

Abstract
Methylmercury is among the most potentially toxic species to which human populations are exposed, both at high levels through poisonings and at lower levels through consumption of fish and other seafood. However, the molecular mechanisms of methylmercury toxicity in humans remain poorly understood. We used synchrotron X-ray absorption spectroscopy (XAS) to study mercury chemical forms in human brain tissue. Individuals poisoned with high levels of methylmercury species showed elevated cortical selenium with significant proportions of nanoparticulate mercuric selenide plus some inorganic mercury and methylmercury bound to organic sulfur. Individuals with a lifetime of high fish consumption showed much lower levels of mercuric selenide and methylmercury cysteineate. Mercury exposure did not perturb organic selenium levels. These results elucidate a key detoxification pathway in the central nervous system and provide new insights into the appropriate methods for biological monitoring.
Keywords: Methylmercury, methylmercury toxicology, human brain, neurotoxicology, X-ray absorption spectroscopy, X-ray fluorescence imaging
Methylmercury species are among the most potentially, problematic compounds to which human populations are exposed (1). These compounds, which all contain the CH3Hg− group, are known to cross the blood−brain barrier and to be highly neurotoxic. Neurological signs depend on the dose but can include paresthesia, ataxia, tremors, impairment of hearing, and other sensory disturbances (1). One of the most puzzling aspects of methylmercury toxicity is the latency in onset of symptoms, which may not appear for as long as 150 days after exposure in humans (1). Methylmercury compounds are present in most fish species at some level and at higher levels in predatory marine fish such as swordfish and shark, with comparable levels in other predatory fish such as large tuna; a joint US EPA/FDA advisory recommended that pregnant women and nursing mothers should completely avoid high-mercury fish (2). Still higher levels are present in predatory marine mammals (3,4), which may be a cause for concern for populations that consume these organisms. The United Nations Food and Agriculture Organization estimates that over one billion people worldwide depend on fish for daily nutrition (5). Consequently, if methylmercury at levels achieved by fish consumption can adversely affect child development, it would be of substantial global health significance. Thus, the ability of methylmercury species to cross the blood−brain barrier and selectively damage the central nervous system is of significant concern (1,6). Despite its importance, little is known about the molecular mechanisms by which methylmercury causes its toxic effects.
In part, this lack of information of the mechanisms of methylmercury toxicity arises from a lack of techniques suitable for probing chemical form. Synchrotron X-ray absorption spectroscopy (XAS) provides a method for investigating the chemical form of an element in a complex sample. Its advantage is that it probes all of the occurrences of the element in any physical or chemical state without recourse to chemical extractions. Hence, even recalcitrant species can be observed, and quantitative analyses of chemical species can be performed in situ on complex specimens such as brain tissue. Here we use XAS to investigate the molecular nature of mercury in human brain tissue samples taken from five individuals with varying exposure to mercury. Selenium was also investigated because of possible biochemical links between the two elements. The results shed light on the mechanisms of mercury poisoning and detoxification, including the role of selenium.
Results and Discussion
Samples were examined from five individuals, referred to as cases 1−5. Case 1 was a female exposed to high dose of a methylmercury species at age 8 years by consuming contaminated pork (7,8). She survived 21 years in a debilitated state (9). Case 2 was a 48-year-old woman who expired 10 months after skin contact with an unknown volume of dimethylmercury in a research laboratory (10,11). Cases 3 and 4 were a 60-year-old man and a 76-year-old woman, respectively. Both were lifetime residents of the Republic of Seychelles where daily fish consumption is the norm. Neither suffered known adverse effects attributable to mercury. Case 5 was a 67-year-old woman from Rochester, New York, with no known exposure to mercury. For case 1, the available tissue had originally been embedded in histological grade paraffin, whereas in the other cases, biological samples had been preserved in formaldehyde. Additional background information is provided in Table 1.
Table 1. Background Information on Cerebral Cortical Samples.
| case | 1 | 2 | 3 | 4 | 5 |
| gender | F | F | M | F | F |
| age (years) | 29 | 48 | 60 | 76 | 67 |
| cortex | frontal | occipital | occipital | occipital | occipital |
| exposure | acute poisoning | acute poisoning | fish consumption | fish consumption | none known |
| Hg μM [ppb]a | 5.9 [1179] | 13.3 [2670] | 1.6 [324] | 0.6 [120] | 0.06 [12.5] |
| XAS Hg μMb | 30 | 22 | 1.4 | 0.4 | ≤0.2 |
| % inorganic Hgc | 100 | 67 | 19 | 68 | 83 |
| neuropathology | severe cortical atrophy | severe cortical atrophy | normal | normal | normal |
Total mercury was determined using atomic absorption as described in Sample Selection and Characterization. The Hg levels for cases 1 and 2 are taken from the literature and varied significantly between different regions of the brain, so differences in the levels estimated by XAS are expected. In clinical studies, mercury levels are often referred to in weight parts per billion or nanograms per gram, and these are given in square brackets. For an aqueous solution, this is approximately related to the concentration multiplied by the atomic weight of mercury (200.59 g/mol) divided by the density of the sample; human brain tissue is assumed to be approximately 1.05 g/cm312. The Hg levels for cases 3, 4, and 5 were measured from the same block of tissue used for the XAS analysis.
XAS levels of mercury were estimated as described under XAS Quantification of Hg and Se.
The percentage of inorganic mercury was determined using conventional chemical analysis as described in Sample Selection and Characterization.
Total Mercury and Selenium
We observed the highest cortical brain Hg levels in case 1, with case 2 being slightly lower, and cases 3, 4, and 5 showing substantially lower levels (Table 1, 2). For cases 1 and 2, Se levels were significantly elevated and observed to be 9.5 and 7.1 times, respectively, the Se level of case 5. The results of chemical analysis and XAS estimates of mercury are in excellent agreement for samples of cases 3, 4, and 5, but this is not true of cases 1 and 2 (Table 1). This is probably because the conventional chemical analysis values for cases 1 and 2 are literature values obtained from samples measured at different brain locations than the XAS samples, and the differences can be attributed to regional variations in mercury levels.
Table 2. XAS Analysis of Brain Tissue Samplesa.
| concentration (μM) |
Hg speciation (%) |
Se speciation (%) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| case no. | Hg | Se | org-Se | HgSe | Hg(SR)2 | MeHgCys | HgSe | RSeSR′ | RSeR′ |
| 1 | 30(1) | 26(1) | 3.9 | 71(7) | 29(7) | b | 85(2) | 15(2) | b |
| 2 | 22(1) | 11(1) | 3.6 | 29(3) | 16(6) | 55(6) | 67(3) | 7(3) | 26(3) |
| 3 | 1.4(1) | 3.8(2) | 3.3 | 35(5) | b | 65(7) | 14(5) | 45(5) | 41(5) |
| 4 | 0.4(1) | 3.4(2) | 3.4 | c | c | c | b | 58(4) | 42(4) |
| 5 | ≤0.2(1) | 3.6(2) | 3.6 | c | c | c | b | 60(4) | 40(4) |
Effective mercury and selenium concentrations are given in micromolar, estimated relative to standard solutions. org-Se concentrations are estimated from the total Se concentration multiplied by the sum of percentages of RSeSR′ and RSeR′. In cases 1−3, the estimated Hg/Se stoichiometry and the HgSe content estimated from the Hg LIII and Se K-edge data are consistent within the confidence limits of the fits. The values shown in parentheses are estimated standard deviations (precisions) in the last digit obtained from the diagonal elements of the covariance matrix.
Component not detected in linear combination.
The low Hg levels of cases 4 and 5 meant that speciation could not be measured for these samples.
X-ray Absorption Spectroscopy
X-ray absorption spectra arise from excitation of a core electron (e.g., a 1s electron for a K-edge or a 2p3/2 electron for an LIII edge). They can be arbitrarily divided into two overlapping regions: the near-edge spectrum, which is the structured region within about 50 eV of the absorption edge, and the extended X-ray absorption fine structure (EXAFS), which is an oscillatory modulation of the absorption on the high-energy side of the absorption edge and can be interpreted in terms of a local radial structure. Near-edge spectra are comprised of transitions from the core level (1s for a K-edge) to unoccupied molecular orbitals of the system. Intense transitions are dipole-allowed, Δl = ±1, and thus for K and LIII edges are to levels with a lot of p and d orbital character, respectively. Near-edge spectra are therefore sensitive to electronic structure and give a fingerprint of the type of chemical species of the metal or metalloid concerned. XAS determines the presence not of specific molecules but rather of classes of chemical species with similar local environment around the central absorbing atom. Thus, compounds containing selenium coordinated by two aliphatic carbon atoms (e.g., (CH3)2Se and selenomethionine) are essentially indistinguishable; similarly methylmercury and ethylmercury species cannot be discriminated, although in special cases discernible differences may exist. The advantage of the near-edge region of the spectrum is that it can be quickly collected with good signal-to-noise. In contrast, EXAFS is more difficult to collect with adequate signal-to-noise and is not always practical on dilute samples. A unique benefit of XAS is that it requires no pretreatment or extraction and thus provides a tool that can probe chemical species in situ.
Figure 1 shows the Hg LIII and Se K near-edge spectra of selected standard compounds. The differences between individual Hg LIII spectra are more subtle than those for the Se K spectra. In the case of Se K-edges, the valence orbitals have mainly p-character, giving rise to intense dipole-allowed structure in the spectra and rich chemical variability. In contrast for Hg LIII edges, there is only subtle involvement of d-orbitals and correspondingly more subtle variability between the spectra. The consequence of this is that significantly better signal-to-noise (when measured relative to the edge jump) is required for Hg than for Se, but the spectra can nevertheless give the desired information.
Figure 1.

Comparison of Hg LIII and Se K near-edge spectra of selected standard species. Most species were measured as dilute (ca. 1 mM) aqueous solutions buffered at physiological pH (7−7.5) in the presence of 30% v/v glycerol using X-ray fluorescence detection. Exceptions were nano-HgSe, which was run as a colloidal suspension in buffered water with 30% v/v glycerol, and CH3HgCH3, which was run in isopropanol solution due to its low water solubility. CH3HgSe(Cys), (CH3Hg)2Se, and Hg3(PO4)2 were run as solids by monitoring transmittance.
Figure 2 shows the Hg LIII and Se K near-edge XAS of frozen brain tissue samples. Spectra were analyzed by linear combination fitting to the spectra of standard compounds (13) to determine speciation. The predominant Hg species present in case 1 strongly resembled nano-HgSe, a nanoparticulate form of mercuric selenide (tiemannite or HgSe) previously observed in rabbit blood plasma (14). The Hg and Se near-edge spectra of nano-HgSe are very similar to that of crystalline HgSe, except that sharp features in the near-edge are broadened (Figure 3). Quantitative analysis of the Hg LIII near-edge spectrum of case 1 (Figure 2, Table 2) indicates 71% nano-HgSe and 29% mercuric bis-thiolato species [Hg(SR)2] with no observable organic-Hg phases. The Se K near-edge spectrum (Figure 2) indicates 85% nano-HgSe and 15% of an organic selenyl-sulfide species resembling Cys−Se−S−Cys [RSeSR′]. In this case, fits with 8% selenyl sulfide and 7% of an organic selenide resembling selenomethionine [RSeR′] were essentially indistinguishable from the fit giving 15% selenyl sulfide (not illustrated). We attribute this uncertainty to the high percentage of HgSe in this particular sample. The Hg and Se edge analysis estimate the concentration of nano-HgSe to be 21.3 and 22.1 μM, respectively, showing excellent consistency between the two analyses.
Figure 2.

Hg LIII and Se K X-ray absorption near edge spectra of brain samples. Experimental data is shown as points and the solid lines (—) show the linear combination analysis (Table 2). Broken lines show the spectra of individual components, weighted according to their contributions in the fit: for mercury Hg(SR)2 (···), MeHgSCys (−−−), and nano-HgSe (−·−); and for selenium selenomethionine (···), CysSeSCys (−−−), and nano-HgSe (−·−).
Figure 3.

Comparison of Hg LIII and Se K X-ray absorption near edge spectra of crystalline and nanoparticulate mercuric selenide (nano-HgSe). The spectra are very similar except that for the most part sharp features in the nano-HgSe near-edge are broadened.
Extended X-ray absorption fine structure (EXAFS) spectra (Figure 4) provide quantitative support for a significant presence of nano-HgSe. EXAFS curve-fitting (Table 3) yields Hg−Se and Se−Hg contacts at 2.63(1) and 2.61(1) Å, respectively, similar to the 2.63 Å bond length of pure mercuric selenide (15). Crystalline HgSe has the zincblende structure, although for small particle sizes the wurtzite structure is also possible. Both structures exhibit four-coordination of both mercury and selenium. The Cambridge Structural Database (16) indicates typical Hg−Se bond-lengths for two-, three- and four-coordinate Hg of 2.43, 2.53, and 2.65 Å, respectively, the latter being the most common coordination environment. Our observation of 2.63 Å Hg−Se bond lengths in brain thus is diagnostic of four-coordinate mercury. The Se EXAFS (Figure 4) shows well-resolved outer-shell contacts at 4.33 and 4.82 Å (Table 3), again consistent with either zincblende or wurtzite structures for nano-HgSe (14). These peaks are more pronounced than for the ∼200-atom clusters of nano-HgSe previously investigated (14), though not as intense as for bulk HgSe. This suggests that nano-HgSe detected in brain comprises somewhat larger or better ordered clusters than previously observed in blood (14), though still not fully crystalline. Hg outer shells were not observed either for brain (Figure 4) or for nano-HgSe (14), indicating for both an absence of long-range order consistent with nanoparticulate HgSe.
Figure 4.

Hg LIII and Se K EXAFS and corresponding Fourier transforms of brain samples. Solid lines (—) show the experimental data, and broken lines (−−−) the best fit as shown in Table 3. Fourier transforms have been phase-corrected for first-shell interactions.
Table 3. EXAFS Curve Fitting Results for Cases 1 and 2a.
| interaction | N | R | σ2 | ΔE0 | F | |
|---|---|---|---|---|---|---|
| case 1, Hg LIII | Hg−Se | 3.5 (3) | 2.634 (4) | 0.0080 (5) | −11(1) | 0.382 |
| case 1, Se K | Se−Hg | 3.5 (5) | 2.607 (6) | 0.008 (1) | −12(1) | 0.581 |
| Se···Se | 6.4 (2.4) | 4.33 (1) | 0.013 (3) | |||
| Se···Se−Hg | 12.9 (4.8)b | 4.82 (1)b | 0.014 (3)b | |||
| Se···Hg | 6.4 (2.4)b | 5.08 (2)b | 0.013 (3)b | |||
| case 2, Hg LIII | Hg−S | 1.4 (2) | 2.32 (2) | 0.008 (2) | −6(2) | 0.429 |
| Hg−Se | 1.0 (2) | 2.61 (2) | 0.008c |
Coordination numbers (scattering path degeneracies) N, interatomic distances R (Å), mean-square deviation in R (Debye−Waller factor) σ2 (Å2), threshold energy shift ΔE0 (eV) and fit-error function F, which is defined as F = (∑k6(χcalcd − χexptl)2/∑χexptl2)1/2, where the summations are over all data points included in the refinement. The values shown in parentheses are estimated standard deviations (precisions) in the last digit(s) obtained from the diagonal elements of the covariance matrix. The accuracies will be much greater than these values and are generally accepted to be ±0.02 Å for bond lengths and ±20% for coordination numbers and Debye−Waller factors. The near-edge fitting results for case 1 (Table 2) indicate that first-shell Hg−S, Se−S, and Se−C EXAFS should in principle be observed. However, the nano-HgSe species is the dominant component (>70%) and backscattering from S and C is much smaller than from Se and Hg. Because of these considerations EXAFS from Hg−S, Se−S, and Se−C is not expected to be observed. For case 2, the smaller percentage of nano-HgSe allows the additional detection of Hg−S in the first shell, though Hg−C is not expected to be observed due to its lower abundance and smaller backscattering.
Values were linked together to be consistent with a zincblende-like structure.
Value was fixed at the corresponding value of the Hg−Se bond in case 1.
The Hg LIII and Se K near-edge spectra of case 2 are also shown in Figure 2. Here, the dominant mercury species is CH3Hg(SCys) at 55%, with smaller amounts of nano-HgSe and Hg(SR)2 (29% and 16% respectively). The Se K-edge data gave 67% nano-HgSe, 26% selenomethionine-like species, and 7% selenyl-sulfide species resembling Cys−S−Se−Cys. The concentration of nano-HgSe is estimated to be 6.4 and 7.4 μM from the Hg and Se edges, respectively. These values are in excellent agreement with each other and approximately 3-fold lower than case 1. Lower concentrations and smaller EXAFS amplitudes from lighter backscatterers mean that the case 2 Hg LIII EXAFS only extends to k = 9 Å−1 (Figure 4) and Se K-edge EXAFS was not available. Nevertheless, curve-fitting of the Hg EXAFS (Figure 4, Table 3) shows both Hg−S and Hg−Se interactions at 2.32(2) and 2.61(2) Å, respectively, consistent with the near-edge spectra.
Case 2 data show no evidence for significant remaining dimethylmercury, which has a distinctive Hg LIII near-edge spectrum (Figure 1). The lack of dimethylmercury is expected because of its high vapor pressure (58.5 mmHg at 23.7 °C (17)). Consistent with this, mice injected with dimethylmercury have been reported to rapidly exhale 80−90% of the dose, with no detectable dimethylmercury present after 16 h (18), although monomethylmercury species were detected in a number of tissues (18). This indicated that C−Hg bond scission had occurred in vivo during this relatively short time.
Case 3 Hg LIII and Se K near-edge spectra are also shown in Figure 2, and quantitative linear combination analysis indicates that the dominant mercury species is CH3Hg(SCys) at 65%, with 35% nano-HgSe. Linear combination analysis of the Se K-edge data gave 14% nano-HgSe, 41% selenomethionine-like species, and 45% selenyl-sulfide species resembling Cys−S−Se−Cys. Both edges estimate a nano-HgSe concentration of just 0.5 μM, again demonstrating consistency between the two edges. For cases 4 and 5, the levels of Hg were too low (below 1 μM) to give adequate signal-to-noise. Case 4 and case 5 selenium showed a mixture of a selenomethionine-like species and a selenyl-sulfide species but no nano-HgSe. Assuming a similar mercury speciation as in case 3, the Hg/Se ratio predicts less than 4% of Se as nano-HgSe in case 4 and case 5 (equivalent to less than 0.1 μM concentration of nano-HgSe), which is below our detection limit.
X-ray Fluorescence Imaging
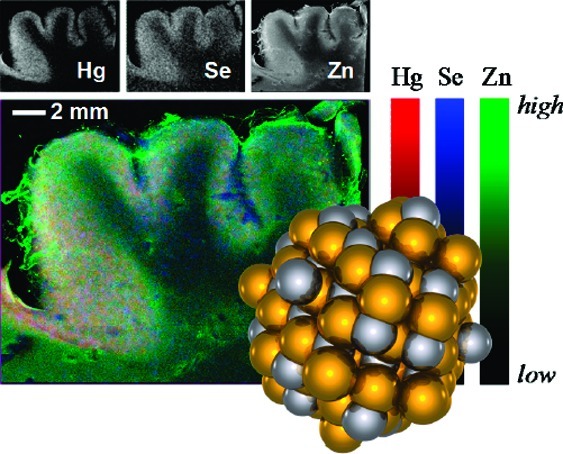
For case 2, samples of sufficiently large dimension were available to measure X-ray fluorescence imaging (19). The results of such measurements are shown in Figure 5 with the Hg localization shown in red, Se in blue, and Zn in green. The majority of the mercury and selenium are colocated in the gray matter, with lower levels of selenium but almost negligible mercury in the white matter.
Figure 5.
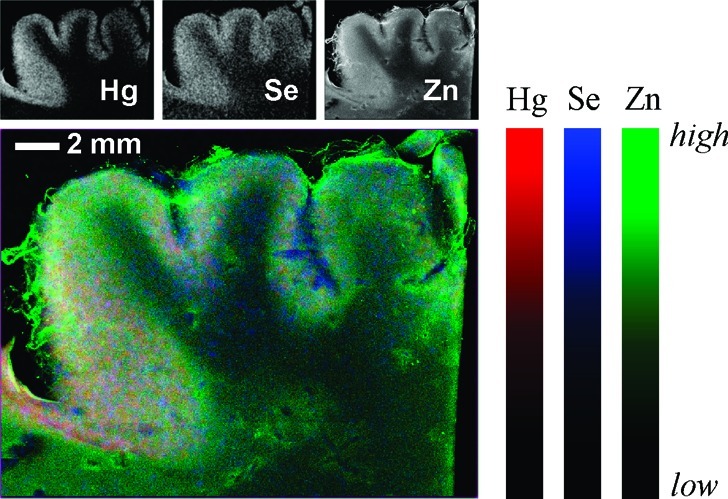
X-ray fluorescence images of cerebral cortex (occipital lobe) from case 2. The large tricolor image overlays Hg, Se, and Zn in red, blue and green, respectively. The small maps show the individual fluorescence images.
Previous work has identified HgSe in whale liver (20), and species with HgSe coordination also have been found in the livers and kidneys of dolphin, seal, and albatross (21). HgSe is a particularly stable and insoluble form of mercury [molar solubility product Ksp ≈ 10−59]. HgSe thus represents an essentially nonbioavailable form, effectively removing any mercury bound to selenide from involvement in biological processes.
In case 1, the poisoned subject lived for 2 decades, whereas in case 2 the subject expired after 10 months. In case 1, the absence of detectible fractions of organomercury species and the presence of predominantly HgSe-like phases suggests the successful detoxification of mercury, contrasting with case 2, which showed lower HgSe levels but significant levels of potentially harmful species resembling methylmercury cysteinate and mercury bis-cysteinate. Case 3 represents exposure but not intoxication; HgSe is present, but the predominant form is organomercury, with the highest fraction of methylmercury species observed. This suggests that methylmercury cysteinate-like species may not be problematic in vivo at the low levels resulting from fish consumption, which were about 1 μM, or more than 13 times lower than in case 2. In all of the data sets, we saw no significant organic mercury bound to selenium forms, such as methylmercury selenocysteinate (13).
We have demonstrated that, after both low and high exposures to methylmercury, mercury in the brain is present in at least two chemically distinct species. It is present in a mobile and potentially toxic form as methylmercury cysteineate (CH3—Hg−SCH2···) and in an inert nontoxic form as mercuric selenide (HgSe). Because these species are present in variable proportions depending on the degree and length of exposure to methylmercury, the levels of total Hg in brain cannot be used as a measure of its toxic potential. In this respect, our findings indicate that biological monitoring must focus on the mobile and potentially toxic species of methylmercury. For example, human head hair avidly accumulates methylmercury via the uptake of methylmercury cysteineate. Total mercury in hair, therefore, serves as a biological indicator of methylmercury cysteineate levels in blood and thereby of the levels of this mercury species in brain (22).
It has been suggested that development of neurotoxic effects following the latency after exposure to methylmercury compounds may be associated with demethylation of mercury in the central nervous system (1,23,24). Indeed, it has been proposed that demethylation combined with mercury’s affinity for selenium is responsible for its neurotoxic effects (24). The Hg−C bond of methylmercury species has remarkable kinetic stability, with almost no cleavage in strong acids at room temperature. Despite this, methylmercury species are demethylated to inorganic forms in vertebrate tissues, and a major question relates to how this proceeds. Density functional theory (DFT) calculations suggest that S coordination of Hg in methylmercury species increases the negative charge on the C bound to Hg, catalyzing proton-mediated C−Hg scission with resulting evolution of methane (25). In agreement with this work, we find that the Mulliken atomic charges from DFT energy-minimized structures (see refs (26 and 27) for methods) predict that dimethylmercury should be inherently more susceptible to C−Hg protonolysis than monomethyl species and also susceptible to R—S− or R—Se−-catalyzed protonolysis via formation of a trigonal [(CH3)2HgS(e)R]− intermediate (not illustrated). This explains the rapid demethylation of dimethylmercury to monomethyl species observed in mice (18) and discussed above. Organoselenium methylmercury complexes are well-known to be unstable over time, degrading to black mercuric selenide and other products. For example, we find that methylmercury-seleno-l-cysteinate [CH3HgSe(Cys)], synthesized according to Carty et al. (28), is stable in anaerobic aqueous solution at very high pH; however at neutral pH, it rapidly forms a milky white precipitate, which turns gray and then black, precipitating HgSe (identified by powder X-ray diffraction), which is initially nanoparticulate in nature. Even as a solid, CH3HgSe(Cys) will degrade on anaerobic storage over a period of months to give black deposits of HgSe. It has been suggested that such selenium-aided demethylation involves formation of bis-methylmercuric selenide [(CH3Hg)2Se] (29). Recently, Kahn and Wang (30) have used nuclear magnetic resonance spectroscopy to detect small amounts of [(CH3Hg)2Se] in solutions of degrading methylmercury seleno-amino acids and have also used mass spectrometry to detect dimethylmercury as an intermediate. These workers have proposed a reaction mechanism involving initial formation of [(CH3Hg)2Se] from the amino acid complex and degradation of this to HgSe and dimethylmercury, which is subsequently demethylated. Examination of the chemical literature indicates that selenium-coordinated mercury species have a tendency to higher metal coordination numbers. Thus, searching the Cambridge Structural Database (16) for mercury compounds with one or more selenium donor indicates that approximately tetrahedral four-coordinate mercury species outnumber the approximately linear two-coordinate ones by about 10:1. A similar search with one or more sulfur donors in place of selenium indicates the reverse trend with an approximate 3-fold predominance of two-coordinate mercury species (16). Mercury organo-selenium complexes have a tendency to form polymeric species (31), and it seems plausible that an initial polymerization step may also be an important aspect of methylmercury demethylation chemistry.
Irrespective of the exact chemistry that occurs in vivo, our observation of substantial amounts of inorganic mercury indicates that the demethylation process must take place in the brain, because inorganic mercury does not cross the blood−brain barrier (22). Our findings also indicate that with release from methylmercury, inorganic mercury is combined with selenium. Overall, this is part of an effective detoxification process because the end product is chemically and physically inert. Although further research is needed on these processes, our results clearly demonstrate, as has been suspected from early experimental work (32), that selenium participates in the detoxification of methylmercury.
Our results may have some bearing on the “selenium depletion hypothesis” (24), which proposes that mercury compounds scavenge selenium in neurological tissues, causing a local deficiency of essential selenium-dependent enzymes (23,24). This seems plausible because of the potentially limiting transport of selenium to the brain (33). Our results indicate that the selenium that is not involved in forming inorganic complexes with mercury is remarkably invariant in the samples studied (see “org-Se” column in Table 2). This is presumably the selenium naturally present fulfilling functional roles in selenoenzymes. In all cases, we find that this organic selenium is close to 3.5 μM and is divided between selenyl sulfides and organic selenides (Table 2). Our value of 3.5 μM is close to total selenium levels in brain previously reported by others (34). If the selenium depletion hypothesis (24) were correct, then some decrease in this organic selenium might be expected in cases 1 and 2. This is not observed, and our results may therefore argue against the selenium depletion hypothesis, although deficiency of a particular enzyme normally present at low levels might not be reflected in our measurements.
In summary, we have introduced a new approach to determine mercury species in human brain that opens up avenues of research. Using methylmercury as an example, we have elucidated a key if not unique detoxification pathway in the central nervous system and have gained new insight into the appropriate methods for biological monitoring.
Methods
Sample Selection and Characterization
Samples of brain adjacent to those analyzed for their X-ray fluorescence emission spectra were analyzed by atomic absorption for total and inorganic mercury by a previously published method (35). All samples were analyzed in the Core Mercury Analysis Laboratory, Department of Environmental Medicine, University of Rochester. Details related to cases 1 and 2 have previously been published (9−11). For cases 1 and 2, levels of mercury were found to be highly elevated in all regions of the brain analyzed; however, there was significant variation in mercury levels in different regions of the brain. Multiple samples of brain from cases 3 and 5 were prepared for light microscopic examination using standard methods for hematoxylin−eosin, Luxol fast blue−cresyl violet, Bodian silver protargol, glial fibrillary acid protein, immunohistochemical expression of CD68 in activated microglia, and autometallographic procedure for mercury. The slides were coded and examined without knowledge of source (Seychelles or USA) by one of the authors. No significant neuropathological lesions were identified, and neither slide set showed autometallographic granules positive for mercury.
The possibility of changes in the sample during preparation and storage should be considered. Studies of fresh tissue would be ideal, but these are unavailable. Insofar as HgSe is concerned, none of the sample treatments is chemically likely to have induced either its formation or its breakdown. Similarly, the other species observed are not expected to be modified by the treatment of the samples. We cannot, however, exclude the possibility that some mercury-containing species have degraded due to formaldehyde fixative or other treatments of the samples.
Case 1 was a female exposed at age 8 years by consuming pork that was previously contaminated with methylmercury compounds. The hog had been fed grain treated with the fungicide Panogen (methylmercury dicyandiamide). It was slaughtered and consumed by a farmer and his family prior to its developing signs of methylmercury intoxication (7,8). The individual studied survived a further 21 years with signs that included muteness, dementia, quadriparesis, blindness, choreoathetosis, and seizures (9).
Case 2 received therapy with oral meso-dimercaptosuccinic acid, which was begun shortly after symptoms appeared 5 months after exposure to dimethylmercury. Both cases 1 and 2 died as a result of medical complications of their intoxications, including pneumonia. Clinical and neuropathological changes observed for cases 1 and 2 have been described previously (9−11).
The cause of death for cases 3, 4, and 5 were cirrhosis with septicaemia, pulmonary embolism, and arrhythmia due to acute myocardial infarction, respectively. Case 3 was chosen for analysis from among 17 adult (age 37−76 yrs) Seychellois brains as it had the highest total mercury content in the occipital lobe (range 46−324 ng/g of brain for all 17 brains considered). Case 5 was chosen for analysis at random from cases presenting for autopsy without evidence of neurological disease.
Sample Preparation for X-ray Absorption Spectroscopy
Samples of approximate dimension 20 × 3 × 2 mm3 were cut from predominantly gray matter of the cerebral cortex, loaded into acrylic sample cuvettes, and frozen in liquid nitrogen. Samples were coded and examined by X-ray absorption spectroscopy without prior knowledge of their source.
X-ray Absorption Spectroscopy
X-ray absorption spectroscopic (XAS) measurements were conducted at the Stanford Synchrotron Radiation Lightsource (SSRL). Mercury LIII-edge and selenium K-edge data were collected on the structural molecular biology XAS beamline 9-3 operating with a wiggler field of 2 T and employing a Si(220) double-crystal monochromator. Beamline 9-3 is equipped with a vertical collimating mirror upstream of the monochromator, and a downstream bent-cylindrical focusing mirror; both are rhodium-coated. Harmonic rejection was accomplished by setting the cutoff angle of the mirrors to reject energies above 15 keV. To minimize radiation damage and thermal vibrations, samples were maintained at a temperature of approximately 10 K in a liquid helium flow cryostat (Oxford Instruments, Abingdon, U.K.). X-ray absorption spectra were measured as the Se Kα1,2 or Hg Lα1,2 fluorescence excitation spectra using a 30-element germanium array detector (36) with analog electronics (Canberra Corporation, Meriden, CT, U.S.A.) employing an amplifier shaping time of 0.125 μs. To avoid problems with nonlinearity of the detector due to high count rates, X-ray filters (made of elemental As for Se and Ga2O3 for Hg) were used to preferentially absorb scattered radiation, with silver Soller-slits (EXAFS Co., Pinoche Nevada) optimally positioned between the sample and the detector. In previous work, we have shown that it is possible to extend the Hg LIII EXAFS data through the Se K-edge using Ga2O3 filters and careful electronic windowing (37), and we employed these same methods here.
Incident and transmitted XAS intensities were measured using nitrogen-filled ionization chambers. The mercury spectra were energy-calibrated with reference to LIII-edge spectrum of Hg−Sn amalgam foil measured simultaneously with the data, the lowest energy inflection of which was assumed to be 12285.0 eV. The selenium spectra were similarly energy calibrated with reference to the lowest energy inflection of hexagonal elemental selenium, which was assumed to be at 12658.0 eV. XAS data were processed using standard techniques and employing the EXAFSPAK program suite (38). Near-edge spectra were fitted to linear combinations of standard spectra using the EXAFSPAK program DATFIT using the procedures and criteria previously described (13). EXAFS oscillations were analyzed using the EXAFSPAK program OPT employing ab initio theoretical phase and amplitude functions calculated using the program FEFF v7.02 (39,40). For both DATFIT and OPT analyses, estimated standard deviations were obtained from the diagonal elements of the covariance matrix.
XAS Quantification of Hg and Se
Quantitative estimation of relative Se and Hg contents used examination of the Hg Lα1,2 peak areas (corrected for differences in fluorescence yield) from emission spectra and the Hg LIII and Se K absorption edge jumps. Approximate elemental concentrations were obtained by comparing the edge jumps from un-normalized background-subtracted spectra with edge jumps measured from spectra of solutions containing the element of interest (either Se or Hg) at known concentrations measured under identical conditions. Estimated standard deviations were determined from 20 data points above the edge in a region of the scan where the absorption was relatively flat and subtracting a least-squares cubic polynomial to account for any variation in absorption. Somewhat larger systematic errors in the concentration estimates arise from thickness differences between the samples but are expected to be <5% of the concentration estimate.
X-ray Fluorescence Imaging
X-ray fluorescence imaging data were collected on SSRL beamline 10-2 with microfocus X-ray beams generated with a tapered glass monocapillary displaced from the focal point to give an X-ray beam diameter of approximately 40 μm (determined by scanning a blade through the beam). Samples were imaged by rastering through the beam using continuous scan mode (41) with an effective pixel size of 40 × 40 μm2 and an effective dwell time of 128 ms/pixel. X-ray fluorescence was monitored used a Vortex-90EX silicon drift detector (SII NanoTechnology USA Inc. Northridge CA, U.S.A.) connected to a DXP-XMAP digital signal processing unit (XIA LLC, Hayward CA, U.S.A.). Full X-ray fluorescence emission spectra were collected for every pixel and analyzed to obtain peak areas for Hg Lα1,2, Se Kα1,2, and other fluorescence lines, using specially developed computer software for removal of Compton scattering.
Acknowledgments
We thank Dr. Rubell Brewer (Victoria Hospital and the Ministry of Health, Republic of Seychelles), Dr. Larry E. Davis (Neurology Service, New Mexico Veterans Affairs Health Care System), and Dr. David W. Nierenberg (Department of Medicine and Pharmacology/Toxicology, Dartmouth Medical School) for providing brain specimens for analysis. We thank members of the George and Pickering research groups and the staff at the Stanford Synchrotron Radiation Lightsource (SSRL) for their assistance with synchrotron experiments.
All authors contributed toward design of the research, in interpretation of results, and in writing the paper. Drs. Korbas, Watson, Singh, Pickering, and George performed X-ray absorption spectroscopy and X-ray fluorescence mapping. Dr. O’Donoghue prepared the samples and performed conventional microscopic examinations and Drs. O’Donoghue and Watson performed conventional chemical analyses.
This work was supported by the Canadian Institutes of Health Research (Grant FRN68849), Canada Research Chair awards (G.N.G. and I.J.P.), by the Natural Sciences and Engineering Research Council of Canada (I.J.P.), and by the National Institutes of Environmental Health Sciences (Environmental Health Sciences Center Grant P30 ES01247). Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource (SSRL), a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program.
Funding Statement
National Institutes of Health, United States
References
- Clarkson T. W.; Magos L. (2006) The toxicology of mercury and its chemical compounds. Crit. Rev. Toxicol. 36, 609–662. [DOI] [PubMed] [Google Scholar]
- http://www.epa.gov/mercury/.
- Wagemann R.; Trebacz U, E.; Boila G.; Lockhart W. L. (1998) Methylmercury and total mercury in tissues of arctic marine mammals. Sci. Total Environ. 218, 19–31. [DOI] [PubMed] [Google Scholar]
- Weihe P.; Grandjean P.; Debes F.; White R. (1996) Health implications for Faroe islanders of heavy metals and PCBs from pilot whales. Sci. Total Environ. 186, 141–148. [DOI] [PubMed] [Google Scholar]
- World Health Organization, Global and regional food consumption patterns and trends. Availability and consumption of fish. Available at http://www.who.int/nutrition/topics/3_foodconsumption/en/index5.html (2009).
- Choi A. L.; Grandjean P. (2008) Methylmercury exposure and health effects in humans. Environ. Chem. 5, 112–120. [Google Scholar]
- Curley A.; Sedlak V. A.; Girling E. F.; Hawk R. E.; Barthel W. F.; Pierce P. E.; Likosky W. H. (1971) Organic mercury identified as the cause of poisoning in humans and hogs. Science 172, 65–67. [DOI] [PubMed] [Google Scholar]
- Piper R. C.; Miller V. L.; Dickinson E. O. (1971) Toxicity and distribution of mercury in pigs with acute methylmercurialism. Am. J. Vet. Res. 32, 263–273. [PubMed] [Google Scholar]
- Davis L. E.; Kornfeld M.; Mooney H. S.; Fiedler K. J.; Haaland K. Y.; Orrison W. W.; Cernichiari E.; Clarkson T. W. (1994) Methylmercury poisoning: Long-term clinical, radiological, toxicological, and pathological studies of an affected family. Ann. Neurol. 35, 680–688. [DOI] [PubMed] [Google Scholar]
- Siegler R. W.; Nierenberg D. W.; Hickey W. F. (1999) Fatal poisoning from liquid dimethylmercury: A neuropathologic study. Hum. Pathol. 30, 720–723. [DOI] [PubMed] [Google Scholar]
- Nierenberg D. W.; Nordgren R. E.; Chang M. B.; Siegler R. W.; Blayney M. B.; Hochberg F.; Toribara T. Y.; Cernichiari E.; Clarkson T. (1998) Delayed cerebellar disease and death after accidental exposure to dimethylmercury. N. Engl. J. Med. 338, 1672–1676. [DOI] [PubMed] [Google Scholar]
- Pellicer A.; Gayá F.; Madero R.; Quero J.; Cabañas F. (2002) Noninvasive continuous monitoring of the effects of head position on brain hemodynamics in ventilated infants. Pediatrics 109, 434–440. [DOI] [PubMed] [Google Scholar]
- George G. N.; Singh S. P.; Prince R. C.; Pickering I. J. (2008) The chemical forms of mercury and selenium in fish following digestion with simulated gastric fluid. Chem. Res. Toxicol. 21, 2106–2110. [DOI] [PubMed] [Google Scholar]
- Galier J.; George G. N.; Pickering I. J.; Madden S; Prince R. C.; Yu E. Y.; Denton M. B.; Younis H. S.; Aposhian H. V. (2000) The structural basis of the antagonism between inorganic mercury and selenium in mammals. Chem. Res. Toxicol. 13, 1135–1142. [DOI] [PubMed] [Google Scholar]
- Early J. W. (1950) Description of the selenide minerals. Am. Mineral. 35, 337–364. [Google Scholar]
- Allen F. H.; Kennard O.; Watson D. G. (1994) Crystallographic databases: Search and retrieval of information from the Cambridge Structural Database. Struct. Correl. 1, 71–110. [Google Scholar]
- Long L. H.; Cattanach J. (1961) Antoine vapor-pressure equations and heats of vaporization for the dimethyls of zinc, cadmium, and mercury. J. Inorg. Nucl. Chem. 20, 340–342. [Google Scholar]
- Ostlund K. (1969) Metabolism of methyl mercury and dimethyl mercury in mice. Acta Pharmacol. Toxicol. 27(Suppl), 1–132. [PubMed] [Google Scholar]
- Korbas M.; Blechinger S.; Krone P. H.; Pickering I. J.; George G. N. (2008) Localizing organomercury uptake and accumulation in zebrafish larvae at the tissue and cellular level. Proc. Natl. Acad. Sci. U.S.A. 105, 12108–12112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martoja R.; Berry J. P. (1980) Identification of Tiemannite as a probable product of demethylation of mercury by selenium in cetaceans. A complement to the scheme of the biological cycle of mercury. Vie Milieu 30, 7–10. [Google Scholar]
- Arai T.; Ikemoto T.; Hokura A.; Terada Y.; Kunito T.; Tanabe S.; Nakai I. (2004) Chemical forms of mercury and cadmium accumulated in marine mammals and seabirds as determined by XAFS analysis. Environ. Sci. Technol. 38, 6468–6474. [DOI] [PubMed] [Google Scholar]
- Cernichiari E.; Myers G. J.; Ballatori N.; Zareba G.; Vyas J.; Clarkson T. (2007) The biological monitoring of prenatal exposure to methylmercury. Neurotoxicology 28, 1015–1022. [DOI] [PubMed] [Google Scholar]
- Watanabe C.; Yin K.; Kasanuma Y.; Satoh H. (1999) In utero exposure to methylmercury and Se deficiency converge on the neurobehavioral outcome in mice. Neurotoxicol. Tetratol. 21, 83–88. [DOI] [PubMed] [Google Scholar]
- Ralston N. V. C.; Blackwell J. L. III; Raymond L. J. (2007) Importance of molar ratios in selenium-dependent protection against methylmercury toxicity. Biol. Trace Elem. Res. 119, 255–268. [DOI] [PubMed] [Google Scholar]
- Ni B.; Kramer J. R.; Bell R. A.; Werstiuk N. H. (2006) Protonolysis of the Hg-C Bond of chloromethylmercury and dimethylmercury. A DFT and QTAIM study. J. Phys. Chem. A 110, 9451–9458. [DOI] [PubMed] [Google Scholar]
- George G. N.; Prince R. C.; Gailer J.; Buttigieg G. A.; Denton M. B.; Harris H. H.; Pickering I. J. (2004) Mercury binding to the chelation therapy agents DMSA and DMPS, and the rational design of custom chelators for mercury. Chem. Res. Toxicol. 17, 999–1006. [DOI] [PubMed] [Google Scholar]
- Hoffmeyer R.; Singh S. P.; Doonan C. J.; Ross A. R. S.; Hughes R. J.; Pickering I. J.; George G. N. (2006) Molecular mimicry in mercury toxicology. Chem. Res. Toxicol. 19, 753–759. [DOI] [PubMed] [Google Scholar]
- Carty A. J.; Malone S. F.; Taylor N. J.; Canty A. J. (1983) Synthesis, spectroscopic, and X-ray structural characterization of methylmercury-D,L-selenocysteinate monohydrate, a key model for the methylmercury(II)-selenoprotein interaction. J. Inorg. Biochem. 18, 291–300. [Google Scholar]
- Naganuma A.; Imura N. (1981) New method for bis(methylmercuric) selenide synthesis from methylmercuric chloride, sodium selenite and reduced glutathione. Chemosphere 10, 441–443. [Google Scholar]
- Kahn M. A. K.; Wang F. (2010) Chemical demethylation of methylmercury by selenoamino acids. Chem. Res. Toxicol. 23, 1202–1206. [DOI] [PubMed] [Google Scholar]
- Arnold A. P.; Canty A. J.; Skelton B. W.; White A. H. (1982) Mercury(II) selenolates. Crystal structures of polymeric Hg(SeMe)2 and the tetrameric pridinates [{HgCl(py)(SeEt)}4] and [HgCl(py)0.5(SeBut)}4]. J. Chem. Soc. Dalton 607–613. [Google Scholar]
- Ganther H. E.; Goudie C.; Sunde M. L.; Kopecky M. J.; Wagner P. (1972) Selenium: Relation to decreased toxicity of methylmercury added to diets containing tuna. Science 175, 1122–1124. [DOI] [PubMed] [Google Scholar]
- Nakayama A.; Hill K. E.; Austin L. M.; Motley A. K.; Burk R. F. (2007) All regions of mouse brain are dependent on selenoprotein P for maintenance of selenium. J. Nutr. 137, 690–693. [DOI] [PubMed] [Google Scholar]
- Ejima A.; Watanabe C.; Koyama H.; Matsuno K.; Satoh H. (1996) Determination of selenium in the human brain by graphite furnace atomic absorption spectrometry. Biol. Trace. Elem. Res. 54, 9–21. [DOI] [PubMed] [Google Scholar]
- Cernichiari E.; Toribara T. Y.; Liang L.; Marsh D. O.; Berlin M. W.; Myers G. J.; Cox C.; Shamlaye C. F.; Choisy O.; Davidson P.; Clarkson T. W. (1995) The biological monitoring of mercury in the Seychelles study. Neurotoxicology 16, 613–628. [PubMed] [Google Scholar]
- Cramer S. P.; Tench O.; Yocum M.; George G. N. (1998) A 13-element Ge detector for fluorescence EXAFS. Nucl. Instrum. Meth. A266, 586–591. [Google Scholar]
- Korbas M.; Percy A. J.; Gailer J.; George G. N. (2008) A possible molecular link between the toxicologies of arsenic, selenium and methyl-mercury: methyl mercury(II) seleno bis(S-glutathionyl)arsenic (III). J. Biol. Inorg. Chem. 13, 461–470. [DOI] [PubMed] [Google Scholar]
- http://ssrl.slac.stanford.edu/exafspak.html.
- Rehr J. J.; Albers R. C.; Zabinsky S. I. (1992) High-order multiple-scattering calculations of X-ray-absorption fine structure. Phys. Rev. Lett. 69, 3397–3400. [DOI] [PubMed] [Google Scholar]
- Rehr J. J.; Mustre de Leon J.; Zabinsky S. I.; Albers R. C. (1991) Theoretical X-ray absorption fine structure standards. J. Am. Chem. Soc. 113, 5135–5140. [Google Scholar]
- Popescu B. F. G.; George M. J.; Bergmann U.; Garachtchenko A. V.; Kelly M. E.; McCrea R. P. E.; Lüning K.; Devon R. M.; George G. N.; Hanson A. D.; Harder S. M.; Chapman L. D.; Pickering I. J.; Nichol H. (2009) Mapping metals in Parkinson’s and normal brain using rapid-scanning X-ray fluorescence. Phys. Med. Biol. 54, 651–663. [DOI] [PubMed] [Google Scholar]