

Abstract
Aims
The transient outward potassium current (Ito) plays important roles in action potential (AP) morphology and dynamics; however, its role in the genesis of early afterdepolarizations (EADs) is not well understood. We aimed to study the effects and mechanisms of Ito on EAD genesis in cardiac cells using combined experimental and computational approaches.
Methods and results
We first carried out patch-clamp experiments in isolated rabbit ventricular myocytes exposed to H2O2 (0.2 or 1 mM), in which EADs were induced at a slow pacing rate. EADs were eliminated by either increasing the pacing rate or blocking Ito with 2 mM 4-aminopyridine. In addition to enhancing the L-type calcium current (ICa,L) and the late sodium current, H2O2 also increased the conductance, slowed inactivation, and accelerated recovery from the inactivation of Ito. Computer simulations showed that Ito promoted EADs under the condition of reduced repolarization reserve, consistent with the experimental observations. However, EADs were only promoted in the intermediate ranges of the Ito conductance and the inactivation time constant. The underlying mechanism is that Ito lowers the AP plateau voltage into the range at which the time-dependent potassium current (namely IKs) activation is further slowed and ICa,L is available for reactivation, leading to voltage oscillations to manifest EADs. Further experimental studies in cardiac cells of other species validated the theoretical predictions.
Conclusion
In cardiac cells, Ito, with a proper conductance and inactivation speed, potentiates EADs by setting the AP plateau into the voltage range where ICa,L reactivation is facilitated and IKs activation is slowed.
Keywords: Transient outward current, Early afterdepolarization, Cardiac arrhythmias, Computer model, Dynamic mechanisms
1. Introduction
The transient outward potassium (K) current (Ito) has been shown to play important roles in the action potential (AP) morphology and AP duration (APD) alternans,1–3 which has been linked to arrhythmogenesis in Brugada syndrome.4 In a study by Guo et al.,5 it was shown that the elimination of Ito in mouse ventricular myocytes increased the propensity of early afterdepolarizations (EADs). EADs are secondary depolarizations or oscillations during the AP plateau or repolarizing phase, which are associated with arrhythmogenesis in cardiac diseases, such as long QT syndrome.6,7 It is well accepted that EADs are facilitated under conditions of reduced repolarization reserve,8,9 in which APDs are prolonged by either increasing inward currents or decreasing outward currents, or both. It should be noted that although a reduced repolarization reserve prolongs the APD, simply lengthening the APD does not necessarily lead to EADs. In a recent theoretical study,10 Tran et al. showed that in addition to a reduced repolarization reserve, the activation and inactivation kinetics of ICa,L must be properly matched to cause the voltage oscillations underlying EADs. In addition, the activation time of the slow time-dependent K current (namely IKs) must be slow enough to allow the voltage oscillations to manifest during the plateau or repolarizing phase. More specifically, the membrane voltage must decline to <0 mV quickly enough before IKs has sufficient time to fully activate so that ICa,L can be reactivated while the repolarization reserve is still low. This leads us to hypothesize that Ito, by lowering the AP plateau voltage into the range at which IKs activation is slowed and ICa,L is available for reactivation, can lead to voltage oscillations to manifest EADs. In our previous study,11 we showed that exogenous addition of H2O2 to myocytes induces EADs, which is probably mediated by the activation of Ca/calmodulin kinase II (CaMK II),12 and the consequent activation of the L-type calcium current (ICa,L) and the late sodium current (INa).13–16 In the present study, we used this H2O2-induced EAD model and confirmed our hypothesis via experimental and theoretical studies.
2. Methods
2.1. Patch-clamp experiments
This investigation conforms with the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health (NIH Publication No. 85–23, Revised 1996). All animal experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Medicine and Dentistry of New Jersey-New Jersey Medical School, by the Ethical Committee of Xi'an Jiaotong University, and by the Institutional Animal Care and Use Committee at Columbia University.
2.1.1. Single cell isolation
Myocytes were enzymatically isolated from the left ventricles of adult rabbit, rat, and mouse hearts in Langendorff fashion at 37°C with collagenase and protease.11,17 Canine epicardial ventricular myocytes were isolated from the LV sections. Canine Purkinje cells were isolated from the Purkinje fibres of both right and left ventricles. The animals were euthanized with Euthasol (0.22 mL/kg, i.e. pentobarbital 86 mg/kg + phenytoin 11 mg/kg; iv in rabbits and ip in rats and mice) or anaesthetized with propofol (5–15 mg/kg; iv in dogs). Immediately after the cessation of breathing (in rabbits, rats, mice) or the induction into a surgical plane of anaesthesia (in dogs, the jawbone, pupil reflexes, paw relaxes, and tongue rigidity were continually monitored during the procedure), the chest was opened through a left thoracotomy. The heart was quickly excised from the chest. The detailed protocols for heart perfusion and single cell dispersion are described in Supplementary material online.
2.1.2. Patch clamp
Myocytes were patch-clamped using the whole-cell configuration in either current-clamp (for AP recording) or voltage (or AP)-clamp mode (for Ito or ICa,L recording) as in our previous publications.11,18–20 To record Ito, the ICa,L blocker Cd3+ (0.3 mM) and Na current blocker TTX (10 µM) were added to Tyrode's solution. Cells were paced at a pacing cycle length (PCL) of 1 and 6 s to study the rate dependence of Ito and EADs. All experiments were carried out at 34–36°C.
2.1.3. Pacing protocols
A dynamic pacing protocol was used to measure APD restitution. The myocytes were paced at PCLs of 5, 2, 1 s, 700 ms for 6 beats each, and then at PCL 400 ms, decremented every 12 beats by 20 ms (from 400 to 300 ms), 10 ms (from 300 to 200 ms), or 5 ms (from 200 to 150 ms), or until a 2:1 block occurred. Recovery of Ito from the inactivation was investigated using a conventional two-pulse protocol: an inactivating pulse depolarizing to +40 mV for 400 ms (P1) followed by a variable recovery interval (5, 10, 20, 50, 100, 300, 600 ms; 1, 3, 6, 10, 15 s, respectively) and subsequent +40 mV test pulse (P2). The interpulse potential was set at – 80 mV.
2.2. Computer simulation and theoretical analysis
Simulations were carried out in single myocyte models with the following governing equation for voltage (V): CmdV/dt = −Iion+ Isti, where Cm= 1 μF/cm2, Iion is the total ionic current density, and Isti is the stimulation current density. Iion formulation was taken from the rabbit ventricular myocyte AP model by Mahajan et al.21 with modifications. Modifications to the model were made to better fit the APD restitution and EAD properties observed in the experimental studies. The major changes include substitution of ICa,L by the Hodgkin-Huxley type of formulation as in the 1994 Luo and Rudy model (LR2)22 and Ito by the formulation modified from the one by Dong et al.23 Therefore, the ionic current density of the rabbit ventricular model became: 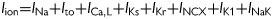 . The effects of H2O2 on ion channels were modelled by increasing ICa,L, late INa, Na-Ca exchange current (INCX), and Ito, based on previous11,13,14,17 and present experimental studies. 4-AP is known to inhibit different components of Ito.24 For simplicity, however, we modelled the 4-AP effect by only reducing the maximum conductance of Ito with the goal of reducing 4-AP sensitive currents. Details of the modifications and formulations of the ionic currents are presented in Supplementary material online, Methods and Figures S2–6.
. The effects of H2O2 on ion channels were modelled by increasing ICa,L, late INa, Na-Ca exchange current (INCX), and Ito, based on previous11,13,14,17 and present experimental studies. 4-AP is known to inhibit different components of Ito.24 For simplicity, however, we modelled the 4-AP effect by only reducing the maximum conductance of Ito with the goal of reducing 4-AP sensitive currents. Details of the modifications and formulations of the ionic currents are presented in Supplementary material online, Methods and Figures S2–6.
To study the general mechanism of Ito promoting EADs and for the convenience of theoretical analysis, we used the 1991 Luo and Rudy (LR1) model25 and added an Ito to the LR1 model, with Ito modified also from the formulation by Dong et al.23 (see Supplementary material online, Methods and Figures S7–8). The total ionic current density was
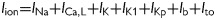 . The rationale for choosing LR1 is because it is much simpler than the rabbit ventricular model and it has the minimum requirements for EADs to occur, as shown in the previous study.10 Using this simple model, we can follow the same method as in the previous study10 to perform bifurcation analysis to understand how Ito promotes EADs (see Supplementary material online, Figure S11).
. The rationale for choosing LR1 is because it is much simpler than the rabbit ventricular model and it has the minimum requirements for EADs to occur, as shown in the previous study.10 Using this simple model, we can follow the same method as in the previous study10 to perform bifurcation analysis to understand how Ito promotes EADs (see Supplementary material online, Figure S11).
3. Results
3.1. Ito promotes EADs in rabbit ventricular myocytes
EADs are usually promoted by bradycardia because during the longer diastolic intervals26 the delayed rectifier K channels enter the states that are deeply closed, thus reducing outward currents and lengthening the APD. However, in rabbit ventricular myocytes, the APD first increases as the heart rate decreases, but then becomes shorter at very slow heart rates (i.e. the APD restitution curve is biphasic) (Figure 1A and B). This has been attributed to the slow recovery of Ito in the rabbit cells.27 However, it is at these very slow heart rates (PCL > 3 s) that exposure to H2O2 induces EADs, but not at the intermediate heart rates at which the APD is normally longer (Figure 1C).11,19 We assessed the role of Ito in the EAD formation by applying 4-AP to block Ito. As shown in Figure 1D and E, 4-AP (2 mM) alone prolonged APD at all PCL, to a greater extent at the long PCL (e.g. 6 s), and to a less extent at short PCLs (e.g. 1 s). Therefore, the restitution curve was transformed from a biphasic shape to a monophasic one. Despite prolonging the APD at very slow heart rates, 4-AP paradoxically prevented overt EADs in response to 200 μM H2O2 (Figure 1F). The same phenomenon was observed in 20 cells from 8 rabbits. It should be noted that some cells from rabbits showed a less negative slope in the biphasic restitution curve, suggesting a lower Ito level. However, these cells exhibited similar responses to H2O2 for inducing EADs and to 4-AP for attenuating EADs (data not shown).
Figure 1.

Dependence of APD and EAD genesis on Ito. (A) The APD restitution curve obtained from a rabbit ventricular myocyte showing a biphasic change of the APD. The expanded region between 0 and 300 ms is shown in the insets. DI, diastolic interval. (B) APs recorded at PCL 6 s (grey solid) and 1 s (dashed). (C) H2O2 (200 μM, perfusion for 6 min) induced EADs at PCL = 6 s. (D–F) Same as (A)–(C), but in the presence of 2 mM 4-AP. Note that 4-AP eliminated the EADs (F). Five consecutive APs are shown in panels (C) and (F).
3.2. Effects of H2O2 on Ito kinetics and conductance
Under our experimental conditions, H2O2 enhanced Ito in a time course comparable with that for the EAD onset.11 This was attenuated by 4-AP (Figure 2A and B). H2O2 increased both the peak amplitude of Ito and the magnitude of the current at the late phase under the 400 ms-pulse clamp (we refer to the late component as Ilate, including the slow and non-inactivating components) (Figure 2C). Finally, H2O2 slowed the Ito inactivation by decreasing the time constants of both the slow (τs,in) and fast components (τf in) (Figure 2D).
Figure 2.

Effect of H2O2 Ito in rabbit ventricular myocytes. (A–D) Effects of H2O2 on Ito amplitude and inactivation: Ito was elicited by depolarization to + 60 mV from a holding potential of −70 mV every 10 s. (A) The time course showing the effect of H2O2 and 4-AP on the Ito amplitude. The current values measured at 100 ms after the depolarization pulse [as indicated by the dashed line in (B)]. The current traces at point a, b, c are shown in (B). (C) Bar graph summarizing the effect of H2O2 on peak and late currents (measured at the end of 400 -ms pulse). (D) H2O2 slowed down the inactivation of both the fast and slow components of Ito. **P < 0.01 compared with control (n = 7). (E–H) Effect of H2O2 on the recovery from inactivation: (E) representative Ito traces following a standard two-pulse protocol (supplements) to measure the recovery from inactivation under control in the absence of H2O2 (Ctl). P2/P1 is the ratio of the test pulse current/inactivating pulse current amplitudes. The inactivated current amplitude was measured by the difference between the peak inactivating pulse current or the test pulse current and the current at the end of the inactivating pulse. The inter-pulse potential was set at –80 mV. (F) The same as (E), in the presence of 1 mM H2O2. (G) Summarized peak Ito values fitted to a two-term exponential function. (H) The effect of H2O2 on time constants for slow (τslow) and fast (τfast) components.
Under control conditions and consistent with APD shortening at PCL > 3 s, Ito displayed a slow recovery from inactivation, as shown in Figure 2E and G. Ito recovery from the inactivation was best fit with a double exponential equation (see Supplementary material online, Methods) where the averaged time constants for the fast (τf, re) and slow (τs, re) components are 832 ± 222 ms and 5.8 ± 2.1 s, respectively. H2O2 accelerated the recovery from the inactivation of Ito, mainly by decreasing τf, re to 240 ± 61 ms, while τs, re showed no significant change (Figure 2F–H). The amplitudes of the fast and slow components (Af: 0.22 ± 0.08 and As: 0.79 ± 0.14 under control conditions) were significantly affected by H2O2, (Af: 0.40 ± 0.05 and As: 0.61 ± 0.04) (P < 0.05, n = 6).
3.3. Effect of Ito on ICa,L during the AP during H2O2 exposure
Since ICa,L reactivation plays an essential role in EAD genesis, we compared the effects of an AP with or without Ito on ICa,L during an AP clamp. When the cells were clamped with the AP morphology corresponding to the presence of Ito (i.e. an AP pre-recorded at a PCL 6 s), ICa,L was significantly larger both in the absence and presence of H2O2, than those recorded during the AP morphology corresponding to the absence of Ito (i.e. rectangular AP pre-recorded at PCL 1 s) ( see Supplementary material online, Figure S1). This result indicates that the presence of Ito lowers the AP plateau voltage into a voltage range that produces a larger inward ICa,L, known to be a key factor in EAD formation.28,29
3.4. Ito promotes EADs in an AP model of rabbit ventricular myocytes
To understand how Ito promotes EAD genesis, we carried out simulations using a rabbit ventricular AP model,21 modified to include the property of slow recovery of Ito observed experimentally. The effects of H2O2 were modelled by increasing the late INa, ICa,L, and Ito (see Supplementary material online, Methods and Figures S2–6). The APD restitution curve for the control (Figure 3A and B) was biphasic, similar to experimental data. When the effects of H2O2 on ionic currents were simulated, EADs occurred only at slow pacing rates (Figure 3C). When we removed Ito from the model to simulate the effects of blocking Ito with 4-AP, the APD restitution curve became monotonic (Figure 3D and E), and no EADs were observed (Figure 3F). These data agree well with the experimental observations shown in Figure 1.
Figure 3.

APD restitution and H2O2-induced EADs in the rabbit ventricular model in the presence of Ito. The plots are an analogy to Figure 1. (A) The APD restitution curve of the control cases. (B) Control APs for PCLs of 6 s (grey solid) and 1 s (dashed). (C) Simulated H2O2-induced EADs. (D–F) The same as (A)–(C), but in the presence of 4-AP and Ito blocked.
To show how Ito interacts with other currents to potentiate EADs, we plotted voltages along with Ito, ICa,L, IKs, and INCX under different conditions in Figure 4. In the control case (Figure 4A and B), Ito was larger during slower pacing (PCL 6 s) due to its slow recovery, which lowered the voltage at the plateau phase. This in turn caused a larger peak ICa,L, a larger INCX, and a very small IKs; however, the APD was shorter. When the effects of H2O2 were simulated (Figure 4C and D), the AP and the ionic currents at PCL = 1 s were similar to those of the control at PCL = 1 s. At PCL = 6 s (Figure 4D), EADs were induced by H2O2. In this case, a larger Ito resulted in a lower AP plateau voltage that caused a larger peak ICa,L and a larger INCX. Concomitantly, the activation of IKs was much slower due to the lower AP plateau voltage (here the activation time constant of IKs vs. voltage is reproduced in Supplementary material online, Figure S9), which also played an important role in allowing the inward currents to reverse repolarization and cause EADs. When Ito was partially blocked (Figure 4E), the AP plateau voltage was elevated, resulting in a smaller peak ICa,L, a smaller INCX, and a much larger IKs, which prevented EADs from forming.
Figure 4.

Ito-induced ionic current changes in the rabbit ventricular model. AP, Ito, ICa,L, IKs, and INCX were plotted under different conditions. The dashed line in the top row indicates the plateau level of AP under the control condition. (A and B) The cell was paced at PCLs of 1 and 6 s, respectively, in the absence of H2O2. (C and D) The cell was paced at PCLs of 1 and 6 s, respectively, in the presence of H2O2. Note that EADs were induced by H2O2 at PCL 6 s. (E) H2O2-induced EADs were eliminated by partially blocking Ito.
3.5. Mechanisms by which Ito promotes EADs
As shown in Figure 4, Ito did not directly cause EADs; rather, its effects on voltage caused complex changes in other currents, making it difficult to sort out the general underlying mechanism. To simplify the analysis, we used the LR1 model with modifications to reduce repolarization reserve (see Supplementary material online for detailed changes). Under this (control) condition, the APD is long with no EAD presence (Figure 5A). Consistent with the results from myocyte experiments and the rabbit ventricular AP model, adding an Ito to the control APinduced EADs (Figure 5B). In this case, ICa,L was increased and the activation of the time-dependent K current (IK, similar to IKs in the rabbit ventricular AP model) was slowed due to the lowering of the plateau voltage, similar to the more detailed rabbit ventricular AP model. When Ito was increased to a higher value, the APD became very short and no EADs could form (Figure 5C).
Figure 5.

Effects of Ito on EAD genesis in the LR1 model. APs, Ito, ICa,L, and IK were plotted for three different conditions. (A) Control, no Ito. (B) A proper Ito added to the control model induced EADs. (C) A larger Ito shortened the APD and eliminated EADs. (D) Phase diagram showing the EAD region (coloured) in the Ito conductance (Gto) and τh0 parameter space. τh0 is the constant component of the inactivation time constant τh of Ito, i.e. τh= τh0+ τh(V) [see equation (17) in the Supplementary material online, Methods]. (E) The APD distribution in the Ito conductance and the τh0 parameter space.
In Figure 5D and E, we scanned the parameters of Ito conductance (Gto) and its inactivation time constant for EADs and the APD, respectively. EADs only occurred in the intermediate ranges of conductance and the inactivation time constant, whereas small or large conductance and slow or fast inactivation suppressed EADs. When Ito inactivation was fast, an increase in Ito conductance first increased the APD (and EADs) but then caused a sudden transition to a short APD when the Ito conductance was greater than a critical value (colour changes from green or red to blue suddenly in Figure 5E). When Ito inactivation was slow, the APD decreased continuously as the Ito conductance increased. These effects of Ito on the APD agree with those observed in previous studies.23,30 In fact, a very slow-inactivating Ito component suppresses EADs (see Supplementary material online, Figure S10).
To further understand how Ito promotes EADs, we used non-linear dynamics and bifurcation theory following the method in a previous study by Tran et al.10 Details of this analysis and discussion are presented in Supplementary material online (Section 2.3 and Figure S11). The major finding is that Ito, with a proper conductance and inactivation speed, can bring the membrane voltage into the window to undergo the bifurcation that is required for voltage oscillations and thus EADs.
3.6. The role of Ito in EAD formation in cardiac cells of other species
It is well known that the molecular composition, current density, and kinetics (e.g. inactivation and recovery from inactivation) of Ito vary in different species and locations in the heart. To validate the general predictions from the general theoretical analysis (Figure 5D–E), we carried out additional experiments in ventricular cells from rats, mice, and canine epicardium, as well as canine Purkinje cells. Experimental results are shown in detail in Supplementary material online, Figures S12–15 and summarized and compared qualitatively with simulations in Figure 6.
Figure 6.

Roles of Ito in EAD genesis in different species. (A) The EAD distribution in the ICa,L and Ito conductance (GCa and Gto) space, using the same model as in Figure 5. The inactivation time constant of the Ito is the intermediate value (τh0= 200 ms) as revealed by Figure 5D. The EAD occurrence region is marked in grey. (B) APs recorded in different species as indicated (a–d). The black and coloured symbols mark the putative locations of the APs in the distribution diagram (A). (C) Summarized bar graphs showing the incidence of EADs within 20 APs (n ≥ 3 cells). ns, not significant; **P < 0.01 by Fisher's exact test. (D) The same as (A), except that the inactivation time constant of the Ito is fast (τh0= 20 ms), which is similar to that of canine ventricular myocytes. (E). APs recorded from canine ventricular myocytes. The symbols mark the putative locations of the APs in the distribution diagram (C).
Since we showed that H2O2 increases both ICa,L and Ito and in order to compare with experimental results, we first scanned the Ito conductance and ICa,L conductance on EAD formation in the LR1 model for slow and fast Ito inactivation (Figure 6A and D). Figure 6A was obtained for an Ito inactivation time constant in the intermediate range as shown in Figure 5D. In our model, EADs occurred in large ICa,L conductance and intermediate Ito conductance (grey region). For rabbit (Figure 6Ba and C) and rat ventricular myocytes (Figure 6Bb), H2O2 increased ICa,L and Ito, caused the AP to exhibit EADs. Blocking Ito by 4-AP (at either a low or high concentration) eliminated EADs. This is illustrated in Figure 6A by the transition from the black open circle (no EAD region) to the red closed circle (EAD region) and then to the blue open circle (no EAD region). For mouse ventricular myocytes (Figure 6Bc) and canine Purkinje cells (Figure 6Bd and C); however, H2O2 did not promote EADs. But partially blocking Ito by a low dose of 4-AP (50–100 µM) promoted EADs, which were eliminated by a higher dose of 4-AP (2 mM). This indicates that either Ito was already high in the control and/or H2O2 activated too much additional Ito. This scenario is illustrated in Figure 6A by the transitions from black open triangle (no EAD region) to the open red triangle (no EAD region), then to the red closed circle (EAD region), and finally to the blue open circle (no EAD region). In canine epicardial myocytes, the inactivation of Ito is very fast (∼19.9 ms). Based on our theoretical analysis, the propensity for EADs is greatly reduced for fast Ito inactivation (Figures 5D and 6D), and indeed we were not able to induced EADs in canine ventricular myocytes by H2O2 (Figure 6E). This is illustrated by the transitions of Figure 6D. In summary, the experimental results agree qualitatively well with the theoretical predictions for all the species and cell types analysed. Thus Ito with a proper conductance and inactivation time constant can promote EADs in cardiac myocytes, independent of species.
4. Discussion
Ito has been shown to play important roles in AP morphology and APD alternans.1–3 In this study, we carried out both experiments and simulations to study the effects of Ito on EAD genesis in cardiac cells exposed to H2O2. In rabbit ventricular myocytes, EADs occurred only at slow pacing rates and were eliminated by either shortening PCL or blocking Ito with 4-AP. The experimental observations were confirmed in computer simulations of an AP model of rabbit ventricular myocytes. We have shown in general that Ito with a proper conductance and inactivation time constant can bring membrane voltage into the window allowing ICa,L reactivation before IKs is fully activated to repolarize the myocytes. In addition, this lowering of the AP plateau also brings IKs into a voltage range where its activation is the slowest, further decreasing the repolarization reserve. These two effects together potentiate voltage oscillations and thus EAD generation. When Ito is weak or inactivates too quickly, the AP is long but no EADs can be induced. When Ito is too strong or inactivates too slowly, the AP is short and no EADs occur either. This general prediction of the effects of Ito on EAD genesis was validated in cardiac cells isolated from other species.
4.1. Reduced repolarization reserve and EAD genesis
The repolarization reserve is a concept articulated originally by Roden31 stating that normal ventricles have a redundancy in total repolarizing currents (mainly K currents), ensuring rapid repolarization. However, when this reserve is reduced, the cell loses the ability of normal repolarization and exhibits an excessive prolongation of APD. Based on the dynamical analysis by Tran et al.10 and the present study (see Supplementary material online, Figure S11), we show that membrane voltage needs to decrease ‘fast’ enough to enter an oscillatory window before slowly activating repolarization currents, such as IKs, grow too large. In the study by Tran et al.,10 EADs were induced by increasing the maximum conductance of ICa,L and slowing the activation of the time-dependent K current to reduce repolarization reserve. In the present study, we did not slow the activation of the repolarizing currents directly, but rather added Ito, which accelerated the early repolarization phase of the AP, bringing the voltage to the window range to undergo the bifurcation to promote EADs. However, it should be noted that when Ito becomes very large (such as would occur with a specific Ito activator), it causes sufficient AP shortening to suppress EAD formation. Therefore, strong repolarization (enhanced repolarization reserve) is needed to bring the voltage into the window for ICa,L reactivation, but once the voltage is in this window, weak repolarization (reduced repolarization reserve) is needed to maintain the voltage at this window for oscillations and thus EADs. Notably a recent study has shown that coupling to fibroblasts can also speed up the early repolarization phase of a myocyte AP and promote EAD generation.32
4.2. Roles of different Ito components in EAD genesis
At least two major Ito components, i.e. fast (Ito,f) and slow (Ito,s) components are present in adult myocytes.33 It has been suggested that pore-forming α subunits for Kv4.2 and Kv4.3 encode Ito,f, whereas Kv1.4 encodes Ito,s. They differ in the kinetics of inactivation and, in particular, the kinetics of recovery from inactivation. Ito,f recovers very rapidly with time constants in the range from tens to hundreds of milliseconds, while Ito,s recovers slowly with time constants in the range of seconds.34
The Ito components and their density are different in different species as well as different regions in the same heart. The kinetic properties of Ito in rabbit ventricular myocytes are substantially different from those of canine/human ventricular myocytes. The rabbit ventricular Ito is at least 10 times slower to inactivate and recover from inactivation than canine/human ventricular Ito.34 The epicardial myocytes from a canine left ventricle only express the high level of Ito,f.35 In contrast, both Ito,f and Ito,s are present in canine and human Purkinje cells.36–39
Different components of Ito have different effects on AP properties. For example, in canine ventricular cells that only have the fast component, the presence of a moderate Ito prolongs AP, whereas a high Ito substantially shortens AP, resulting in a sudden discontinuous change in the APD as Ito increases.40 In rabbit myocyte and canine Purkinje cells, Ito inactivates slowly with a non-inactivating steady-state component,38 whose presence always shortens the AP. This was demonstrated in Figure 1 in which the rabbit APD was shortened as the pacing rate decreased due to the slow recovery (τslow, re = 5.8 ± 2.1 s) of Ito. The 4-AP-induced APD prolongation (Figure 1E and F) was at least partially due to the blockage of this non-inactivating component.
As to the roles of the multiple components of Ito in EAD genesis, we showed that EADs only occurred in a certain range of Ito conductance and inactivation speed (Figure 5). In fact, a very slow-inactivating Ito component suppresses EADs (see Supplementary material online, Figure S10). This may also explain why a moderate decrease in Ito density results in the elevation of the AP plateau and the enhancement of EADs in mouse myocytes and canine Purkinje cells (Figure 6 and see Supplementary material online, Figures S13 and 14). On the other hand, if the Ito inactivation is too fast, such as in canine ventricular myocytes, it is not sufficient to promote EADs and is cardioprotective.
4.3. Role of H2O2 in EAD genesis
It has been shown that H2O2 activates late INa and increases ICa,L; both are considered to be the major ionic mechanisms for a H2O2-induced EAD.11,13–15 Here, we show that H2O2 also enhances the conductance of Ito, slows its inactivation, and accelerates its recovery from inactivation. This resembles the effect of CaMKII on the mammalian A-type K+ channel KV1.4.41 The findings of the present study indicate that H2O2-mediated activation of Ito also plays an important role in EAD genesis. Han et al.36 showed that H2O2 only activated Ito in canine Purkinje cells, but not in ventricular myocytes. This explains why H2O2 first caused further suppression of the low plateau in Purkinje cells before EAD generation. H2O2 only generated a dome in canine ventricular myocytes presumably by purely reactivation of ICa,L.
While more direct evidence is needed to validate the involvement of ROS in EAD genesis in vivo, a growing body of evidence has comprehensively established the link between an elevated ROS level and high propensity for cardiac arrhythmias in a number of circumstances, such as coronary heart disease, heart failure, and ageing.42–46 Moreover, a recent whole-heart study has shown that H2O2 causes EADs and focal ventricular arrhythmias in the aged fibrotic hearts.47
4.4. Limitations
It should be noted that in either experiments or simulations, Ito, an outward current per se cannot induce EADs. Other conditions (such as applying H2O2 in the experiments to activate inward currents, i.e. INa and ICa,L) must be satisfied. In addition, the mechanisms by which Ito contributes to EADs may be more complicated than those revealed in the simple model. For example, lowering the AP plateau promotes the Ca influx into the cell due to the reactivation of ICa,L, elevating intracellular Ca concentration and thus INCX, which could be the primary factor potentiating EADs. The elevation of Ca concentration may also affect CaMKII signalling, which affects many targets (such as ICa,L, late INa, and RyR).11,14,48 In fact, the prolonged treatment by H2O2 occasionally induced both DADs and EADs, or even sustained the depolarization of the membrane potential, most likely due to intracellular Ca overload.11 In a recent study49 we have demonstrated that the ionic mechanisms for EAD generation can be dominated by either ICa,L reactivation or spontaneous Ca release (Ca waves)-induced increase of INCX in different models. This study focuses on EADs primarily caused by the reactivation of ICa,L. We would expect that Ito would play a less important role in Ca wave-induced DADs or EADs.
Nevertheless, by a combination of experiments and simulations, we show here that the presence of Ito plays an important role in EAD genesis under the condition of reduced repolarization reserve. This may provide useful insights into the development of novel therapeutic strategies for EAD-related arrhythmias. Our results suggest that, in general, blocking Ito helps to prevent EADs under the condition of reduced repolarization reserve, such as the clinical settings of the long QT syndrome and heart failure. However, it should be noted that complex changes (remodelling) occur in heart failure and many changes in Ca cycling are arrhythmogenic.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This study is supported by a Postdoctoral Fellowship from American Heart Association Western states affiliate (Y.X.), HL 66140 (P.A.B.), NIH/NHLBI P01 HL078931 and the Laubisch endowments (Z.Q.), and NIH/NHLBI R01 HL97979 (L.H.X.).
Supplementary Material
Acknowledgements
The authors wish to thank Dr James N. Weiss for critical reading of the manuscript and his continuous support and encouragement.
Conflict of interest: none declared.
References
- 1.Maoz A, Krogh-Madsen T, Christini DJ. Instability in action potential morphology underlies phase 2 reentry: a mathematical modeling study. Heart Rhythm. 2009;6:813–822. doi: 10.1016/j.hrthm.2009.02.043. doi:10.1016/j.hrthm.2009.02.043. [DOI] [PubMed] [Google Scholar]
- 2.Qu Z, Xie Y, Garfinkel A, Weiss JN. T-wave alternans and arrhythmogenesis in cardiac diseases. Frontiers Physiol. 2010;1:154. doi: 10.3389/fphys.2010.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lukas A, Antzelevitch C. Phase 2 reentry as a mechanism of initiation of circus movement reentry in canine epicardium exposed to simulated ischemia. Cardiovasc Res. 1996;32:593–603. [PubMed] [Google Scholar]
- 4.Antzelevitch C, Nof E. Brugada syndrome: recent advances and controversies. Curr Cardiol Rep. 2008;10:376–383. doi: 10.1007/s11886-008-0060-y. doi:10.1007/s11886-008-0060-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo W, Li H, London B, Nerbonne JM. Functional consequences of elimination of Ito,f and Ito,s: early afterdepolarizations, atrioventricular block, and ventricular arrhythmias in mice lacking Kv1.4 and expressing a dominant-negative Kv4 α subunit. Circ Res. 2000;87:73–79. doi: 10.1161/01.res.87.1.73. [DOI] [PubMed] [Google Scholar]
- 6.Antzelevitch C. Ionic, molecular, and cellular bases of QT-interval prolongation and torsade de pointes. Europace. 2007;9(Suppl 4):iv4–15. doi: 10.1093/europace/eum166. doi:10.1093/europace/eum166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moss AJ, Kass RS. Long QT syndrome: from channels to cardiac arrhythmias. J Clin Invest. 2005;115:2018–2024. doi: 10.1172/JCI25537. doi:10.1172/JCI25537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biliczki P, Virag L, Iost N, Papp JG, Varro A. Interaction of different potassium channels in cardiac repolarization in dog ventricular preparations: role of repolarization reserve. Br J Pharmacol. 2002;137:361–368. doi: 10.1038/sj.bjp.0704881. doi:10.1038/sj.bjp.0704881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roden DM. Long QT syndrome: reduced repolarization reserve and the genetic link. J Intern Med. 2006;259:59–69. doi: 10.1111/j.1365-2796.2005.01589.x. doi:10.1111/j.1365-2796.2005.01589.x. [DOI] [PubMed] [Google Scholar]
- 10.Tran DX, Sato D, Yochelis A, Weiss JN, Garfinkel A, Qu Z. Bifurcation and chaos in a model of cardiac early afterdepolarizations. Phys Rev Lett. 2009;102:258103. doi: 10.1103/PhysRevLett.102.258103. doi:10.1103/PhysRevLett.102.258103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie LH, Chen F, Karagueuzian HS, Weiss JN. Oxidative-stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ Res. 2009;104:79–86. doi: 10.1161/CIRCRESAHA.108.183475. doi:10.1161/CIRCRESAHA.108.183475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048. doi:10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song YH, Cho H, Ryu SY, Yoon JY, Park SH, Noh CI, et al. L-type Ca2+ channel facilitation mediated by H2O2-induced activation of CaMKII in rat ventricular myocytes. J Mol Cell Cardiol. 2010;48:773–780. doi: 10.1016/j.yjmcc.2009.10.020. doi:10.1016/j.yjmcc.2009.10.020. [DOI] [PubMed] [Google Scholar]
- 14.Ward CA, Giles WR. Ionic mechanism of the effects of hydrogen peroxide in rat ventricular myocytes. J Physiol. 1997;500(Pt 3):631–642. doi: 10.1113/jphysiol.1997.sp022048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song Y, Shryock JC, Wagner S, Maier LS, Belardinelli L. Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J Pharmacol Exp Ther. 2006;318:214–222. doi: 10.1124/jpet.106.101832. doi:10.1124/jpet.106.101832. [DOI] [PubMed] [Google Scholar]
- 16.Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T, et al. Reactive oxygen species-activated Ca/calmodulin kinase IIdelta is required for late INa augmentation leading to cellular Na and Ca overload. Circ Res. 2011;108:555–565. doi: 10.1161/CIRCRESAHA.110.221911. doi:10.1161/CIRCRESAHA.110.221911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldhaber JI, Parker JM, Weiss JN. Mechanisms of excitation-contraction coupling failure during metabolic inhibition in guinea-pig ventricular myocytes. J Physiol(Lond) 1991;443:371–386. doi: 10.1113/jphysiol.1991.sp018838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldhaber JI, Xie LH, Duong T, Motter C, Khuu K, Weiss JN. Action potential duration restitution and alternans in rabbit ventricular myocytes: the key role of intracellular calcium cycling. Circ Res. 2005;96:459–466. doi: 10.1161/01.RES.0000156891.66893.83. doi:10.1161/01.RES.0000156891.66893.83. [DOI] [PubMed] [Google Scholar]
- 19.Sato D, Xie LH, Sovari AA, Tran DX, Morita N, Xie F, et al. Synchronization of chaotic early afterdepolarizations in the genesis of cardiac arrhythmias. Proc Natl Acad Sci USA. 2009;106:2983–2988. doi: 10.1073/pnas.0809148106. doi:10.1073/pnas.0809148106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie LH, Weiss JN. Arrhythmogenic consequences of intracellular calcium waves. Am J Physiol Heart Circ Physiol. 2009;297:H997–H1002. doi: 10.1152/ajpheart.00390.2009. doi:10.1152/ajpheart.00390.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mahajan A, Shiferaw Y, Sato D, Baher A, Olcese R, Xie LH, et al. A rabbit ventricular action potential model replicating cardiac dynamics at rapid heart rates. Biophys J. 2008;94:392–410. doi: 10.1529/biophysj.106.98160. doi:10.1529/biophysj.106.98160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luo CH, Rudy Y. A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ Res. 1994;74:1071–1096. doi: 10.1161/01.res.74.6.1071. [DOI] [PubMed] [Google Scholar]
- 23.Dong M, Sun X, Prinz AA, Wang HS. Effect of simulated Ito on guinea pig and canine ventricular action potential morphology. Am J Physiol Heart Circ Physiol. 2006;291:H631–H637. doi: 10.1152/ajpheart.00084.2006. doi:10.1152/ajpheart.00084.2006. [DOI] [PubMed] [Google Scholar]
- 24.Brouillette J, Clark RB, Giles WR, Fiset C. Functional properties of K+ currents in adult mouse ventricular myocytes. J Physiol. 2004;559:777–798. doi: 10.1113/jphysiol.2004.063446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo CH, Rudy Y. A model of the ventricular cardiac action potential. Depolarization, repolarization, and their interaction. Circ Res. 1991;68:1501–1526. doi: 10.1161/01.res.68.6.1501. [DOI] [PubMed] [Google Scholar]
- 26.Silva J, Rudy Y. Subunit interaction determines IKs participation in cardiac repolarization and repolarization reserve. Circulation. 2005;112:1384–1391. doi: 10.1161/CIRCULATIONAHA.105.543306. doi:10.1161/CIRCULATIONAHA.105.543306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bassani RA, Altamirano J, Puglisi JL, Bers DM. Action potential duration determines sarcoplasmic reticulum Ca2+ reloading in mammalian ventricular myocytes. J Physiol. 2004;559:593–609. doi: 10.1113/jphysiol.2004.067959. doi:10.1113/jphysiol.2004.067959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.January CT, Riddle JM. Early afterdepolarizations: mechanism of induction and block. A role for L-type Ca2+ current. Circ Res. 1989;64:977–990. doi: 10.1161/01.res.64.5.977. [DOI] [PubMed] [Google Scholar]
- 29.Tanskanen AJ, Greenstein JL, O'Rourke B, Winslow RL. The role of stochastic and modal gating of cardiac L-type Ca2+ channels on early after-depolarizations. Biophys J. 2005;88:85–95. doi: 10.1529/biophysj.104.051508. doi:10.1529/biophysj.104.051508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greenstein JL, Wu R, Po S, Tomaselli GF, Winslow RL. Role of the calcium-independent transient outward current Ito1 in shaping action potential morphology and duration. Circ Res. 2000;87:1026–1033. doi: 10.1161/01.res.87.11.1026. [DOI] [PubMed] [Google Scholar]
- 31.Roden DM. Taking the ‘idio’ out of ‘idiosyncratic’: predicting torsades de pointes. Pacing Clin Electrophysiol. 1998;21:1029–1034. doi: 10.1111/j.1540-8159.1998.tb00148.x. doi:10.1111/j.1540-8159.1998.tb00148.x. [DOI] [PubMed] [Google Scholar]
- 32.Nguyen TP, Xie Y, Garfinkel A, Qu Z, Weiss JN. Arrhythmogenic consequences of myofibroblast-myocyte coupling. Cardiovasc Res. 2012;93:242–251. doi: 10.1093/cvr/cvr292. doi:10.1093/cvr/cvr292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nerbonne JM. Molecular basis of functional voltage-gated K+ channel diversity in the mammalian myocardium. J Physiol. 2000;525(Pt 2)):285–298. doi: 10.1111/j.1469-7793.2000.t01-1-00285.x. doi:10.1111/j.1469-7793.2000.t01-1-00285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patel SP, Campbell DL. Transient outward potassium current, Ito, phenotypes in the mammalian left ventricle: underlying molecular, cellular and biophysical mechanisms. J Physiol. 2005;569:7–39. doi: 10.1113/jphysiol.2005.086223. doi:10.1113/jphysiol.2005.086223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu DW, Gintant GA, Antzelevitch C. Ionic bases for electrophysiological distinctions among epicardial, midmyocardial, and endocardial myocytes from the free wall of the canine left ventricle. Circ Res. 1993;72:671–687. doi: 10.1161/01.res.72.3.671. [DOI] [PubMed] [Google Scholar]
- 36.Han W, Wang Z, Nattel S. A comparison of transient outward currents in canine cardiac Purkinje cells and ventricular myocytes. Am J Physiol Heart Circ Physiol. 2000;279:H466–H474. doi: 10.1152/ajpheart.2000.279.2.H466. [DOI] [PubMed] [Google Scholar]
- 37.Han W, Zhang L, Schram G, Nattel S. Properties of potassium currents in Purkinje cells of failing human hearts. Am J Physiol Heart Circ Physiol. 2002;283:H2495–2503. doi: 10.1152/ajpheart.00389.2002. [DOI] [PubMed] [Google Scholar]
- 38.Jeck C, Pinto J, Boyden P. Transient outward currents in subendocardial Purkinje myocytes surviving in the infarcted heart. Circulation. 1995;92:465–473. doi: 10.1161/01.cir.92.3.465. [DOI] [PubMed] [Google Scholar]
- 39.Dun W, Boyden PA. The Purkinje cell; 2008 style. J Mol Cell Cardiol. 2008;45:617–624. doi: 10.1016/j.yjmcc.2008.08.001. doi:10.1016/j.yjmcc.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun X, Wang HS. Role of the transient outward current Ito in shaping canine ventricular action potential-a dynamic clamp study. J Physiol. 2005;564:411–419. doi: 10.1113/jphysiol.2004.077263. doi:10.1113/jphysiol.2004.077263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roeper J, Lorra C, Pongs O. Frequency-dependent inactivation of mammalian A-type K+ channel KV1.4 regulated by Ca2+/calmodulin-dependent protein kinase. J Neurosci. 1997;17:3379–3391. doi: 10.1523/JNEUROSCI.17-10-03379.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lakatta EG, Sollott SJ. The ‘heartbreak’ of older age. Mol Interv. 2002;2:431–446. doi: 10.1124/mi.2.7.431. doi:10.1124/mi.2.7.431. [DOI] [PubMed] [Google Scholar]
- 43.Slezak J, Tribulova N, Pristacova J, Uhrik B, Thomas T, Khaper N, et al. Hydrogen peroxide changes in ischemic and reperfused heart. Cytochemistry and biochemical and X-ray microanalysis. Am J Pathol. 1995;147:772–781. [PMC free article] [PubMed] [Google Scholar]
- 44.Dhalla NS, Duhamel TA. The paradoxes of reperfusion in the ischemic heart. Heart Metab. 2007;37:31–34. [Google Scholar]
- 45.Xing D, Chaudhary AK, Miller FJ, Jr, Martins JB. Free radical scavenger specifically prevents ischemic focal ventricular tachycardia. Heart Rhythm. 2009;6:530–536. doi: 10.1016/j.hrthm.2008.12.032. doi:10.1016/j.hrthm.2008.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tomaselli GF, Barth AS. Sudden cardio arrest: oxidative stress irritates the heart. Nat Med. 2010;16:648–649. doi: 10.1038/nm0610-648. doi:10.1038/nm0610-648. [DOI] [PubMed] [Google Scholar]
- 47.Morita N, Sovari AA, Xie Y, Fishbein MC, Mandel WJ, Garfinkel A, et al. Increased susceptibility of aged hearts to ventricular fibrillation during oxidative stress. Am J Physiol Heart Circ Physiol. 2009;297:H1594–1605. doi: 10.1152/ajpheart.00579.2009. doi:10.1152/ajpheart.00579.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hashambhoy YL, Greenstein JL, Winslow RL. Role of CaMKII in RyR leak, EC coupling and action potential duration: a computational model. J Mol Cell Cardiol. 2010;49:617–624. doi: 10.1016/j.yjmcc.2010.07.011. doi:10.1016/j.yjmcc.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao Z, Wen H, Fefelova N, Allen C, Baba A, Matsuda T, et al. Revisiting the ionic mechanisms of early afterdepolarizations in cardiomyocytes: predominant by Ca waves or Ca currents? Am J Physiol Heart Circ Physiol. 2012;302:H1636–1644. doi: 10.1152/ajpheart.00742.2011. doi:10.1152/ajpheart.00742.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.