908(85)90070-x. [DOI] [PubMed] [Google Scholar]
- Cutsuridis V, Perantonis S. A neural network model of Parkinson's disease bradykinesia. Neural Netw. 2006;19:354–374. doi: 10.1016/j.neunet.2005.08.016. [DOI] [PubMed] [Google Scholar]
- Day M, Wang Z, Ding J, An X, Ingham CA, Shering AF, Wokosin D, Ilijic E, Sun Z, Sampson AR, Mugnaini E, Deutch AY, Sesack SR, Arbuthnott GW, Surmeier DJ. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat Neurosci. 2006;9:251–259. doi: 10.1038/nn1632. [DOI] [PubMed] [Google Scholar]
- Dayan P, Abbott LF. Theoretical Neuroscience. Cambridge, MA: The MIT Press; 2001. [Google Scholar]
- Degos B, Deniau JM, Chavez M, Maurice N. Chronic but not acute dopaminergic transmission interruption promotes a progressive increase in cortical beta frequency synchronization: relationships to vigilance state and akinesia. Cereb Cortex. 2009;19:1616–1630. doi: 10.1093/cercor/bhn199. [DOI] [PubMed] [Google Scholar]
- Dejean C, Hyland B, Arbuthnott G. Cortical effects of subthalamic stimulation correlate with behavioral recovery from dopamine antagonist induced akinesia. Cereb Cortex. 2009;19:1055–1063. doi: 10.1093/cercor/bhn149. [DOI] [PubMed] [Google Scholar]
- DeLong MR. Activity of pallidal neurons during movement. J. Neurophys. 1971;34:414–427. doi: 10.1152/jn.1971.34.3.414. [DOI] [PubMed] [Google Scholar]
- DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends in Neurosciences. 1990;13:281–285. doi: 10.1016/0166-2236(90)90110-v. [DOI] [PubMed] [Google Scholar]
- DeLong MR, Crutcher MD, Georgopoulos AP. Primate globus pallidus and subthalamic nucleus: functional organization. J.Neurophysiol. 1985;53:530–543. doi: 10.1152/jn.1985.53.2.530. [DOI] [PubMed] [Google Scholar]
- Deniau JM, Chevalier G. Disinhibition as a basic process in the expression of striatal functions. II. The striato-nigral influence on thalamocortical cells of the ventromedial thalamic nucleus. Brain Res. 1985;334:227–233. doi: 10.1016/0006-8993(85)90214-8. [DOI] [PubMed] [Google Scholar]
- Destexhe A, Contreras D, Steriade M. Mechanisms underlying the synchronizing action of corticothalamic feedback through inhibition of thalamic relay cells. J. Neurophys. 1998;79:999–1016. doi: 10.1152/jn.1998.79.2.999. [DOI] [PubMed] [Google Scholar]
- Devergnas A, Wichmann T. Cortical potentials evoked by deep brain stimulation in the subthalamic area. Front Syst Neurosci. 2011;5:30. doi: 10.3389/fnsys.2011.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVito JL, Anderson ME. An autoradiographic study of efferent connections of the globus pallidus in Macaca mulatta. Exp. Brain Res. 1982;46:107–117. doi: 10.1007/BF00238104. [DOI] [PubMed] [Google Scholar]
- Dorval AD, Kuncel AM, Birdno MJ, Turner DA, Grill WM. Deep brain stimulation alleviates parkinsonian bradykinesia by regularizing pallidal activity. J. Neurophys. 2010;104:911–921. doi: 10.1152/jn.00103.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudet DJ, Gross C, Arluison M, Bioulac B. Modifications of precentral cortex discharge and EMG activity in monkeys with MPTP-induced lesions of DA nigral neurons. Exp. Brain Res. 1990;80:177–188. doi: 10.1007/BF00228859. [DOI] [PubMed] [Google Scholar]
- Doyle LM, Kuhn AA, Hariz M, Kupsch A, Schneider GH, Brown P. Levodopa-induced modulation of subthalamic beta oscillations during self-paced movements in patients with Parkinson's disease. Eur. J. Neurosci. 2005a;21:1403–1412. doi: 10.1111/j.1460-9568.2005.03969.x. [DOI] [PubMed] [Google Scholar]
- Doyle LM, Yarrow K, Brown P. Lateralization of event-related beta desynchronization in the EEG during pre-cued reaction time tasks. Clin Neurophysiol. 2005b;116:1879–1888. doi: 10.1016/j.clinph.2005.03.017. [DOI] [PubMed] [Google Scholar]
- Elder CM, Vitek JL. The motor thalamus: Alteration of neuronal activity in the parkinsonian state. In: Kultas-Ilinsky K, Ilinsky IA, editors. Basal Ganglia and Thalamus in Health and Movement Disorders. New York: Kluwer Academic; 2001. [Google Scholar]
- Emborg ME, Carbon M, Holden JE, During MJ, Ma Y, Tang C, Moirano J, Fitzsimons H, Roitberg BZ, Tuccar E, Roberts A, Kaplitt MG, Eidelberg D. Subthalamic glutamic acid decarboxylase gene therapy: changes in motor function and cortical metabolism. J Cereb Blood Flow Metab. 2007;27:501–509. doi: 10.1038/sj.jcbfm.9600364. [DOI] [PubMed] [Google Scholar]
- Ermentrout GB, Terman DH. The Mathematical Foundations of Neuroscience. New York, NY: Springer Verlag; 2010. [Google Scholar]
- Eusebio A, Pogosyan A, Wang S, Averbeck B, Gaynor LD, Cantiniaux S, Witjas T, Limousin P, Azulay JP, Brown P. Resonance in subthalamo-cortical circuits in Parkinson's disease. Brain. 2009;132:2139–2150. doi: 10.1093/brain/awp079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filion M, Tremblay L. Abnormal spontaneous activity of globus pallidus neurons in monkeys with MPTP-induced parkinsonism. Brain Res. 1991;547:142–151. [PubMed] [Google Scholar]
- Filion M, Tremblay L, Bedard PJ. Abnormal influences of passive limb movement on the activity of globus pallidus neurons in parkinsonian monkeys. Brain Res. 1988;444:165–176. doi: 10.1016/0006-8993(88)90924-9. [DOI] [PubMed] [Google Scholar]
- Foffani G, Priori A. Deep brain stimulation in Parkinson's disease can mimic the 300 Hz subthalamic rhythm. Brain. 2006;129:e59. doi: 10.1093/brain/awl209. author reply e60. [DOI] [PubMed] [Google Scholar]
- Foffani G, Priori A, Egidi M, Rampini P, Tamma F, Caputo E, Moxon KA, Cerutti S, Barbieri S. 300-Hz subthalamic oscillations in Parkinson's disease. Brain. 2003;126:2153–2163. doi: 10.1093/brain/awg229. [DOI] [PubMed] [Google Scholar]
- Fogelson N, Williams D, Tijssen M, van Bruggen G, Speelman H, Brown P. Different Functional Loops between Cerebral Cortex and the Subthalmic Area in Parkinson's Disease. Cerebr. Cortex. 2005 doi: 10.1093/cercor/bhi084. [DOI] [PubMed] [Google Scholar]
- Freyaldenhoven TE, Ali SF, Schmued LC. Systemic administration of MPTP induces thalamic neuronal degeneration in mice. Brain Res. 1997;759:9–17. doi: 10.1016/s0006-8993(97)00045-0. [DOI] [PubMed] [Google Scholar]
- Galvan A, Wichmann T. Pathophysiology of parkinsonism. Clin. Neurophysiol. 2008;119:1459–1474. doi: 10.1016/j.clinph.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia L, Audin J, D'Alessandro G, Bioulac B, Hammond C. Dual effect of high-frequency stimulation on subthalamic neuron activity. J. Neurosci. 2003;23:8743–8751. doi: 10.1523/JNEUROSCI.23-25-08743.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia L, D'Alessandro G, Bioulac B, Hammond C. High-frequency stimulation in Parkinson's disease: more or less? Trends in Neurosciences. 2005;28:209–216. doi: 10.1016/j.tins.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Gatev P, Darbin O, Wichmann T. Oscillations in the basal ganglia under normal conditions and in movement disorders. Mov Disord. 2006;21:1566–1577. doi: 10.1002/mds.21033. [DOI] [PubMed] [Google Scholar]
- Gatev PG, Wichmann T. Changes In Arousal Alter Neuronal Activity In Primate Basal Ganglia. Soc. Neurosci. Abstr. 2003;29 [Google Scholar]
- Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ, Jr, Sibley DR. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250:1429–1432. doi: 10.1126/science.2147780. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghorayeb I, Fernagut PO, Hervier L, Labattu B, Bioulac B, Tison F. A 'single toxin-double lesion' rat model of striatonigral degeneration by intrastriatal 1-methyl-4-phenylpyridinium ion injection: a motor behavioural analysis. Neurosci. 2002;115:533–546. doi: 10.1016/s0306-4522(02)00401-3. [DOI] [PubMed] [Google Scholar]
- Gill SS, Heywood P. Bilateral dorsolateral subthalamotomy for advanced Parkinson's disease. Lancet. 1997;350:1224. doi: 10.1016/s0140-6736(05)63455-1. [DOI] [PubMed] [Google Scholar]
- Goldberg JA, Boraud T, Maraton S, Haber SN, Vaadia E, Bergman H. Enhanced synchrony among primary motor cortex neurons in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine primate model of Parkinson's disease. J. Neurosci. 2002;22:4639–4653. doi: 10.1523/JNEUROSCI.22-11-04639.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JA, Rokni U, Boraud T, Vaadia E, Bergman H. Spike synchronization in the cortex/basal-ganglia networks of Parkinsonian primates reflects global dynamics of the local field potentials. J. Neurosci. 2004;24:6003–6010. doi: 10.1523/JNEUROSCI.4848-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golomb D, Wang XJ, Rinzel J. Synchronization properties of spindle oscillations in a thalamic reticular nucleus model. J Neurophysiol. 1994;72:1109–1126. doi: 10.1152/jn.1994.72.3.1109. [DOI] [PubMed] [Google Scholar]
- Gradinaru V, Mogri M, Thompson KR, Henderson JM, Deisseroth K. Optical deconstruction of parkinsonian neural circuitry. Science. 2009;324:354–359. doi: 10.1126/science.1167093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill WM, Snyder AN, Miocinovic S. Deep brain stimulation creates an informational lesion of the stimulated nucleus. Neuroreport. 2004;15:1137–1140. doi: 10.1097/00001756-200405190-00011. [DOI] [PubMed] [Google Scholar]
- Guehl D, Pessiglione M, Francois C, Yelnik J, Hirsch EC, Feger J, Tremblay L. Tremor-related activity of neurons in the 'motor' thalamus: changes in firing rate and pattern in the MPTP vervet model of parkinsonism. Eur. J. Neurosci. 2003;17:2388–2400. doi: 10.1046/j.1460-9568.2003.02685.x. [DOI] [PubMed] [Google Scholar]
- Guo Y, Rubin JE, McIntyre CC, Vitek JL, Terman D. Thalamocortical relay fidelity varies across subthalamic nucleus deep brain stimulation protocols in a data-driven computational model. J. Neurophys. 2008;99:1477–1492. doi: 10.1152/jn.01080.2007. [DOI] [PubMed] [Google Scholar]
- Hahn PJ, McIntyre CC. Modeling shifts in the rate and pattern of subthalamopallidal network activity during deep brain stimulation. J Comput Neurosci. 2010;28:425–441. doi: 10.1007/s10827-010-0225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn PJ, Russo GS, Hashimoto T, Miocinovic S, Xu W, McIntyre CC, Vitek JL. Pallidal burst activity during therapeutic deep brain stimulation. Exp. Neurol. 2008;211:243–251. doi: 10.1016/j.expneurol.2008.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C, Bergman H, Brown P. Pathological synchronization in Parkinson's disease: networks, models and treatments. Trends Neurosci. 2007;30:357–364. doi: 10.1016/j.tins.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Hanajima R, Ashby P, Lozano AM, Lang AE, Chen R. Single pulse stimulation of the human subthalamic nucleus facilitates the motor cortex at short intervals. J. Neurophys. 2004;92:1937–1943. doi: 10.1152/jn.00239.2004. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Elder CM, Okun MS, Patrick SK, Vitek JL. Stimulation of the subthalamic nucleus changes the firing pattern of pallidal neurons. J. Neurosci. 2003;23:1916–1923. doi: 10.1523/JNEUROSCI.23-05-01916.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauptmann C, Tass PA. Restoration of segregated, physiological neuronal connectivity by desynchronizing stimulation. J Neural Eng. 2010;7 doi: 10.1088/1741-2560/7/5/056008. 056008. [DOI] [PubMed] [Google Scholar]
- Heimer G, Bar-Gad I, Goldberg JA, Bergman H. Dopamine replacement therapy reverses abnormal synchronization of pallidal neurons in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine primate model of parkinsonism. J. Neurosci. 2002;22:7850–7855. doi: 10.1523/JNEUROSCI.22-18-07850.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimer G, Rivlin-Etzion M, Bar-Gad I, Goldberg JA, Haber SN, Bergman H. Dopamine replacement therapy does not restore the full spectrum of normal pallidal activity in the 1-methyl-4-phenyl-1,2,3,6-tetra-hydropyridine primate model of Parkinsonism. J. Neurosci. 2006;26:8101–8114. doi: 10.1523/JNEUROSCI.5140-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson JM, Carpenter K, Cartwright H, Halliday GM. Degeneration of the centre median-parafascicular complex in Parkinson's disease. Ann. Neurol. 2000a;47:345–352. [PubMed] [Google Scholar]
- Henderson JM, Carpenter K, Cartwright H, Halliday GM. Loss of thalamic intralaminar nuclei in progressive supranuclear palsy and Parkinson's disease: clinical and therapeutic implications. Brain. 2000b;123:1410–1421. doi: 10.1093/brain/123.7.1410. [DOI] [PubMed] [Google Scholar]
- Henderson JM, Schleimer SB, Allbutt H, Dabholkar V, Abela D, Jovic J, Quinlivan M. Behavioural effects of parafascicular thalamic lesions in an animal model of parkinsonism. Behav Brain Res. 2005;162:222–232. doi: 10.1016/j.bbr.2005.03.017. [DOI] [PubMed] [Google Scholar]
- Hershey T, Revilla FJ, Wernle AR, McGee-Minnich L, Antenor JV, Videen TO, Dowling JL, Mink JW, Perlmutter JS. Cortical and subcortical blood flow effects of subthalamic nucleus stimulation in PD. Neurology. 2003;61:816–821. doi: 10.1212/01.wnl.0000083991.81859.73. [DOI] [PubMed] [Google Scholar]
- Holgado AJ, Terry JR, Bogacz R. Conditions for the generation of beta oscillations in the subthalamic nucleus-globus pallidus network. J Neurosci. 2010;30:12340–12352. doi: 10.1523/JNEUROSCI.0817-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover JE, Strick PL. Multiple output channels in the basal ganglia. Science. 1993;259:819–821. doi: 10.1126/science.7679223. [DOI] [PubMed] [Google Scholar]
- Hornykiewicz O, Kish SJ. Biochemical pathophysiology of Parkinson's disease. Adv. Neurol. 1987;45:19–34. [PubMed] [Google Scholar]
- Hoshi E, Tremblay L, Feger J, Carras PL, Strick PL. The cerebellum communicates with the basal ganglia. Nature Neuroscience. 2005;8:1491–1493. doi: 10.1038/nn1544. [DOI] [PubMed] [Google Scholar]
- Hutchison WD, Lozano AM, Davis K, Saint-Cyr JA, Lang AE, Dostrovsky JO. Differential neuronal activity in segments of globus pallidus in Parkinson's disease patients. Neuroreport. 1994;5:1533–1537. doi: 10.1097/00001756-199407000-00031. [DOI] [PubMed] [Google Scholar]
- Inase M, Buford JA, Anderson ME. Changes in the control of arm position, movement, and thalamic discharge during local inactivation in the globus pallidus of the monkey. J. Neurophys. 1996;75:1087–1104. doi: 10.1152/jn.1996.75.3.1087. [DOI] [PubMed] [Google Scholar]
- Ingham CA, Hood SH, Arbuthnott GW. Spine density on neostriatal neurones changes with 6-hydroxydopamine lesions and with age. Brain Res. 1989;503:334–338. doi: 10.1016/0006-8993(89)91686-7. [DOI] [PubMed] [Google Scholar]
- Izhikevich EM. Dynamical Systems in Neuroscience: The Geometry of Excitability and Bursting. Cambridge, MA: The MIT Press; 2007. [Google Scholar]
- Jahnsen H, Llinas R. Electrophysiological properties of guinea-pig thalamic neurones: an in vitro study. J.Physiol. 1984a;349:205–226. doi: 10.1113/jphysiol.1984.sp015153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnsen H, Llinas R. Ionic basis for the electro-responsiveness and oscillatory properties of guinea-pig thalamic neurones in vitro. J Physiol (Lond) 1984b;349:227–247. doi: 10.1113/jphysiol.1984.sp015154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jech R, Urgosik D, Tintera J, Nebuzelsky A, Krasensky J, Liscak R, Roth J, Ruzicka E. Functional magnetic resonance imaging during deep brain stimulation: a pilot study in four patients with Parkinson's disease. Mov Disord. 2001;16:1126–1132. doi: 10.1002/mds.1217. [DOI] [PubMed] [Google Scholar]
- Jones EG. The thalamus. New York, NY: Cambridge University Press; 2007. [Google Scholar]
- Kaneoke Y, Vitek JL. Burst and oscillation as disparate neuronal properties. J. Neurosci. Methods. 1996;68:211–223. doi: 10.1016/0165-0270(96)00081-7. [DOI] [PubMed] [Google Scholar]
- Kempf F, Brucke C, Salih F, Trottenberg T, Kupsch A, Schneider GH, Doyle Gaynor LM, Hoffmann KT, Vesper J, Wohrle J, Altenmuller DM, Krauss JK, Mazzone P, Di Lazzaro V, Yelnik J, Kuhn AA, Brown P. Gamma activity and reactivity in human thalamic local field potentials. Eur. J. Neurosci. 2009;29:943–953. doi: 10.1111/j.1460-9568.2009.06655.x. [DOI] [PubMed] [Google Scholar]
- Kempf F, Kuhn AA, Kupsch A, Brucke C, Weise L, Schneider GH, Brown P. Premovement activities in the subthalamic area of patients with Parkinson's disease and their dependence on task. Eur J Neurosci. 2007;25:3137–3145. doi: 10.1111/j.1460-9568.2007.05536.x. [DOI] [PubMed] [Google Scholar]
- Kliem MA, Maidment NT, Ackerson LC, Chen S, Smith Y, Wichmann T. Activation of nigral and pallidal dopamine D1-like receptors modulates basal ganglia outflow in monkeys. J. Neurophys. 2007;98:1489–1500. doi: 10.1152/jn.00171.2007. [DOI] [PubMed] [Google Scholar]
- Kravitz AV, Freeze BS, Parker PR, Kay K, Thwin MT, Deisseroth K, Kreitzer AC. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature. 2010;466:622–626. doi: 10.1038/nature09159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota S, Rubin JE. NMDA-induced burst firing in a model subthalamic nucleus neuron. J Neurophysiol. 2011;106:527–537. doi: 10.1152/jn.01127.2010. [DOI] [PubMed] [Google Scholar]
- Kuhn AA, Doyle L, Pogosyan A, Yarrow K, Kupsch A, Schneider GH, Hariz MI, Trottenberg T, Brown P. Modulation of beta oscillations in the subthalamic area during motor imagery in Parkinson's disease. Brain. 2006;129:695–706. doi: 10.1093/brain/awh715. [DOI] [PubMed] [Google Scholar]
- Kuhn AA, Kempf F, Brucke C, Gaynor Doyle L, Martinez-Torres I, Pogosyan A, Trottenberg T, Kupsch A, Schneider GH, Hariz MI, Vandenberghe W, Nuttin B, Brown P. High-frequency stimulation of the subthalamic nucleus suppresses oscillatory beta activity in patients with Parkinson's disease in parallel with improvement in motor performance. J. Neurosci. 2008;28:6165–6173. doi: 10.1523/JNEUROSCI.0282-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn AA, Trottenberg T, Kivi A, Kupsch A, Schneider GH, Brown P. The relationship between local field potential and neuronal discharge in the subthalamic nucleus of patients with Parkinson's disease. Exp. Neurol. 2005;194:212–220. doi: 10.1016/j.expneurol.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Kuhn AA, Tsui A, Aziz T, Ray N, Brucke C, Kupsch A, Schneider GH, Brown P. Pathological synchronisation in the subthalamic nucleus of patients with Parkinson's disease relates to both bradykinesia and rigidity. Exp Neurol. 2009;215:380–387. doi: 10.1016/j.expneurol.2008.11.008. [DOI] [PubMed] [Google Scholar]
- Kuhn AA, Williams D, Kupsch A, Limousin P, Hariz M, Schneider GH, Yarrow K, Brown P. Event-related beta desynchronization in human subthalamic nucleus correlates with motor performance. Brain. 2004;127:735–746. doi: 10.1093/brain/awh106. [DOI] [PubMed] [Google Scholar]
- Kuriakose R, Saha U, Castillo G, Udupa K, Ni Z, Gunraj C, Mazzella F, Hamani C, Lang AE, Moro E, Lozano AM, Hodaie M, Chen R. The nature and time course of cortical activation following subthalamic stimulation in Parkinson's disease. Cereb Cortex. 2010;20:1926–1936. doi: 10.1093/cercor/bhp269. [DOI] [PubMed] [Google Scholar]
- Laitinen LV. Pallidotomy for Parkinson's diesease. Neurosurg.Clin.N.Am. 1995;6:105–112. [PubMed] [Google Scholar]
- Lalo E, Thobois S, Sharott A, Polo G, Mertens P, Pogosyan A, Brown P. Patterns of bidirectional communication between cortex and basal ganglia during movement in patients with Parkinson disease. J. Neurosci. 2008;28:3008–3016. doi: 10.1523/JNEUROSCI.5295-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblois A, Boraud T, Meissner W, Bergman H, Hansel D. Competition between feedback loops underlies normal and pathological dynamics in the basal ganglia. J. Neurosci. 2006;26:3567–3583. doi: 10.1523/JNEUROSCI.5050-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblois A, Meissner W, Bioulac B, Gross CE, Hansel D, Boraud T. Late emergence of synchronized oscillatory activity in the pallidum during progressive Parkinsonism. Eur. J. Neurosci. 2007;26:1701–1713. doi: 10.1111/j.1460-9568.2007.05777.x. [DOI] [PubMed] [Google Scholar]
- Lee JI, Shin HJ, Nam DH, Kim JS, Hong SC, Park K, Eoh W, Kim JH, Lee WY. Increased burst firing in substantia nigra pars reticulata neurons and enhanced response to selective D2 agonist in hemiparkinsonian rats after repeated administration of apomorphine. J. Korean Med. Sci. 2001;16:636–642. doi: 10.3346/jkms.2001.16.5.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz FA, Tasker RR, Kwan HC, Schnider S, Kwong R, Murayama Y, Dostrovsky JO, Murphy JT. Single unit analysis of the human ventral thalamic nuclear group: correlation of thalamic "tremor cells" with the 3–6 Hz component of parkinsonian tremor. J. Neurosci. 1988;8:754–764. doi: 10.1523/JNEUROSCI.08-03-00754.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leocani L, Toro C, Manganotti P, Zhuang P, Hallett M. Event-related coherence and event-related desynchronization/synchronization in the 10 Hz and 20 Hz EEG during self-paced movements. Electroencephalogr Clin Neurophysiol. 1997;104:199–206. doi: 10.1016/s0168-5597(96)96051-7. [DOI] [PubMed] [Google Scholar]
- Levy R, Ashby P, Hutchison WD, Lang AE, Lozano AM, Dostrovsky JO. Dependence of subthalamic nucleus oscillations on movement and dopamine in Parkinson's disease. Brain. 2002a;125:1196–1209. doi: 10.1093/brain/awf128. [DOI] [PubMed] [Google Scholar]
- Levy R, Dostrovsky JO, Lang AE, Sime E, Hutchison WD, Lozano AM. Effects of apomorphine on subthalamic nucleus and globus pallidus internus neurons in patients with Parkinson's disease. J. Neurophys. 2001a;86:249–260. doi: 10.1152/jn.2001.86.1.249. [DOI] [PubMed] [Google Scholar]
- Levy R, Hutchison WD, Lozano AM, Dostrovsky JO. High-frequency synchronization of neuronal activity in the subthalamic nucleus of parkinsonian patients with limb tremor. J. Neurosci. 2000;20:7766–7775. doi: 10.1523/JNEUROSCI.20-20-07766.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy R, Hutchison WD, Lozano AM, Dostrovsky JO. Synchronized neuronal discharge in the basal ganglia of parkinsonian patients is limited to oscillatory activity. J. Neurosci. 2002b;22:2855–2861. doi: 10.1523/JNEUROSCI.22-07-02855.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy R, Lang AE, Dostrovsky JO, Pahapill P, Romas J, Saint-Cyr J, Hutchison WD, Lozano AM. Lidocaine and muscimol microinjections in subthalamic nucleus reverse Parkinsonian symptoms. Brain. 2001b;124:2105–2118. doi: 10.1093/brain/124.10.2105. [DOI] [PubMed] [Google Scholar]
- Lewitt PA, Rezai AR, Leehey MA, Ojemann SG, Flaherty AW, Eskandar EN, Kostyk SK, Thomas K, Sarkar A, Siddiqui MS, Tatter SB, Schwalb JM, Poston KL, Henderson JM, Kurlan RM, Richard IH, Van Meter L, Sapan CV, During MJ, Kaplitt MG, Feigin A. AAV2-GAD gene therapy for advanced Parkinson's disease: a double-blind, sham-surgery controlled, randomised trial. Lancet Neurol. 2011;10:309–319. doi: 10.1016/S1474-4422(11)70039-4. [DOI] [PubMed] [Google Scholar]
- Li S, Arbuthnott GW, Jutras MJ, Goldberg JA, Jaeger D. Resonant antidromic cortical circuit activation as a consequence of high-frequency subthalamic deep-brain stimulation. J. Neurophys. 2007;98:3525–3537. doi: 10.1152/jn.00808.2007. [DOI] [PubMed] [Google Scholar]
- Lieberman DM, Corthesy ME, Cummins A, Oldfield EH. Reversal of experimental parkinsonism by using selective chemical ablation of the medial globus pallidus. J. Neurosurg. 1999;90:928–934. doi: 10.3171/jns.1999.90.5.0928. [DOI] [PubMed] [Google Scholar]
- Lozano AM, Lang AE, Galvez-Jimenez N, Miyasaki J, Duff J, Hutchinson WD, Dostrovsky JO. Effect of GPi pallidotomy on motor function in Parkinson's disease. Lancet. 1995;346:1383–1387. doi: 10.1016/s0140-6736(95)92404-3. [DOI] [PubMed] [Google Scholar]
- Luo J, Kaplitt MG, Fitzsimons HL, Zuzga DS, Liu Y, Oshinsky ML, During MJ. Subthalamic GAD gene therapy in a Parkinson's disease rat model. Science. 2002;298:425–429. doi: 10.1126/science.1074549. [DOI] [PubMed] [Google Scholar]
- Ma Y, Wichmann T. Disruption of motor performance by basal ganglia stimulation. Society of Neuroscience Annual Meeting Abstracts. 2004 [Google Scholar]
- MacKinnon CD, Webb RM, Silberstein P, Tisch S, Asselman P, Limousin P, Rothwell JC. Stimulation through electrodes implanted near the subthalamic nucleus activates projections to motor areas of cerebral cortex in patients with Parkinson's disease. Eur. J. Neurosci. 2005;21:1394–1402. doi: 10.1111/j.1460-9568.2005.03952.x. [DOI] [PubMed] [Google Scholar]
- Magill PJ, Bolam JP, Bevan MD. Dopamine regulates the impact of the cerebral cortex on the subthalamic nucleus-globus pallidus network. Neurosci. 2001;106:313–330. doi: 10.1016/s0306-4522(01)00281-0. [DOI] [PubMed] [Google Scholar]
- Magill PJ, Sharott A, Bevan MD, Brown P, Bolam JP. Synchronous unit activity and local field potentials evoked in the subthalamic nucleus by cortical stimulation. J. Neurophys. 2004a;92:700–714. doi: 10.1152/jn.00134.2004. [DOI] [PubMed] [Google Scholar]
- Magill PJ, Sharott A, Bolam JP, Brown P. Brain state-dependency of coherent oscillatory activity in the cerebral cortex and basal ganglia of the rat. J. Neurophys. 2004b;92:2122–2136. doi: 10.1152/jn.00333.2004. [DOI] [PubMed] [Google Scholar]
- Magnin M, Morel A, Jeanmonod D. Single-unit analysis of the pallidum, thalamus and subthalamic nucleus in parkinsonian patients. Neurosci. 2000;96:549–564. doi: 10.1016/s0306-4522(99)00583-7. [DOI] [PubMed] [Google Scholar]
- Mallet N, Pogosyan A, Sharott A, Csicsvari J, Bolam JP, Brown P, Magill PJ. Disrupted dopamine transmission and the emergence of exaggerated beta oscillations in subthalamic nucleus and cerebral cortex. J. Neurosci. 2008;28:4795–4806. doi: 10.1523/JNEUROSCI.0123-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden JF, Limousin-Dowsey P, Ashby P, Pollak P, Brown P. Subthalamic nucleus, sensorimotor cortex and muscle interrelationships in Parkinson's disease. Brain. 2001;124:378–388. doi: 10.1093/brain/124.2.378. [DOI] [PubMed] [Google Scholar]
- Masilamoni GJ, Bogenpohl JW, Alagille D, Delevich K, Tamagnan G, Votaw JR, Wichmann T, Smith Y. Metabotropic glutamate receptor 5 antagonist protects dopaminergic and noradrenergic neurons from degeneration in MPTP-treated monkeys. Brain. 2011;134:2057–2073. doi: 10.1093/brain/awr137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathai A, Ma Y, Wichmann T, Smith Y. Glutamatergic inputs to the subthalamic nucleus degenerate in experimental parkinsonism. Soc. Neurosci. Abstracts. 2011 [Google Scholar]
- McCairn KW, Turner RS. Deep brain stimulation of the globus pallidus internus in the parkinsonian primate: local entrainment and suppression of low-frequency oscillations. J. Neurophys. 2009;101:1941–1960. doi: 10.1152/jn.91092.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MM, Moore-Kochlacs C, Gu X, Boyden ES, Han X, Kopell N. Striatal origin of the pathologic beta oscillations in Parkinson's disease. Proc Natl Acad Sci U S A. 2011;108:11620–11625. doi: 10.1073/pnas.1107748108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre CC, Grill WM, Sherman DL, Thakor NV. Cellular effects of deep brain stimulation: model-based analysis of activation and inhibition. J. Neurophys. 2004;91:1457–1469. doi: 10.1152/jn.00989.2003. [DOI] [PubMed] [Google Scholar]
- Meijer HG, Krupa M, Cagnan H, Lourens MA, Heida T, Martens HC, Bour LJ, van Gils SA. From Parkinsonian thalamic activity to restoring thalamic relay using deep brain stimulation: new insights from computational modeling. J Neural Eng. 2011;8 doi: 10.1088/1741-2560/8/6/066005. 066005. [DOI] [PubMed] [Google Scholar]
- Meissner W, Leblois A, Hansel D, Bioulac B, Gross CE, Benazzouz A, Boraud T. Subthalamic high frequency stimulation resets subthalamic firing and reduces abnormal oscillations. Brain. 2005;128:2372–2382. doi: 10.1093/brain/awh616. [DOI] [PubMed] [Google Scholar]
- Middleton FA, Strick PL. Basal ganglia and cerebellar loops: motor and cognitive circuits. Brain Res Rev. 2000;31:236–250. doi: 10.1016/s0165-0173(99)00040-5. [DOI] [PubMed] [Google Scholar]
- Miller WC, DeLong MR. Parkinsonian symptomatology. An anatomical and physiological analysis. Ann. N. Y. Acad. Sci. 1988;515:287–302. doi: 10.1111/j.1749-6632.1988.tb32998.x. [DOI] [PubMed] [Google Scholar]
- Modolo J, Legros A, Thomas AW, Beuter A. Model-driven therapeutic treatment of neurological disorders: reshaping brain rhythms with neuromodulation. Interface Focus. 2011;1:61–74. doi: 10.1098/rsfs.2010.0509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnar GF, Pilliar A, Lozano AM, Dostrovsky JO. Differences in neuronal firing rates in pallidal and cerebellar receiving areas of thalamus in patients with Parkinson's disease, essential tremor, and pain. J. Neurophys. 2005;93:3094–3101. doi: 10.1152/jn.00881.2004. [DOI] [PubMed] [Google Scholar]
- Montgomery EB, Jr, Baker KB. Mechanisms of deep brain stimulation and future technical developments. Neurol Res. 2000;22:259–266. doi: 10.1080/01616412.2000.11740668. [DOI] [PubMed] [Google Scholar]
- Moroney R, Heida C, Geelen J. Increased bradykinesia in Parkinson's disease with increased movement complexity: elbow flexion-extension movements. J Comput Neurosci. 2008;25:501–519. doi: 10.1007/s10827-008-0091-9. [DOI] [PubMed] [Google Scholar]
- Nambu A, Mori S, Stuart DG, Wiesendanger M. Progress in Brain Research. Elsevier; 2004. A new dynamic model of the cortico-basal ganglia loop; pp. 461–466. [DOI] [PubMed] [Google Scholar]
- Ni ZG, Gao DM, Benabid AL, Benazzouz A. Unilateral lesion of the nigrostriatal pathway induces a transient decrease of firing rate with no change in the firing pattern of neurons of the parafascicular nucleus in the rat. Neurosci. 2000;101:993–999. doi: 10.1016/s0306-4522(00)00337-7. [DOI] [PubMed] [Google Scholar]
- Nini A, Feingold A, Slovin H, Bergman H. Neurons in the globus pallidus do not show correlated activity in the normal monkey, but phase-locked oscillations appear in the MPTP model of parkinsonism. J. Neurophys. 1995;74:1800–1805. doi: 10.1152/jn.1995.74.4.1800. [DOI] [PubMed] [Google Scholar]
- Ogura M, Kita H. Dynorphin exerts both postsynaptic and presynaptic effects in the Globus pallidus of the rat. J Neurophysiol. 2000;83:3366–3376. doi: 10.1152/jn.2000.83.6.3366. [DOI] [PubMed] [Google Scholar]
- Ohara S, Ikeda A, Kunieda T, Yazawa S, Baba K, Nagamine T, Taki W, Hashimoto N, Mihara T, Shibasaki H. Movement-related change of electrocorticographic activity in human supplementary motor area proper. Brain. 2000;123:1203–1215. doi: 10.1093/brain/123.6.1203. [DOI] [PubMed] [Google Scholar]
- Orieux G, Francois C, Feger J, Hirsch EC. Consequences of dopaminergic denervation on the metabolic activity of the cortical neurons projecting to the subthalamic nucleus in the rat. J Neurosci. 2002;22:8762–8770. doi: 10.1523/JNEUROSCI.22-19-08762.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahapill PA, Lozano AM. The pedunculopontine nucleus and Parkinson's disease. Brain. 2000;123:1767–1783. doi: 10.1093/brain/123.9.1767. [DOI] [PubMed] [Google Scholar]
- Parent M, Parent A. Axon collateralization in primate basal ganglia and related thalamic nuclei. Thalamus & Related Systems. 2002;2:71–86. [Google Scholar]
- Pasquereau B, Turner RS. Primary Motor Cortex of the Parkinsonian Monkey: Differential Effects on the Spontaneous Activity of Pyramidal Tract-Type Neurons. Cereb Cortex. 2010 doi: 10.1093/cercor/bhq217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquereau B, Turner RS. Primary motor cortex of the parkinsonian monkey: differential effects on the spontaneous activity of pyramidal tract-type neurons. Cereb Cortex. 2011;21:1362–1378. doi: 10.1093/cercor/bhq217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul G, Reum T, Meissner W, Marburger A, Sohr R, Morgenstern R, Kupsch A. High frequency stimulation of the subthalamic nucleus influences striatal dopaminergic metabolism in the naive rat. Neuroreport. 2000;11:441–444. doi: 10.1097/00001756-200002280-00003. [DOI] [PubMed] [Google Scholar]
- Pessiglione M, Guehl D, Rolland AS, Francois C, Hirsch EC, Feger J, Tremblay L. Thalamic neuronal activity in dopamine-depleted primates: evidence for a loss of functional segregation within basal ganglia circuits. J. Neurosci. 2005;25:1523–1531. doi: 10.1523/JNEUROSCI.4056-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfurtscheller G, Neuper C. Simultaneous EEG 10 Hz desynchronization and 40 Hz synchronization during finger movements. Neuroreport. 1992;3:1057–1060. doi: 10.1097/00001756-199212000-00006. [DOI] [PubMed] [Google Scholar]
- Pirini M, Rocchi L, Sensi M, Chiari L. A computational modelling approach to investigate different targets in deep brain stimulation for Parkinson's disease. J Comput Neurosci. 2009;26:91–107. doi: 10.1007/s10827-008-0100-z. [DOI] [PubMed] [Google Scholar]
- Plenz D, Kitai S. A basal ganglia pacemaker formed by the subthalamic nucleus and external globus pallidus. Nature. 1999;400:677–682. doi: 10.1038/23281. [DOI] [PubMed] [Google Scholar]
- Priori A, Foffani G, Pesenti A, Tamma F, Bianchi AM, Pellegrini M, Locatelli M, Moxon KA, Villani RM. Rhythm-specific pharmacological modulation of subthalamic activity in Parkinson's disease. Exp. Neurol. 2004;189:369–379. doi: 10.1016/j.expneurol.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Raeva S, Vainberg N, Tikhonov Y, Tsetlin I. Analysis of evoked activity patterns of human thalamic ventrolateral neurons during verbally ordered voluntary movements. Neurosci. 1999;88:377–392. doi: 10.1016/s0306-4522(98)00230-9. [DOI] [PubMed] [Google Scholar]
- Raz A, Feingold A, Zelanskaya V, Vaadia E, Bergman H. Neuronal synchronization of tonically active neurons in the striatum of normal and parkinsonian primates. J. Neurophys. 1996;76:2083–2088. doi: 10.1152/jn.1996.76.3.2083. [DOI] [PubMed] [Google Scholar]
- Raz A, Frechter-Mazar V, Feingold A, Abeles M, Vaadia E, Bergman H. Activity of pallidal and striatal tonically active neurons is correlated in mptp-treated monkeys but not in normal monkeys. J. Neurosci. 2001;21:RC128. doi: 10.1523/JNEUROSCI.21-03-j0006.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitsma P, Doiron B, Rubin JE. Correlation transfer from basal ganglia to thalamus in Parkinson's disease. Frontiers Computational Neuroscience. 2011;5 doi: 10.3389/fncom.2011.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivlin-Etzion M, Marmor O, Heimer G, Raz A, Nini A, Bergman H. Basal ganglia oscillations and pathophysiology of movement disorders. Curr. Opin. Neurobiol. 2006;16:629–637. doi: 10.1016/j.conb.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Rivlin-Etzion M, Marmor O, Saban G, Rosin B, Haber SN, Vaadia E, Prut Y, Bergman H. Low-pass filter properties of basal ganglia cortical muscle loops in the normal and MPTP primate model of parkinsonism. J. Neurosci. 2008;28:633–649. doi: 10.1523/JNEUROSCI.3388-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland AS, Herrero MT, Garcia-Martinez V, Ruberg M, Hirsch EC, Francois C. Metabolic activity of cerebellar and basal ganglia-thalamic neurons is reduced in parkinsonism. Brain. 2007;130:265–275. doi: 10.1093/brain/awl337. [DOI] [PubMed] [Google Scholar]
- Rommelfanger KS, Wichmann T. Extrastriatal dopaminergic circuits of the Basal Ganglia. Front Neuroanat. 2010;4:139. doi: 10.3389/fnana.2010.00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum M, Pikovsky A. Delayed feedback control of collective synchrony: an approach to suppression of pathological brain rhythms. Physical review. E, Statistical, nonlinear, and soft matter physics. 2004a;70 doi: 10.1103/PhysRevE.70.041904. 041904. [DOI] [PubMed] [Google Scholar]
- Rosenblum MG, Pikovsky AS. Controlling synchronization in an ensemble of globally coupled oscillators. Phys Rev Lett. 2004b;92 doi: 10.1103/PhysRevLett.92.114102. 114102. [DOI] [PubMed] [Google Scholar]
- Rosin B, Slovik M, Mitelman R, Rivlin-Etzion M, Haber SN, Israel Z, Vaadia E, Bergman H. Closed-loop deep brain stimulation is superior in ameliorating parkinsonism. Neuron. 2011;72:370–384. doi: 10.1016/j.neuron.2011.08.023. [DOI] [PubMed] [Google Scholar]
- Ruberg M, Rieger F, Villageois A, Bonnet AM, Agid Y. Acetylcholinesterase and butyrylcholinesterase in frontal cortex and cerebrospinal fluid of demented and non-demented patients with Parkinson's disease. Brain Res. 1986;362:83–91. doi: 10.1016/0006-8993(86)91401-0. [DOI] [PubMed] [Google Scholar]
- Rubin JE, Terman D. High Frequency Stimulation of the Subthalamic Nucleus Eliminates Pathological Thalamic Rhythmicity in a Computational Model. Journal of Computational Neuroscience. 2004;16:211–235. doi: 10.1023/B:JCNS.0000025686.47117.67. [DOI] [PubMed] [Google Scholar]
- Rye DB, Bliwise DL, Dihenia B, Gurecki P. FAST TRACK: daytime sleepiness in Parkinson's disease. J Sleep Res. 2000;9:63–69. doi: 10.1046/j.1365-2869.2000.00201.x. [DOI] [PubMed] [Google Scholar]
- Scatton B, Dennis T, L'Heureux RL, Monfort J-C, Duyckaerts C, Javoy-Agid F. Degeneration of noraadrenergic and serotonergic ut not dopaminergic neurones in the lumbar spinal cord of parkinsonian patients. Brain Res. 1986;380:181–185. doi: 10.1016/0006-8993(86)91446-0. [DOI] [PubMed] [Google Scholar]
- Schneider JS, Rothblat DS. Alterations in intralaminar and motor thalamic physiology following nigrostriatal dopamine depletion. Brain Res. 1996;742:25–33. doi: 10.1016/s0006-8993(96)00988-2. [DOI] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321:848–851. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman SM. Tonic and burst firing: dual modes of thalamocortical relay. Trends in Neurosciences. 2001;24:122–126. doi: 10.1016/s0166-2236(00)01714-8. [DOI] [PubMed] [Google Scholar]
- Sherman SM, Guillery RW. The role of the thalamus in the flow of information to the cortex. Philos Trans R Soc Lond B Biol Sci. 2002;357:1695–1708. doi: 10.1098/rstb.2002.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberstein P, Pogosyan A, Kuhn AA, Hotton G, Tisch S, Kupsch A, Dowsey-Limousin P, Hariz MI, Brown P. Cortico-cortical coupling in Parkinson's disease and its modulation by therapy. Brain. 2005;128:1277–1291. doi: 10.1093/brain/awh480. [DOI] [PubMed] [Google Scholar]
- Silva GA. The need for the emergence of mathematical neuroscience: beyond computation and simulation. Frontiers in computational neuroscience. 2011;5:51. doi: 10.3389/fncom.2011.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares J, Kliem MA, Betarbet R, Greenamyre JT, Yamamoto B, Wichmann T. Role of external pallidal segment in primate parkinsonism: comparison of the effects of MPTP-induced parkinsonism and lesions of the external pallidal segment. J. Neurosci. 2004;24:6417–6426. doi: 10.1523/JNEUROSCI.0836-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sochurkova D, Rektor I. Event-related desynchronization/synchronization in the putamen. An SEEG case study. Exp Brain Res. 2003;149:401–404. doi: 10.1007/s00221-003-1371-2. [DOI] [PubMed] [Google Scholar]
- Sohal V, Huguenard J. Reciprocal inhibition controls the oscillatory state in thalamic networks. Neurocomputation. 2002;44:653–659. [Google Scholar]
- Stanford IM, Cooper AJ. Presynaptic mu and delta opioid receptor modulation of GABAA IPSCs in the rat globus pallidus in vitro. J. Neurosci. 1999;19:4796–4803. doi: 10.1523/JNEUROSCI.19-12-04796.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr PA, Kang GA, Heath S, Shimamoto S, Turner RS. Pallidal neuronal discharge in Huntington's disease: support for selective loss of striatal cells originating the indirect pathway. Exp. Neurol. 2008;211:227–233. doi: 10.1016/j.expneurol.2008.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr PA, Rau GM, Davis V, Marks WJ, Jr, Ostrem JL, Simmons D, Lindsey N, Turner RS. Spontaneous pallidal neuronal activity in human dystonia: comparison with Parkinson's disease and normal macaque. J. Neurophys. 2005;93:3165–3176. doi: 10.1152/jn.00971.2004. [DOI] [PubMed] [Google Scholar]
- Steiner H, Kitai ST. Regulation of rat cortex function by D1 dopamine receptors in the striatum. J Neurosci. 2000;20:5449–5460. doi: 10.1523/JNEUROSCI.20-14-05449.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner H, Kitai ST. Unilateral striatal dopamine depletion: time-dependent effects on cortical function and behavioural correlates. Eur J Neurosci. 2001;14:1390–1404. doi: 10.1046/j.0953-816x.2001.01756.x. [DOI] [PubMed] [Google Scholar]
- Steriade M, Llinas RR. The functional states of the thalamus and the associated neuronal interplay. Physiol Rev. 1988;68:649–742. doi: 10.1152/physrev.1988.68.3.649. [DOI] [PubMed] [Google Scholar]
- Tass PA. Phase Resetting in Medicine and Biology: Stochastic Modelling and Data Analysis. Berlin: Springer Verlag; 2006. [Google Scholar]
- Tass PA. Long-lasting neuronal desynchronization caused by coordinated reset stimulation. BMC Neuroscience. 2011;12(Suppl 1):K3. [Google Scholar]
- Tass PA, Majtanik M. Long-term anti-kindling effects of desynchronizing brain stimulation: a theoretical study. Biol Cybern. 2006;94:58–66. doi: 10.1007/s00422-005-0028-6. [DOI] [PubMed] [Google Scholar]
- Terman D, Rubin JE, Yew AC, Wilson CJ. Activity patterns in a model for the subthalamopallidal network of the basal ganglia. J. Neurosci. 2002;22:2963–2976. doi: 10.1523/JNEUROSCI.22-07-02963.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermann L, Wojtecki L, Gross J, Lehrke R, Voges J, Maarouf M, Treuer H, Sturm V, Schnitzler A. Ten-Hertz stimulation of subthalamic nucleus deteriorates motor symptoms in Parkinson's disease. Mov Disord. 2004;19:1328–1333. doi: 10.1002/mds.20198. [DOI] [PubMed] [Google Scholar]
- Titcombe MS, Edwards R, Beuter A. Mathematical modelling of parkinsonian tremor. Nonlinear Studies. 2004;11:363–384. [Google Scholar]
- Toro C, Deuschl G, Thatcher R, Sato S, Kufta C, Hallett M. Event-related desynchronization and movement-related cortical potentials on the ECoG and EEG. Electroencephalography and Clinical Neurophysiology/Evoked Potentials Section. 1994;93:380–389. doi: 10.1016/0168-5597(94)90126-0. [DOI] [PubMed] [Google Scholar]
- Tseng KY, Riquelme LA, Belforte JE, Pazo JH, Murer MG. Substantia nigra pars reticulata units in 6-hydroxydopamine-lesioned rats: responses to striatal D2 dopamine receptor stimulation and subthalamic lesions. Eur. J. Neurosci. 2000;12:247–256. doi: 10.1046/j.1460-9568.2000.00910.x. [DOI] [PubMed] [Google Scholar]
- Tukhlina N, Rosenblum M, Pikovsky A, Kurths J. Feedback suppression of neural synchrony by vanishing stimulation. Physical review. E, Statistical, nonlinear, and soft matter physics. 2007;75 doi: 10.1103/PhysRevE.75.011918. 011918. [DOI] [PubMed] [Google Scholar]
- Vila M, Levy R, Herrero MT, Ruberg M, Faucheux B, Obeso JA, Agid Y, Hirsch EC. Consequences of nigrostriatal denervation on the functioning of the basal ganglia in human and nonhuman primates: an in situ hybridization study of cytochrome oxidase subunit I mRNA. J. Neurosci. 1997;17:765–773. doi: 10.1523/JNEUROSCI.17-02-00765.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila M, Perier C, Feger J, Yelnik J, Faucheux B, Ruberg M, Raisman-Vozari R, Agid Y, Hirsch EC. Evolution of changes in neuronal activity in the subthalamic nucleus of rats with unilateral lesion of the substantia nigra assessed by metabolic and electrophysiological measurements. Eur. J. Neurosci. 2000;12:337–344. doi: 10.1046/j.1460-9568.2000.00901.x. [DOI] [PubMed] [Google Scholar]
- Villalba RM, Smith Y. Striatal spine plasticity in Parkinson's disease. Front Neuroanat. 2010;4:133. doi: 10.3389/fnana.2010.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba RM, Smith Y. Differential structural plasticity of corticostriatal and thalamostriatal axo-spinous synapses in MPTP-treated Parkinsonian monkeys. J Comp Neurol. 2011;519:989–1005. doi: 10.1002/cne.22563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba RM, Wichmann T, Smith Y. Significant degeneration of the intralaminar thalamic nuclei (CM/Pf) in MPTP-treated parkinsonian monkeys. Soc. Neurosci. Abstr. 2011 [Google Scholar]
- Vitek JL. Mechanisms of deep brain stimulation: excitation or inhibition. Mov Disord. 2002;17(Suppl 3):S69–S72. doi: 10.1002/mds.10144. [DOI] [PubMed] [Google Scholar]
- Vitek JL, Ashe J, DeLong MR, Alexander GE. Physiologic properties and somatotopic organization of the primate motor thalamus. J.Neurophysiol. 1994;71:1498–1513. doi: 10.1152/jn.1994.71.4.1498. [DOI] [PubMed] [Google Scholar]
- Vitek JL, Bakay RA, Freeman A, Evatt M, Green J, McDonald W, Haber M, Barnhart H, Wahlay N, Triche S, Mewes K, Chockkan V, Zhang JY, DeLong MR. Randomized trial of pallidotomy versus medical therapy for Parkinson's disease. Ann. Neurol. 2003;53:558–569. doi: 10.1002/ana.10517. [DOI] [PubMed] [Google Scholar]
- Wang HC, Lees AJ, Brown P. Impairment of EEG desynchronisation before and during movement and its relation to bradykinesia in Parkinson's disease. J Neurol Neurosurg Psychiatry. 1999;66:442–446. doi: 10.1136/jnnp.66.4.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts RL, Mandir AS. The role of motor cortex in the pathophysiology of voluntary movement deficits associated with parkinsonism. Neurol Clin. 1992;10:451–469. [PubMed] [Google Scholar]
- Weinberger M, Mahant N, Hutchison WD, Lozano AM, Moro E, Hodaie M, Lang AE, Dostrovsky JO. Beta oscillatory activity in the subthalamic nucleus and its relation to dopaminergic response in Parkinson's disease. J. Neurophys. 2006;96:3248–3256. doi: 10.1152/jn.00697.2006. [DOI] [PubMed] [Google Scholar]
- Wichmann T, Bergman H, DeLong MR. The primate subthalamic nucleus. III. Changes in motor behavior and neuronal activity in the internal pallidum induced by subthalamic inactivation in the MPTP model of parkinsonism. J. Neurophys. 1994;72:521–530. doi: 10.1152/jn.1994.72.2.521. [DOI] [PubMed] [Google Scholar]
- Wichmann T, Bergman H, Starr PA, Subramanian T, Watts RL, DeLong MR. Comparison of MPTP-induced changes in spontaneous neuronal discharge in the internal pallidal segment and in the substantia nigra pars reticulata in primates. Exp. Brain Res. 1999;125:397–409. doi: 10.1007/s002210050696. [DOI] [PubMed] [Google Scholar]
- Wichmann T, Delong MR. Deep brain stimulation for neurologic and neuropsychiatric disorders. Neuron. 2006;52:197–204. doi: 10.1016/j.neuron.2006.09.022. [DOI] [PubMed] [Google Scholar]
- Wichmann T, Soares J. Neuronal firing before and after burst discharges in the monkey basal ganglia is predictably patterned in the normal state and altered in parkinsonism. J. Neurophys. 2006;95:2120–2133. doi: 10.1152/jn.01013.2005. [DOI] [PubMed] [Google Scholar]
- Williams D, Kuhn A, Kupsch A, Tijssen M, van Bruggen G, Speelman H, Hotton G, Loukas C, Brown P. The relationship between oscillatory activity and motor reaction time in the parkinsonian subthalamic nucleus. Eur. J. Neurosci. 2005;21:249–258. doi: 10.1111/j.1460-9568.2004.03817.x. [DOI] [PubMed] [Google Scholar]
- Williams D, Kuhn A, Kupsch A, Tijssen M, van Bruggen G, Speelman H, Hotton G, Yarrow K, Brown P. Behavioural cues are associated with modulations of synchronous oscillations in the human subthalamic nucleus. Brain. 2003;126:1975–1985. doi: 10.1093/brain/awg194. [DOI] [PubMed] [Google Scholar]
- Williams D, Tijssen M, Van Bruggen G, Bosch A, Insola A, Di Lazzaro V, Mazzone P, Oliviero A, Quartarone A, Speelman H, Brown P. Dopamine-dependent changes in the functional connectivity between basal ganglia and cerebral cortex in humans. Brain Cogn. 2002;125:1558–1569. doi: 10.1093/brain/awf156. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Beverlin B, 2nd, Netoff T. Chaotic desynchronization as the therapeutic mechanism of deep brain stimulation. Front Syst Neurosci. 2011;5:50. doi: 10.3389/fnsys.2011.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windels F, Bruet N, Poupard A, Feuerstein C, Bertrand A, Savasta M. Influence of the frequency parameter on extracellular glutamate and gamma-aminobutyric acid in substantia nigra and globus pallidus during electrical stimulation of subthalamic nucleus in rats. J Neurosci Res. 2003;72:259–267. doi: 10.1002/jnr.10577. [DOI] [PubMed] [Google Scholar]
- Windels F, Bruet N, Poupard A, Urbain N, Chouvet G, Feuerstein C, Savasta M. Effects of high frequency stimulation of subthalamic nucleus on extracellular glutamate and GABA in substantia nigra and globus pallidus in the normal rat. Eur. J. Neurosci. 2000;12:4141–4146. doi: 10.1046/j.1460-9568.2000.00296.x. [DOI] [PubMed] [Google Scholar]
- Xu WD, Russo GS, Hashimoto T, Zhang JY, Vitek JL. Subthalamic nucleus stimulation modulates thalamic neuronal activity. J. Neurosci. 2008;28:11916–11924. doi: 10.1523/JNEUROSCI.2027-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Rabin A, Anderson ME. Monosynaptic inhibition of pallidal neurons by axon collaterals of caudatonigral fibers. Exp Brain Res. 1972;15:333–347. doi: 10.1007/BF00234122. [DOI] [PubMed] [Google Scholar]
- Zweig RM, Cardillo JE, Cohen M, Giere S, Hedreen JC. The locus ceruleus and dementia in Parkinson's disease. Neurology. 1993;5:986–991. doi: 10.1212/wnl.43.5.986. [DOI] [PubMed] [Google Scholar]