

Abstract
Indole-3-carbinol (I3C), a phytochemical derived from cruciferous vegetables such as broccoli and Brussels sprouts, has potent anti-proliferative effects in human breast cancer cells and has been shown to decrease metastatic spread of tumors in experimental animals. Using chemotaxis and fluorescent-bead cell motility assays, we demonstrated that I3C significantly decreased the in vitro migration of MDA-MB-231 cells, a highly invasive breast cancer cell line. Immunofluorescence staining of the actin cytoskeleton revealed that concurrent with the loss of cell motility, I3C treatment significantly increased stress fiber formation. Furthermore, I3C induced the localization of the focal adhesion component vinculin and tyrosine-phosphorylated proteins to the cell periphery, which implicates an indole-dependent enhancement of focal adhesions within the outer boundary of the cells. Co-immunoprecipitation analysis of focal adhesion kinase demonstrated that I3C stimulated the dynamic formation of the focal adhesion protein complex without altering the total level of individual focal adhesion proteins. The RhoA-Rho kinase pathway is involved in stress fiber and focal adhesion formation, and I3C treatment stimulated Rho kinase enzymatic activity, and cofilin phosphorylation, which is a downstream target of Rho kinase signaling, but did not increase the level of active GTP-bound RhoA. Exposure of MDA-MB-231 cells to the Rho kinase inhibitor Y-27632, or expression of dominant negative RhoA ablated the I3C induced formation of stress fibers and of peripheral focal adhesions. Expression of constitutively active RhoA mimicked the I3C effects on both processes. Taken together, our data demonstrate that I3C induces stress fibers and peripheral focal adhesions in a Rho kinase-dependent manner that leads to an inhibition of motility in human breast cancer cells.
Keywords: Indole-3-carbinol, breast cancer, migration, focal adhesions, RhoA, Rho kinase
Introduction
The consumption of Brassica (cruciferous) vegetables, such as broccoli, cabbage, and Brussels sprouts, is directly associated with decreased risk of reproductive tissue cancers in humans(1–4) and reduction of mammary tumorigenesis in rodents(5). One of the promising bioactive phytochemicals that is derived from glycobrassicin in Brassica vegetables is Indole-3-carbinol (I3C), which exhibits potent anti-carcinogenic properties in human reproductive cancers such as those of cervical, endometrial, prostate and breast tissues (6–8). I3C tested positive as a chemopreventative agent in a panel of short-term bioassays relevant to carcinogen-induced DNA damage, tumor initiation and promotion, and oxidative stress(9), and has been shown to prevent spontaneous and carcinogen-induced mammary tumors in rodent model systems(10–12). In addition to these cancer preventative effects, there is compelling evidence that I3C has a direct anti-proliferative response in cultured human reproductive cancer cell lines(6–8, 13–23), including breast cancer cells. Furthermore, oral I3C administration to breast cancer patients alters estrogen metabolism, leading to a reduction in breast tumor growth without detectable side effects (24), suggesting that this natural indole could potentially be utilized in anti-cancer therapeutic strategies that target indole-responsive cancers.
Cumulative evidence by us and other groups demonstrates that I3C mediates its anticancer response through several distinct cellular pathways, which can target transcriptional and cell signaling events(6, 7, 13, 16, 20) We have documented that I3C induces a G1 cell cycle arrest of both estrogen sensitive and insensitive human breast cancer cells concomitant with the inhibited expression or activity of CDK6 and CDK2, respectively, and with a marked decrease in endogenous retinoblastoma (Rb) phosphorylation (13–16). I3C down-regulates CDK6 transcription by disrupting Sp1 interactions with the CDK6 promoter (16), and controls CDK2 enzymatic activity by altering cyclin E protein processing (20). In estrogen sensitive breast cancer cells, I3C suppresses estrogen responsiveness (25, 26), down-regulates expression of estrogen receptor-alpha (25), as well as synergizes with the anti-proliferative effects of tamoxifen, an anti-estrogen widely used in breast cancer therapies (15). In nontumorigenic human mammary epithelial cells, I3C can induce the ATM signaling pathway independent of DNA damage to stabilize an active p53 tumor suppressor protein (27).
In addition to its potent anti-proliferative effects, I3C has been shown to effectively suppress in vivo breast cancer cell metastasis (28, 29), as well as inhibit the formation of lung surface metastatic nodules when poorly invasive MCF-7 or highly invasive MDA-MB-468 cells were injected intravenously into mice (30). In vitro, I3C significantly suppressed cellular adhesion, migration, and invasion of T-47D, MCF-7, and MDA-MB-468 human breast cancer cell lines (28, 30), with the concurrent increased expression of the intercellular adhesion molecules E-cadherin and beta-catenin (30). These studies suggest that I3C can act through a distinct set of cellular pathways to induce its anti-metastatic effects in human breast cancer cells, although the mechanism of this indole response has not been fully elucidated.
Directional cell migration is an integral part of cancer cell invasion during metastasis, and involves changes in the cytoskeleton and cell adhesion (31). Migration of cells through an extracellular matrix is a multistep process, and begins with the extension of lamellipodia, cell surface protrusions comprised of actin filaments, which are anchored to the underlying substratum by small integrin-dependent focal adhesions. The polymerization of actin pushes against the plasma membrane and provides the force for forward movement. Within the cell body, actin stress fibers generate contractile forces by pulling against focal adhesions to induce retraction of the rear cell membrane. The bundling of actin filaments into stress fibers clusters and activates integrins, leading to new focal adhesion formation (32, 33). The organization of these multimolecular adhesion complexes is complicated, and includes cytoskeletal proteins such as vinculin, talin and actinin, and several non-receptor protein tyrosine kinases including members of the Src family and FAK (focal adhesion kinase) (34, 35). The targeting of FAK to focal adhesions leads to its autophosphorylation, and the subsequent tyrosine phosphorylation of additional focal complex-associated proteins including adaptor proteins such as p130 Cas, and paxillin (36). FAK has a central role in the dynamic regulation of cell adhesion structures, suggested by the observation that FAK deficient cells display enhanced focal adhesions and impaired migration (37, 38).
The assembly and disassembly of focal adhesions during cell migration is modulated by integrin cell adhesion receptors, and the family of Rho-GTPases (39–41). Rho-GTPases are regulators of actin dynamics and cell-substratum adhesion in migratory cells, and thus play a critical role in tumor metastasis (42, 43). The Rho-GTPases alternate between an active, GTP-bound state, and an inactive, GDP-bound state (44). Rho and Rac, two subgroups of the Rho-GTPases, may have opposing actions on actin polymerization and adhesion dynamics. RhoA induces the formation of stress fibers and mature focal adhesions through activation of its downstream effectors mDia and the Rho kinases (ROCK1 and ROCK2) (45, 46). Rac1 induces lamellipodial extensions, and transient focal complexes that form at the leading edge of a migratory cell, and Rac1 activity has been correlated with increased migration and invasion of reproductive cancer cells (47, 48). Endogenous over-expression of one or both of RhoA and Rac1 is a frequent event in invasive breast cancers (49), and activation of RhoA has been shown to inhibit cell migration through the formation of highly stable focal adhesions that impede cellular motility (50–52). Thus, modulation of Rho-GTPase effector pathways may be an effective therapeutic strategy to inhibit cellular motility, and may represent a potential target pathway for anti-cancer phytochemicals such as I3C. Using the highly metastatic and invasive human breast cancer cell line MDA-MB-231, which is sensitive to anti-proliferative effects of I3C (13, 14) and expresses RhoA (53), we show for the first time that I3C activation of Rho kinase activity is necessary for the indole-regulated formation of stress fibers and focal adhesions that lead to decreased motility of MDA-MB-231 breast cancer cells. Our study implicates a potential mechanism by which I3C can limit the metastastic capability of human breast cancer cells, suggesting its potential therapeutic value in treatment of indole-sensitive tumors.
Material and Methods
Cell lines, cell culture conditions, and plasmids
Breast adenocarcinoma cell line MDA-MB-231 was obtained from the American Type Culture Collection (Manassas, VA). MDA-MB-231 cells were grown in Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 10% fetal bovine serum, 50 units/ml penicillin/streptomycin, and 2 mM L-glutamine (Cambrex Bio Science, Walkersville, MD). Cells were treated for the indicated time points in complete medium with DMSO (Aldrich, Milwaukee, WI), I3C or Tryptophol (LKT Laboratory Inc., St. Paul, Minnesota) dissolved 1000X in DMSO. The medium was changed every 24 h for the duration of each experiment. For cell treatments, I3C was diluted into medium so that the final concentration of DMSO was below 0.5%, a level that has no effect on tumor cells. The ROCK inhibitor Y-27632 (Santa Cruz Biotechnology, Santa Cruz, CA) was used at a final concentration of 10 µM.
The plasmids expressing constitutively active or dominant negative RhoA (pEXV-Myc-RhoA.V14 and pEXV-Myc-RhoA.DN19) and empty vector (pEXV) were a kind gift from Dr. Marc Symons (Onyx Pharmaceuticals, Richmond, CA). To generate stable cell lines, MDA-MB-231 cells were transfected with pEXV, RhoA.V14, or RhoA.DN19 and selected for two weeks using neomycin analog G418. After selection, the clones were selected, expanded, and tested for expression of Myc-tagged RhoA by Western blotting using anti-Myc antibodies (Invitrogen, South San Francisco, CA).
Migration assays
Migration assays were based on a modified Boyden Chamber assay (54, 55). To focus the experiment on motility regulation and minimize the growth inhibitory effects of I3C, the migration assays were performed for duration of no greater than 24h. MDA-MB-231 cells were incubated for 24 h with DMSO, 200 uM Tryptophol, 100 uM I3C, or 200 uM I3C prior to the beginning of the experiment. After 24h, cells were detached with non-enzymatic cell dissociation solution (Sigma Chemical, St. Louis, MO), and resuspended in IMDM with 0.2 mg/ml bovine serum albumin (BSA) (Sigma), 1.0 % FBS, and drug treatment as before. Cells (50,000 cells/well) were seeded in triplicate in the upper chamber of a BD BioCoat Cell Culture Insert (8 mm diameter pore; BD Biosciences, Bedford, MA). The lower chamber contained IMDM with 0.2 mg/ml BSA, and 10% FBS as the chemoattractant. After 24 h, the unmigrated cells in the upper chamber were gently scraped off the filter, and the filters were fixed in 4% formalin. For each well, four fields of migrated cells were photographed and counted using ImageJ software (NIH, Bethesda, MD).
The Fluorescence Cell Motility HitKit was purchased from Cellomics (Pittsburgh, PA). The assays were performed according to manufacturer’s instructions. MDA-MB-231 cells were incubated for 24 h in medium containing with DMSO, 200 µM Tryptophol, 100 µM I3C, or 200 µM I3C prior to the beginning of the experiment. After 16 h, cells were detached with Trypsin-EDTA, washed three times in serum-free IMDM, and then resuspended at 1×104 cells/ml in serum-free IMDM. 50 µl of cell resuspension was gently added per well of a prepared 96-well dish containing a lawn of microscopic blue fluorescent microspheres. Finally, either 50 µl of serum-free IMDM (no serum sample) or 50 µl of IMDM with serum (5% serum final) plus the appropriate treatment was added to the cells. After 24 h, the cells were fixed, permeabilized, and stained with rhodamine-conjugated phalloidin. The cells were visualized using a Q3DM Eidaq 100 high throughput microscope equipped with an ORCA-ER camera. Images were acquired with Cytoshop v2.1 software (Beckman Coulter, Inc., Fullerton, CA), and quantified using Image-Pro Plus (MediaCybernetics, Silver Spring, MD).
Immunofluorescence microscopy
Treated MDA-MB-231 cells, plated on Lab-Tek Permanox slides (Nalge Nunc International, Naperville, IL), were fixed with 3.7% formaldehyde in PBS for 15 min, and permeabilized with 0.5% Triton X-100 in PBS for 10 min. After blocking for 1 h, the cells were incubated with anti-vinculin (Sigma), or anti-phospho-tyrosine (Cell Signaling Technology, Beverly, MA) antibodies for 1 h, followed by incubation for 1 h with Alexa Fluor 488 goat anti-mouse IgG and Texas-red phalloidin (Molecular Probes, Eugene, OR). Coverslips were mounted with Vectashield mounting medium (Vector Laboratories Inc., Burlingame, CA) containing nuclear stain DAPI, and then visualized using a Zeiss Axiophot 381 epifluorescence microscope, with a 3.3 MPix Qimaging MicroPublisher CCD color digital camera.
Immunoprecipitation
MDA-MB-231 cells were washed with ice-cold PBS, harvested in co-immunoprecipitation (co-IP) buffer (150 mM NaCl, 0.1% Triton X-100, 20 mM Tris-HCl, pH 7.5) containing protease inhibitors, and the extracts normalized as for Western blot analysis. 500 mg of protein extract was diluted up to 1 ml in co-IP buffer. Extracts were pre-cleared for 30 min at 4°C with 30 µl of a 1:1 slurry of protein G-beads (Pharmacia Biotech, Sweden). Concurrently, 30 µl of protein G beads and 1 µl of antibodies to FAK (Sigma), were incubated in PBS on a rotating platform for 2 h at 4°C. The protein G-FAK IgG beads were added to each set of precleared extracts, and incubated on a rotating platform for 1.5 h at 4°C. As a negative control, protein G-beads with control IgG were incubated with precleared extract. The beads were washed three times with co-IP buffer, and once with PBS. The beads were then boiled for 3 min in 2X protein loading buffer.
Western blot analysis
Treated MDA-MB-231 cells were harvested in RIPA buffer, and the lysates processed for Western blotting as described previously, with several modifications (27). Blots were incubated with primary antibodies to FAK (Sigma), p130 Cas, paxillin, phospho-MYPT1 (Thr696) (Upstate), phospho-cofilin (Ser3), cofilin, phospho-tyrosine (Cell Signaling Technology), hsp90 (Affinity BioReagents, Golden CO), ROCK1, ROCK2, MYPT1 (BD Biosciences), RhoA, or actin (Santa Cruz Biotechnology, Santa Cruz, CA). Immunoblotting with hsp90 antibodies was used as a loading control.
Rho Kinase Assay
The Rho kinase assay was adapted with some modifications from the protocol of McBeath and colleagues for ROCK2 (56). Treated MDA-MB-231 cells were lysed in immunoprecipitation lysis (IP) buffer (10 mM Tris-HCl at pH 7.5, 1% Triton X-100, 0.5% NP-40, 150mM NaCl, 2mM CaCl2, 0.1 mM sodium orthovanadate, 10 µg/ml aprotinin/leupeptin, and 1 mM PMSF, Sigma), and centrifuged at 14,000 rpm for 4 min. Protein samples were normalized using the Bradford Assay (BioRad, Hercules, CA). Equal aliquots of lysates were incubated with 5 µl of anti-ROCK1, anti-ROCK2 (Santa Cruz Biotechnology), or goat IgG (Sigma), for 30 min on a nutator at 4°C. 100 µl of washed protein G sepharose beads (Amersham Biosciences, Piscataway, NJ) diluted in IP buffer (1:1 slurry) was added to lysate samples, and samples were nutated at 4°C for 60 min. Beads were centrifuged at 2000 rpm for 2 min, and washed four times with IP buffer, then resuspended in kinase assay buffer (50mM HEPES [pH 7.4], 150 mM NaCl, 1mM MgCl2, 1 mM MnCl2, 10mM NaF, 1mM sodium orthovanandate, 5% glycerol, 1% NP-40, 1 mM dithiothreitol, and 1 mM PMSF, Sigma). The ROCK kinase assay was performed using 10 µM ATP (Sigma), and recombinant MYPT1 substrate (Upstate Technologies), and incubated with the bead-kinase assay buffer slurry in a reaction volume of 50 µl at 37°C. To the appropriate samples, 10 µM of the Rho kinase inhibitor Y-27632 (Santa Cruz Biotechnology) was added prior to incubation at 37°C. The reaction was stopped by the addition of SDS-PAGE buffer, and boiling the samples for 10 min at 100°C. Kinase assay activity was detected by SDS-PAGE, followed by Western blotting using anti-phospho-MYPT1 (Upstate Technologies) and anti-MYPT antibodies (BD Biosciences). Data was quantified using ImageJ software (NIH, Bethesda, MD).
RhoA-GTP binding assays
The Rho Activation Assay Biochem Kit (Cytoskeleton, Denver, CO) was utilized to isolate GTP-Rho from cell lysates. Treated MDA-MB-231 cells were harvested in 1X ice-cold lysis buffer, and centrifuged at 8000 rpm for 5 min. The supernatant was immediately added to 45 µg of Rhotekin-RBD beads. 1X protease inhibitor cocktail was added, and the mixture was rotated in the cold for 1 h. The extract and Rhotekin-RBD beads were centrifuged for 3 min at 5000 g, and the supernatant removed. Pelleted beads were subjected to two washes, 1X lysis buffer followed by 1X wash buffer. Beads were resuspended in 10 µl Laemmli dye. The amount of activated Rho is determined by Western blotting of pull-downs using a RhoA specific antibody (Santa Cruz Biotechnology). For the positive control, GTP loading on the cell lysate was performed by adding 1/10th the volume of loading buffer, and 1/100th the volume of GTPγS (200 µM final concentration), and incubation at 30°C for 15 min. The reaction was stopped by transfering the tube to 4°C and adding 1/10th volume of STOP buffer. 45 µg Rhotekin-RBD beads were added, and the protocol continued as above.
Results
I3C decreases motility of MDA-MB-231 breast cancer cells
The effects of I3C on motility of the highly invasive MDA-MB-231 human breast cancer cell line were initially examined using a modified Boyden Chamber assay that monitors cell migration through a filter to the chemoattractant 10% fetal bovine serum (54, 55). Cells were treated with either 100 µM I3C, 200 µM I3C, 200 µM Tryptophol, which is an inactive indole (14, 15) closely structurally related to I3C that contains an ethanol group instead of a methanol group in the 3-carbon position of the indole ring, or with the DMSO vehicle control. Migratory potential was monitored after 24 hours because at this time point MDA-MB-231 cells do not display a cell cycle arrest with I3C. As shown in Figure 1, I3C strongly inhibited chemotactic cell migration. Treatment with 100 µM or 200 µM I3C decreased the number of cells that migrated through the filters by approximately 3-fold and 6-fold, respectively, compared to DMSO or tryptophol treated cells.
Figure 1.
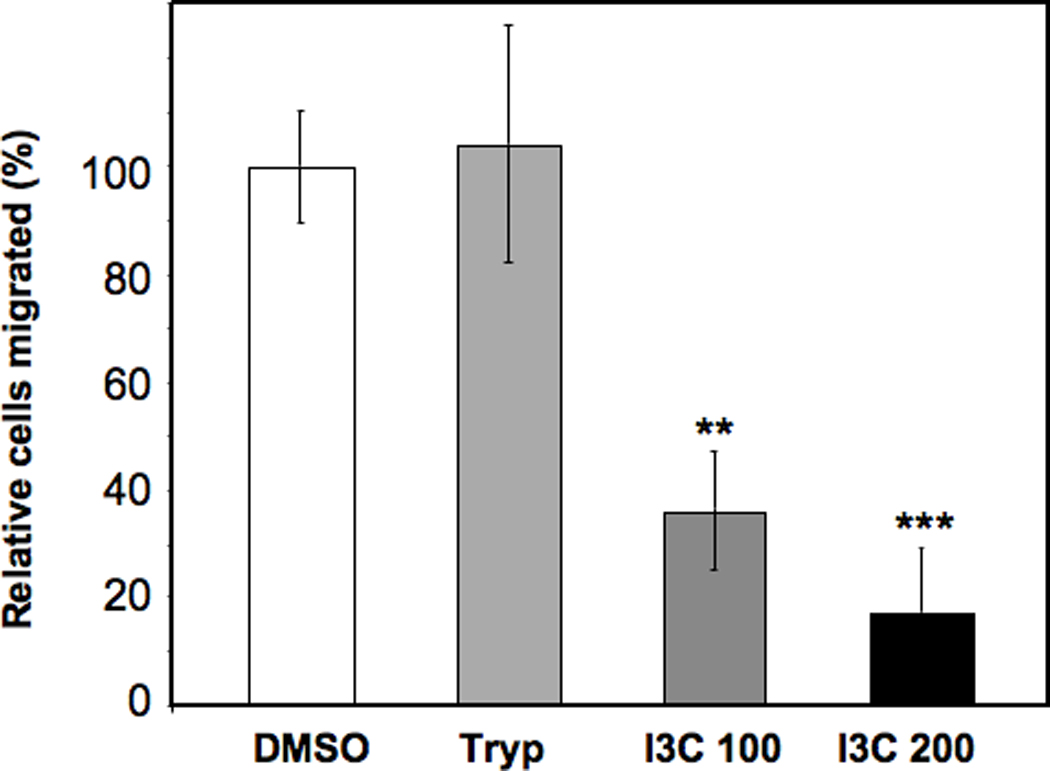
I3C inhibits MDA-MB-231 breast cancer cell migration. Growing MDA-MB-231 cells were treated for 24 h with DMSO, 200 µM Tryptophol (Tryp), 100 µM I3C (I3C 100), or 200 µM I3C, and then plated onto the upper wells of a modified Transwell Boyden Chamber assay chambers and allowed to migrate towards chemoattractant 10% FBS for 24 h. Migrated cells were photographed with a digital camera, and the number of cells that migrated through the upper chamber was quantified relative to cells treated with the DMSO vehicle control. Results are means +/− SD of triplicate samples from three independent experiments (Statistical significance was evaluated by calculating p values using Students’ t test: **P < 0.01, ***P < 0.001 compared to DMSO cells).
The ability of I3C to down-regulate MDA-MD-231 cell migration was further analyzed using a motility assay that does not depend on cell adhesion by measuring the size of tracks made by cells as they migrate through a layer of fluorescent beads. As the cells move, they leave cleared phagokinetic paths behind them, the area of which can be quantified. Figure 2 shows representative images of cells prepared with the Cell Motility HitKit, and the corresponding quantification of areas of the cleared paths from migrating cells tested under the indicated conditions. Treatment of breast cancer cells with either 100 µM or 200 µM I3C decreased the motility path area by approximately 3-fold compared to the DMSO vehicle control or tryptophol treated cells. Strikingly, I3C treatment reduced the level of migration almost to that observed in serum–starved cells, which demonstrates the strong effect of this indole on human breast cancer cell mobility.
Figure 2.
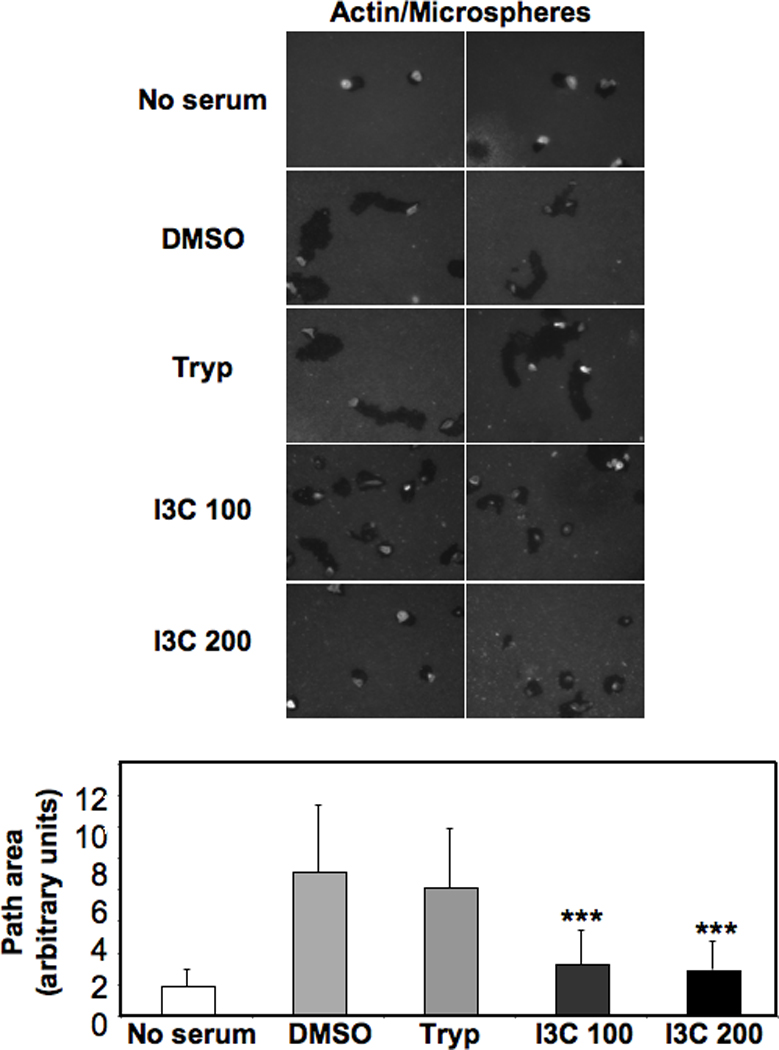
I3C inhibits motility of MDA-MB-231 cells. MDA-MB-231 cells were treated for 24 h with DMSO, 200 µM Tryptophol (Tryp), 100 µM I3C (I3C 100), or 200 µM I3C, and assayed for motility after 16 h by measuring the size of tracks made by cells as they migrate through a layer of fluorescent beads after staining with rhodamine-conjugated phalloidin. For each treatment, representative images from one experiment are shown (n=3). Images are an overlay of the blue fluorescent beads with rhodamine-phalloidin. The path area of cells from each treatment group was quantified: no serum, DMSO, 200 µM Tryptophol (Tryp), 100 µM I3C (I3C 100), and 200 µM I3C (I3C 200) (n = 33, 54, 89, 120, and 76, respectively). Graphed results are means +/− SD from two independent experiments (***P < 0.001 compared to DMSO cells).
I3C induces reorganization of the actin cytoskeleton and formation of stress fibers and focal adhesions
Cell motility is controlled by the organization of the cytoskeleton, whose major components include actin filaments and a network of microtubules (31). To assess potential I3C-regulated changes in specific cytoskeletal components, the localization of F-actin, and distribution of vinculin, a structural component of focal adhesions, were examined by immunofluorescence after 48 hours of I3C treatment compared to DMSO vehicle control and tryptophol treated cells. This time point represents the duration of I3C treatment needed to observe the maximal effects of I3C in the MDA-MB-231 cells. As shown in Figure 3, in DMSO- or tryptophol-treated cells, there was diffuse staining of actin, with few stress fibers and prominent lamellipodia at the leading edge of the migrating cells. In contrast, I3C treatment induced a significant increase in the number of centrally located stress fibers and caused a decrease in detectable leading edge protrusions. The percentage of cells with visible stress fibers increased from 15% or 18% in control cells to 30% or 55% in cells treated with either 100 µM or 200 µM I3C, respectively. I3C treatment of MDA-MB-231 cells did not alter microtubule distribution (data not shown), indicating that I3C is not a general disruptor of the cytoskeleton. The vinculin staining under each tested condition revealed that I3C-treated cells showed an enhancement in the size and number of focal adhesions around the cell periphery, where stress fibers terminate (Figure 3, vinculin panels). The visible difference in the distribution of vinculin in the I3C-treated cells compared to tryptophol or DMSO treated cells was not a result of enhanced protein expression (data not shown).
Figure 3.
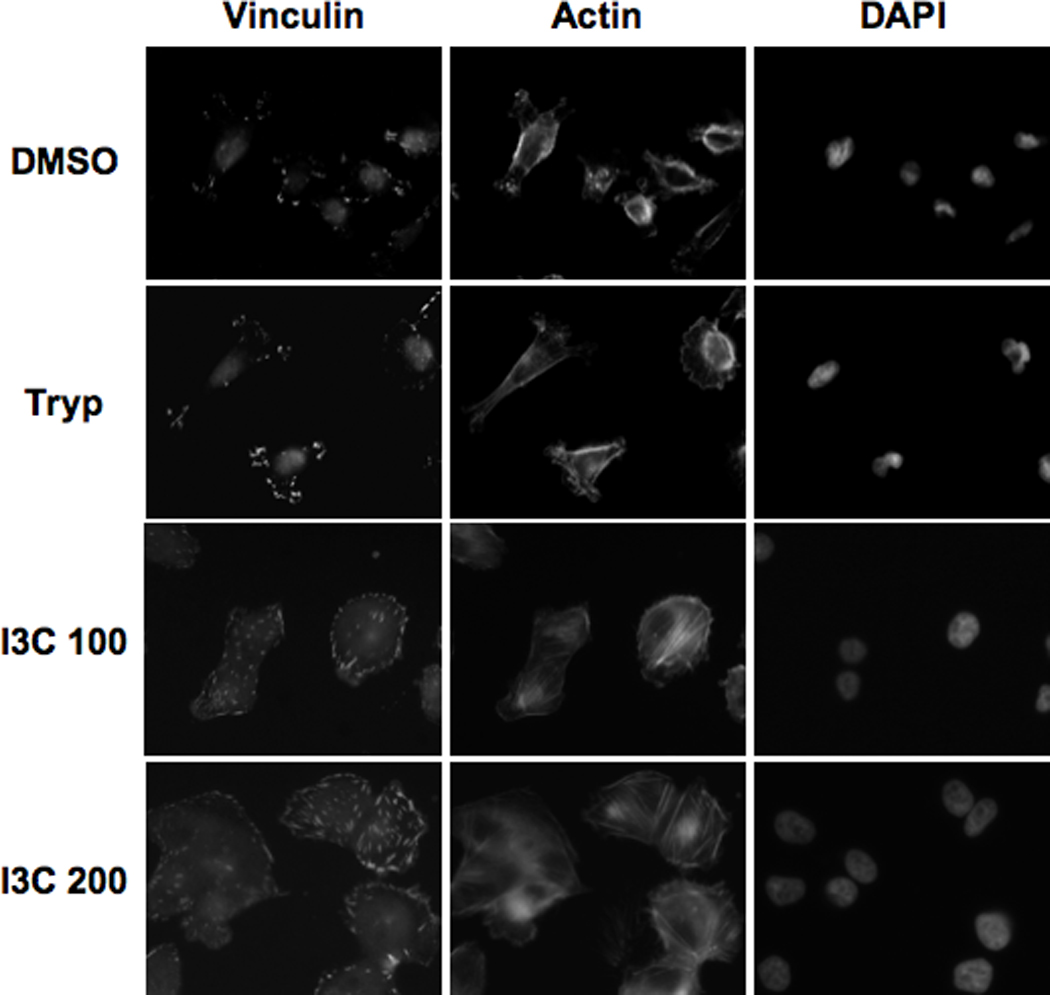
I3C increases formation of actin stress fibers. MDA-MB-231 cells were treated for 48 h with DMSO, 200 µM Tryptophol (Tryp), 100 µM I3C (I3C 100), or 200 µM I3C (I3C 200). The localization of vinculin and actin was examined by indirect immunofluorescence using anti-vinculin or Texas-red phalloidin, or nuclei stained by DAPI as described in the materials and methods section. The micrographs show representative cells treated under each condition.
One potential cellular consequence of the I3C stimulation in stress fiber formation is an enhanced focal adhesion complex formation. Key focal adhesion proteins in the focal adhesion complex include FAK, p130 Cas and Paxillin. Therefore, the effects of I3C on focal adhesion protein expression and protein complex formation were examined in cells treated for 48 hours with 200 µM I3C, 200 µM tryptophol or with DMSO. Western blot analysis showed that I3C had no effect on the total cellular levels of the focal adhesion proteins (Fig 4, top left panels). Focal complex formation was determined by immunoprecipitation with FAK antibodies followed by western blotting of the co-precipitating PAX and Cas as well as for FAK. Compared to the DMSO vehicle control and tryptophol treated cells, I3C significantly increased the level of PAX and Cas that co-precipitates with FAK (Fig 4, top right panels) showing that this indole stimulates the dynamics of focal adhesion complex assembly under conditions in which there is an increase in stress fiber formation. Precipitation and blotting with the same antibody as for the immunoprecipitation confirmed that the cellular protein level of FAK remained unchanged.
Figure 4.
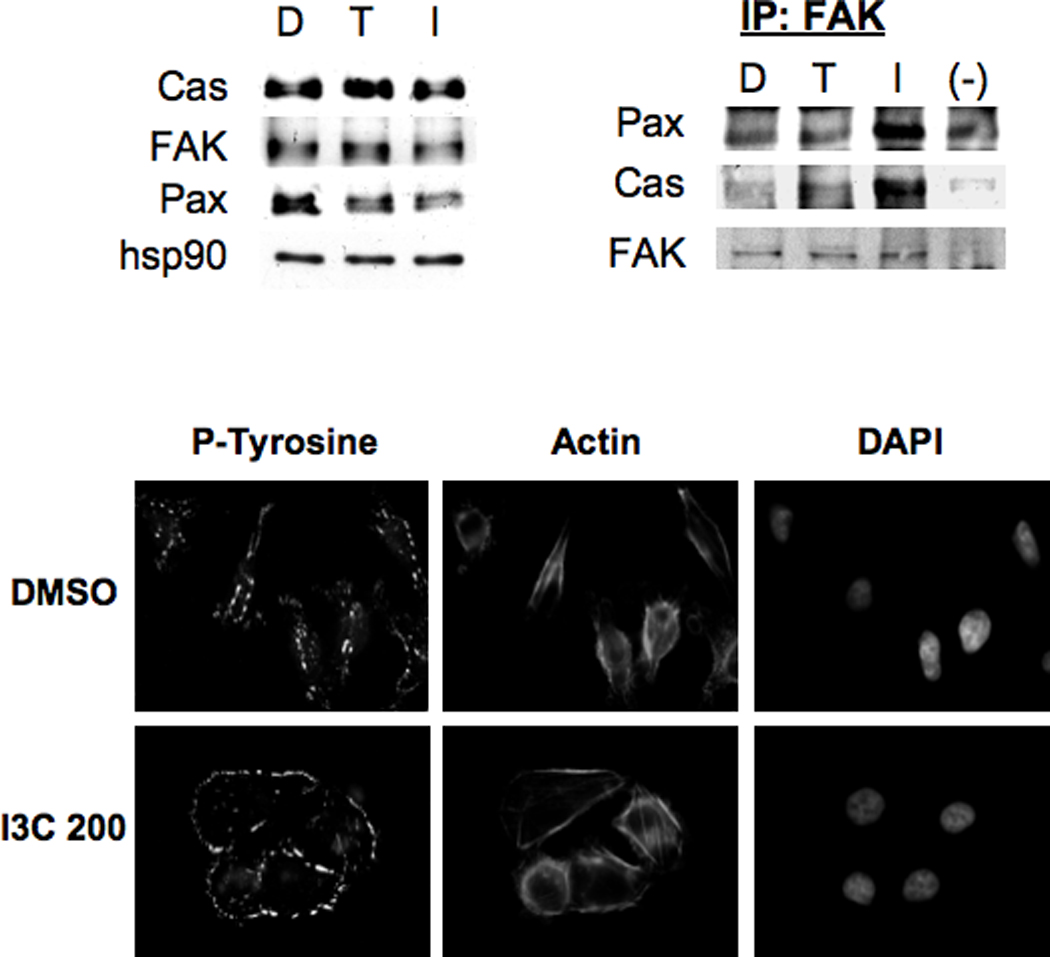
I3C induces formation of focal adhesion complexes. MDA-MB-231 cells were treated for 48 h with DMSO (D), 200 µM Tryptophol (T), or 200 µM I3C (I). Upper Left Panel: Cells were harvested, and extracts were subjected to SDS-PAGE and Western blotting with p130 Cas, FAK, paxillin (Pax), or hsp90 antibodies (loading control). Representative data from one experiment are shown (n=3). Upper Right Panel: Approximately 500 µg of cell extracts from treated cells were subjected to immunoprecipitation with FAK, or control (−) antibodies, and the immune complexes resolved by SDS-PAGE and Western blotting for Pax, Cas, or FAK. Lower Panels: Immunofluorescence was performed using phospho-tyrosine or Texas-red phalloidin for actin staining, or nuclear DNA stained with DAPI as described in the Materials and Methods. Representative cells from one experiment are shown (n=3).
Tyrosine phosphorylation of FAK, p130 Cas, paxillin, and other focal adhesion components regulate the signaling events that occur at focal adhesions (33). To examine tyrosine phosphorylation at focal adhesions, immunofluorescence microscopy was performed using an antibody specific for tyrosine-phosphorylated residues, and compared to actin staining. In DMSO-treated cells, anti-phosphotyrosine staining was concentrated mostly at the leading and trailing cell edges, with some modest staining detected around the cell boundary (Figure 4, fluorescence micrographs). I3C treatment caused the staining of tyrosine-phosphorylated proteins to greatly increase around the cell periphery similar to that observed with vinculin staining (see Fig. 3), which is a characteristic feature of focal adhesion formation. A parallel actin staining shows the I3C-induced stress fiber formation under conditions of this assay.
I3C induces kinase activities of ROCK1 and ROCK2 without activating RhoA
RhoA signaling has been shown to increase stress fiber-dependent adhesions, and negatively influence cell migration (57), suggesting that I3C may be regulating this signaling pathway to mediate its effects on cell migration and stress fiber formation. The Rho kinases ROCK1 and ROCK2 are key RhoA effectors that have multiple substrates including myosin phosphatase (MYPT1), which increases actomyosin contractility, and LIM kinase, which phosphorylates cofilin to abolish its ability to bind and depolymerize actin (42). Western blot analysis of cells treated for 48 hours with 200 µM I3C, 200 µM tryptophol or DMSO vehicle control treated cells revealed no I3C-dependent differences in expression of RhoA, ROCK1 or ROCK2 (Fig 5, top panel). Examination of Rho A downstream effectors revealed that I3C treatment induces production of phosphorylated cofilin but not of myosin phosphatase (MYPT1), suggesting that components of the RhoA signaling pathways are under differential indole control.
Figure 5.
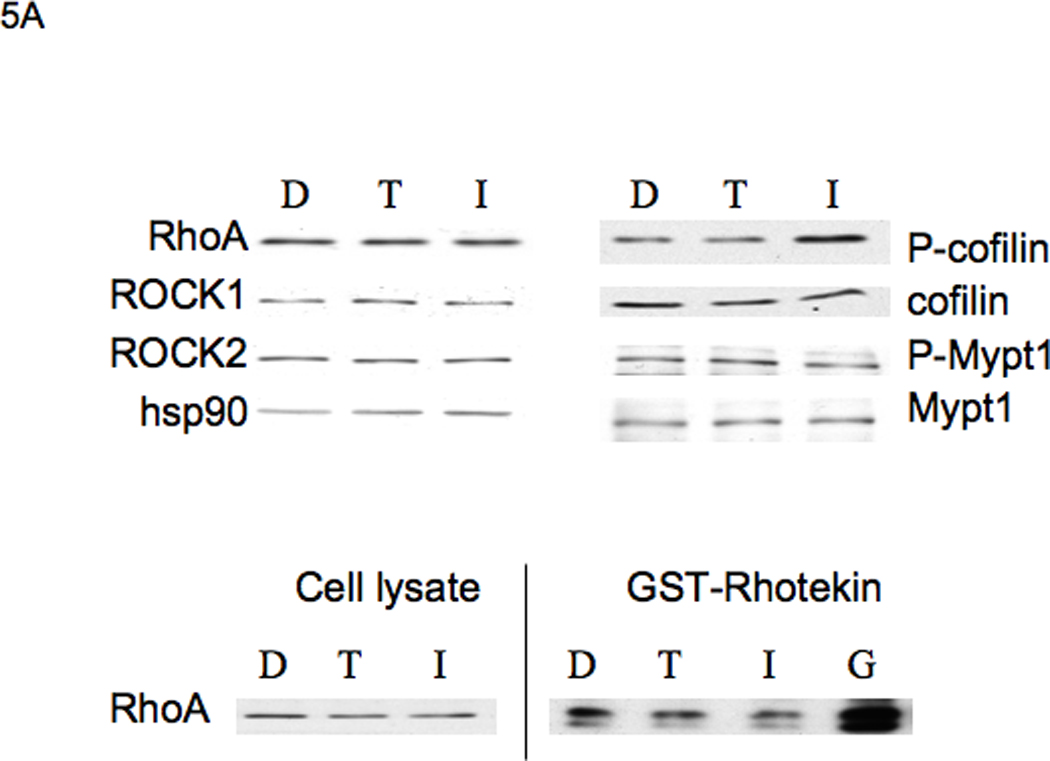
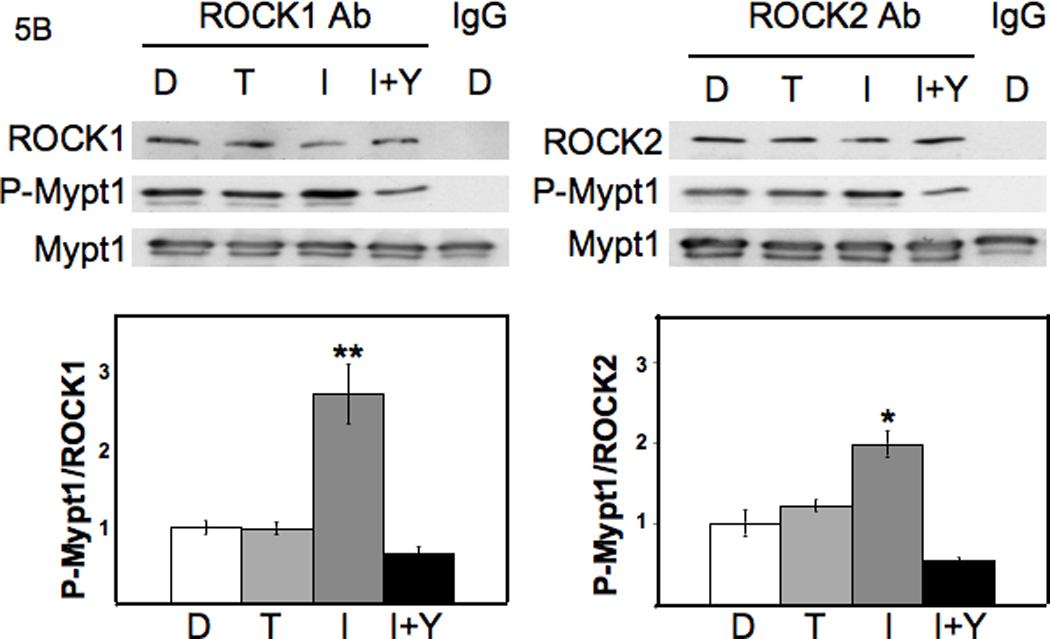
I3C induces ROCK1 and ROCK2 kinase activity, but does not activate RhoA. A: Upper Panels: I3C has no effect on RhoA activity: MDA-MB-231 cells were treated for 48 h with DMSO (D), 200 µM Tryptophol (T), or 200 µM I3C (I), and extracts of harvested cells were subjected to SDS-PAGE and Western blotting with antibodies to components of the RhoA signaling pathway, or hsp90 (loading control). Representative data from one experiment are shown (n=3). Lower Panels: 800 µg of cell extracts from treated cells were subjected to Rho activation assays using Rhotekin affinity beads. As a positive control, a non-hydrolysable GTP analog was incubated with one set of extracts before pull-down (G). Cell lysates (as a control for total RhoA) and Rho-GTP from the pull-down assays were subjected to SDS-PAGE and Western blotting with RhoA antibodies. Representative data from one experiment are shown (n=3). B: Upper Panels: I3C induces ROCK1 and ROCK2 enzymatic activity. Cell lysates were incubated with ROCK1 or ROCK2 polyclonal antibodies bound to Protein G-sepharose beads. Immunoprecipitates were subjected to in vitro kinase assays using MYPT1 as a substrate, and Y-27632 Rho kinase inhibitor (Y) as a control. Lower Panels: The kinase assay mixtures were analyzed by SDS-PAGE, followed by Western blotting with ROCK1, ROCK2, P-MYPT1, or MYPT1 antibodies. The individual bands were quantified using image J software. Graphed results are means +/− SD from two independent experiments (*P < 0.05, **P < 0.01, compared to DMSO cells).
To determine if I3C regulates the activation of the RhoA GTPase, an assay was performed for Rho activation based on specific binding of GTP-bound Rho to the Rho-binding domain of Rhotekin. MDA-MB-231 cells were treated for 48 hours with 200 µM I3C, 200 µM tryptophol, or DMSO and total cell lysates were incubated with Rhotekin affinity beads, to pull down endogenous active, GTP-bound RhoA protein. Western blotting using RhoA antibodies showed that extracts from I3C-treated cells did not have an appreciable difference in levels of RhoA-GTP compared to DMSO- or Tryptophol-treated cells (Figure 5A, lower panels). To show the specificity of the assay, prior to affinity bead pull-down, extracts from I3C-treated cells were incubated with a non-hydrolysable GTP analog to convert RhoA-GDP into a stable form of RhoA-GTP. Western blot results showed a significant level of RhoA signal compared to the direct pull-down assay (Fig 5A, lower right panel, lane G).
Because I3C upregulates the level of phosphorylated cofilin, but does not alter RhoA activation, we tested the effects of I3C treatment on the activities of the two Rho kinases, ROCK1 and ROCK2. MDA-MB-231 cells were treated with 200 µM I3C, 200 µM Tryptophol or the DMSO vehicle control for 48 hours, and then total cell lysates were immunoprecipitated with ROCK1, ROCK2, or control IgG antibodies. In vitro activity of both kinases was determined using MYPT1 as a substrate, which is efficiently phosphorylated in vitro by ROCK1 and ROCK2 on Thr696 (58, 59). For the in vitro kinase assay, immunoprecipitates were incubated with MYPT1 and ATP in the presence or absence of 10 µM of the Rho kinase inhibitor Y-27632. Phosphorylation of MYPT1 was detected by Western blotting of the reactions using antiphospho-MYPT1 antibodies. The kinase specific enzymatic activities were quantified by determining the ratio of phospho-Mypt1 formed in the reactions to the level of either ROCK1 or ROCK2 protein in each reaction (Fig 5B, bar graphs). As shown in Figure 5B, the immunoprecipitated ROCK1 and ROCK2 protein from I3C-treated cells displayed two-to three-fold greater kinase activity compared to cells treated with either tryptophol or DMSO. The phosphorylation of MYPT1 was reduced to below control levels when the Rho kinase inhibitor Y-27632, which inhibits the activities of both ROCK1 and ROCK2 (60, 61), was added to the in vitro kinase reaction (Figure 5B), demonstrating the specificity of the assay. Also, when control IgG was used for the immunoprecipitations, no Rho kinase protein or kinase activities were detected in the assay.
Inhibition of Rho kinase activity reverses the I3C-mediated formation of stress fibers and focal adhesions
To initially determine if the RhoA signaling pathway is required for the cytoskeletal changes observed with I3C treatment, the effects of the Y-27632 Rho kinase inhibitor were examined on the I3C-mediated stress fiber formation. MDA-MB-231 cells were treated with or without 200 µM I3C for 48 h, followed by co-treatment with or without Y-27632 for 2 hours, then the cytoskeletal organization analyzed by indirect immunofluorescence using antibodies to vinculin and phalloidin staining for actin. As shown in Figure 6, exposure of cells to the Y-27632 Rho kinase inhibitor disrupted the formation of thick actin stress fibers induced by I3C (Figure 6, insets) and caused vinculin-stained focal adhesions to diminish in staining intensity to levels resembling cells not treated with I3C. Treatment with Y-27632 alone altered the actin cytoskeleton of MDA-MB-231 cells, as would be expected due to the inhibition of ROCK activity.
Figure 6.
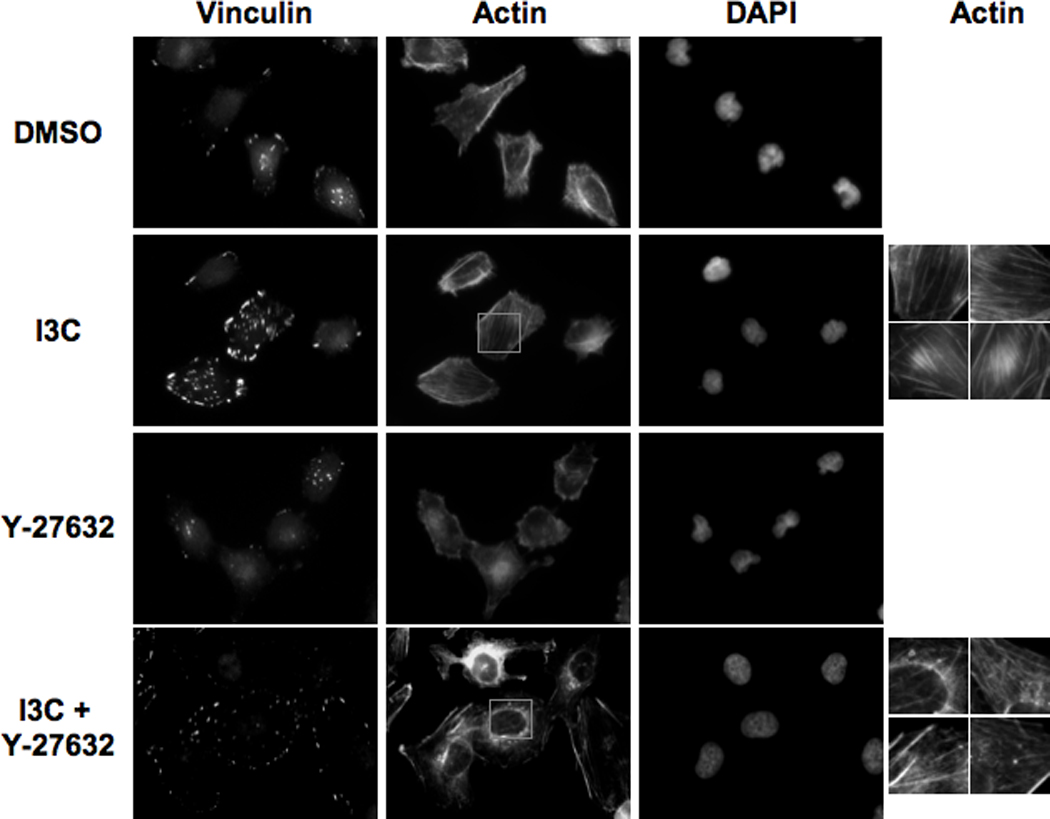
ROCK inhibitor Y-27632 reverses the I3C-mediated formation of stress fibers and stabilization of focal adhesions. MDA-MB-231 cells were treated for 48 h with DMSO or 200 µM I3C, followed by a 2 h incubation in the absence or presence of Rho kinase inhibitor Y-27632. Immunofluorescence was performed using anti-vinculin antibodies, followed by staining with anti-mouse Alexa-fluor 388 secondary antibody or Texas-red phalloidin for actin staining. Representative cells are shown, along with insets (far right column) of 4X-magnified actin filaments from I3C and I3C + Y-27632 treated cells.
RhoA activity is functionally required for the I3C-induced formation of stress fibers and peripheral focal adhesions
To functionally assess the requirement of RhoA signaling for I3C-mediated stress fiber formation and focal adhesion stabilization, MDA-MB-231-derived cell lines that stably express either the constitutively active or dominant negative forms of RhoA were established. The pEXV-RhoA.V14 expression plasmid encodes a constitutively active Myc-tagged RhoA protein, which maintains the GTP-bound form of the protein, whereas pEXV-RhoA.DN19 encodes a Myc-tagged dominant negative RhoA that binds more tightly to Rho-GEFs than the wild type GTPase, and thus prevents activation of endogenous RhoA (44, 62). Individual cell lines were isolated that expresses either the constitutive active RhoA (231.V14), the dominant negative RhoA (231.DN19) or the empty vector (231.neo). The neo cells were transfected with the empty expression vector linked to the neomycin resistance gene and represent the transfection and neomycin selection controls for comparison to the cells expressing either the constitutively active or dominant negative forms of RhoA. Western blot analysis showed that cells expressing the Myc-tagged constitutively active RhoA or the Myc-tagged dominant negative RhoA express high levels of the exogenous genes compared to the empty vector transfected cells (Fig. 7, top panels, V14 vs neo; DN19 vs neo). As expected for activation of the RhoA pathway, the231.V14 cells produce significantly higher levels of phosphorylated cofilin compared to the control neo cells, whereas, 231.DN19 cells had less phospho-cofilin compared to the 231.neo cell clones, indicating an inhibition of RhoA activity (Figure 7). The total levels of cofilin remained approximately the same in the three cell lines.
Figure 7.
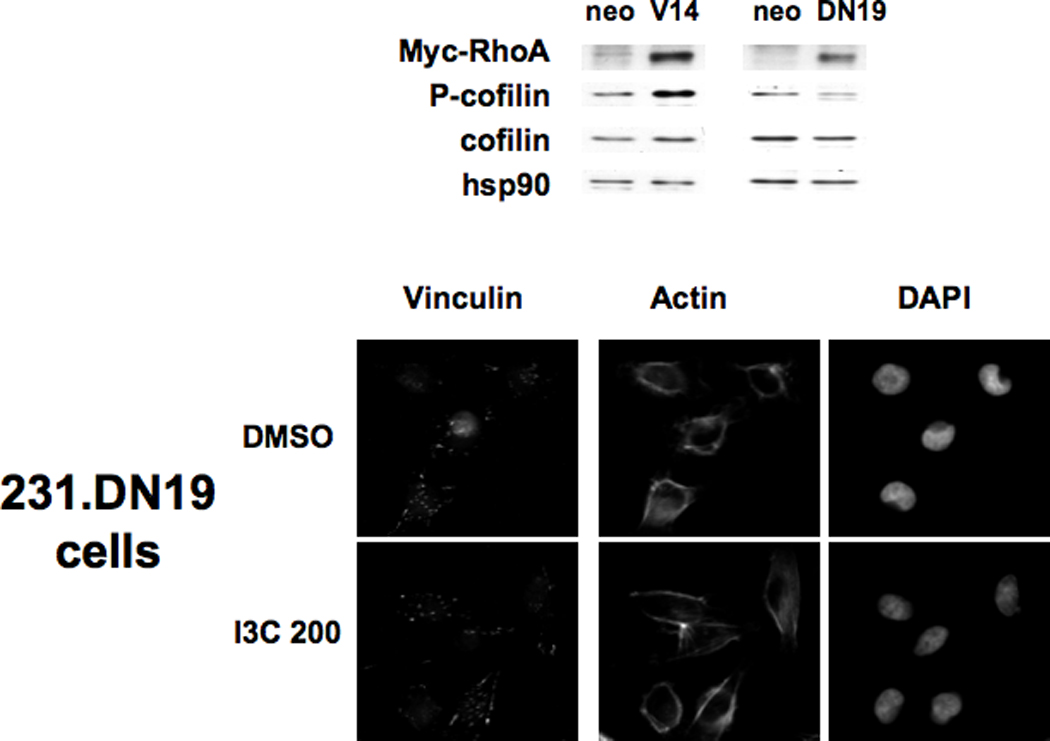
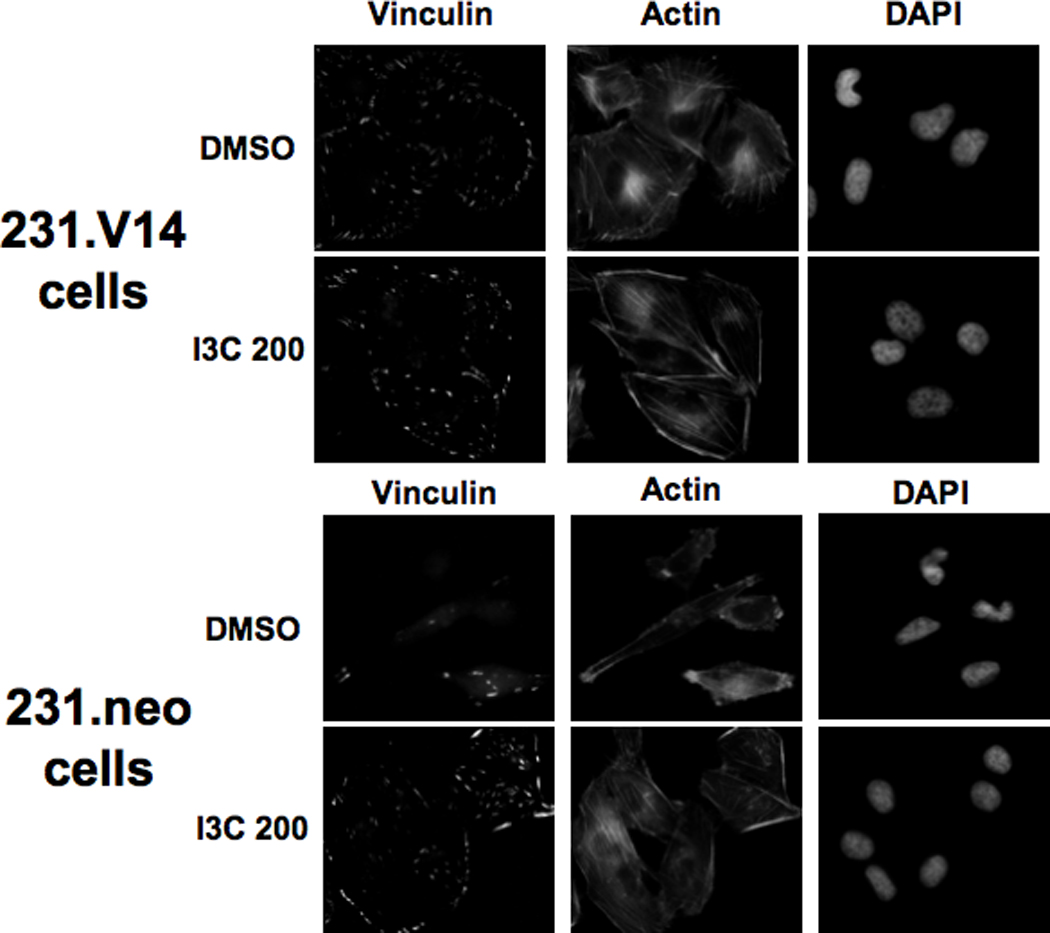
Effects of ectopic expression of dominant negative or constitutively active RhoA on actin stress fibers and vinculin localization. MDA-MB-231 cells were transfected with pEXV empty vector linked to the neomycin resistance gene (neo), the RhoA constitutive expression vector (RhoA.V14), or the dominant negative RhoA (RhoA.DN19) and selected using neomycin analog G418. Upper Panels: The cell clones were tested for expression of Myc-tagged RhoA by Western blotting of extracts using anti-Myc epitope-tag antibodies. Extracts were also tested for expression of phospho-cofilin, cofilin, and hsp90 as a loading control. Micrographs: The stable cell clones expressing empty vector, or a high level of myc-tagged constitutively active or dominant negative RhoA (231.neo, 231.V14, 231.DN19 cells, respectively) were treated for 48 h with DMSO or 200 µM I3C (I3C 200), and immunofluorescence was performed using anti-vinculin antibodies, followed by staining with anti-mouse Alexa-fluor 388 secondary antibody or Texas-red phalloidin. Cell nuclei were stained by DAPI. Representative micrographs of cells are shown.
To determine whether expression of constitutively active or dominant negative RhoA mimicks or disrupts the I3C stimulation of stress fibers and focal adhesion formation, the actin filaments and vinculin localization were examined in I3C treated and untreated 231.neo, 231.V14, and 231.DN19 cells by immunofluorescence microscopy. As shown in Figure 7 (fluorescence micrographs), breast cancer cells expressing dominant negative RhoA (231.DN19 cells) treated with I3C showed diffuse actin staining and thus failed to induce the formation of stress fiber (actin panels), and were unable to stimulate vinculin reorganization or accumulation at the cell periphery (vinculin staining), which is indicative of focal adhesions. Consistent with arole for RhoA signaling in the I3C-activated pathway, expression of constitutively active RhoA mimicked the effects of I3C on formation of both stress fibers and focal adhesions (Fig 7, 231.V14 cells, actin and vinculin panels) in that in the presence or absence I3C the cellsdisplayed a significant amount of stress fibers, filopodia, and focal adhesions at the cell periphery. Control transfected cells were fully indole responsive (Fig 7, 231.neo cells, actin and vinculin panels), showing that the transfection procedure had no deleterious effects on the cell phenotype.
Discussion
Although the anti-proliferative effects of I3C have been well documented (6–8, 13, 14), relatively little is known about the mechanism by which this indole can decrease cell migration and invasion (28, 30). Our results demonstrate that treatment of the invasive MDA-MB-231 human breast cancer cell line with I3C activates the Rho effector kinases ROCK1 and ROCK2, which leads to the stabilization of an extensive network of stress fibers and focal adhesions, an increased phosphorylation of cofilin, and an inhibition of cellular motility. Several lines of evidence show that the I3C induced formation of stress fibers and focal adhesions requires the RhoA/ROCK signaling pathway. First, ectopic expression of dominant negative RhoA in MDA-MB-231 cells prevented the I3C induction of stress fiber and focal adhesion formation. Furthermore, expression of constitutively active RhoA mimicked the indole induction of this phenotype. In vitro assays revealed that the enzymatic activities of ROCK1 and ROCK2 increased over two-fold with I3C treatment, whereas, exposure to the ROCK inhibitor Y-27632 reversed the I3C-induced formation of stress fibers, and decreased the size of focal adhesions. Taken together, our results show that activation of the RhoA/ROCK signaling cascade is critical for the I3C-mediated inhibition of motility of MDA-MB-231 cells.
The balance of activity between RhoA, which induces development of stress fibers that terminate at focal adhesions, and Rac/Cdc42, which promote filopodia and lamellipodia formation observed at the leading edge of migrating cells, is required for contractility during migration of cultured cells (44, 57, 63, 64). When the down-regulation of RhoA signaling is ablated, the breakdown of stress fibers allows the formation of stable focal adhesions which prevents directional motility. Previous studies have shown that hyperactivation of RhoA (65), alteration of integrin signaling (57, 66), or knockout of either FAK (38), cell cycle inhibitor p27 (52), or Fos family member Fra-1 (66) can inhibit cell migration. In MDA-MB-231 cells, treatment with dhMotC, a motuporamine analog from marine sponge, caused significant activation of RhoA, and Rho-dependent induction of stress fibers (50). In these examples, the cells do not form lamellopodia or trailing edges, but instead form extensive stress fiber networks and large stable, peripherally located focal adhesion complexes, analogous to the phenotype observed with I3C treatment. In contrast, knockdown of RhoA by interfering RNA inhibits invasiveness of MDA-MB-231 cells (53), whereas, treatment of these cells with the statin cerivastatin caused disorganization of actin fibers, and disappearance of focal adhesions, by inhibiting Rho transport to the cell membrane (67). These results further implicate a balance of RhoA activity in directing cell migration. In our studies, pull-down assays showed that I3C does not change the level of GTP-bound RhoA, suggesting that I3C does not directly act on RhoA. However, in vitro kinase assays showed a 100% increase in activity of RhoA effector kinases ROCK1 and ROCK2 in I3C-treated cells. Thus, we propose that I3C disrupts the equilibrium between Rho and Rac/Cdc42 activity that leads to the up-regulation of ROCK enzymatic activity. We would predict that the migration of other human breast cancer cells is regulated by I3C through the control of ROCK activity, and we are currently attempting to identify the precise upstream components involved in this indoleregulated signaling cascade.
MDA-MB-231 cells were derived from an aggressive human breast cancer characterized by an oncogenic Ras mutation, and constitutive activation of RhoA (67). Overexpression of Ras has been shown to activate ERK, which mislocalizes and decreases expression of ROCK kinases, thus inhibiting stress fiber formation in the presence of activated RhoA (68) in MDA-MB-231 cells. We found that I3C does not significantly change levels of phosphorylated ERK (data not shown), suggesting that I3C induces ROCK activity and stress fiber formation through a distinct pathway. It is important to point out that although 200 µM I3C was used for our studies, the effective intracellular concentration is significantly lower because only 0.3% of I3C enters the cell, and a fraction of the intracellular I3C is converted into its natural dimer, DIM (13). Also, the concentration of I3C required to inhibit breast cancer cell motility is similar to what is needed to optimally detect indole-mediated changes in cell cycle control of a variety of human reproductive cancer cells.
Previous studies showed that I3C treatment decreased the percentage of dissociated MCF-7 or T-47D breast cancer cells that initially attach to Matrigel basement membrane in vitro, although adhesion was equal by 150 minutes (28, 30). Our current results show a distinct effect on cell adhesion, and may be explained by differences in cell line choice and experimental length. The presence of extensive stress fibers (our unpublished results) and E-cadherin mediated cell-cell adhesion in untreated MCF-7 cells differs greatly from the polarized cell structure of MDA-MB-231 cells, which do not form cell-cell adhesions. In addition, I3C may have differential effects on initial versus sustained adhesion. For example, the initial adhesion of suspended cells to fibronectin can transiently depress RhoA activity (65), but a more prolonged interaction leads to Rho activation whose magnitude is dependent on substrate concentration (57).
Dynamic interactions between focal adhesion complexes and cell adhesion molecules are essential for cell motility (31, 33). We observed that I3C induced the interaction, but not the level, of focal complex proteins FAK, p130 Cas and paxillin, suggesting that I3C increases activation of focal complexes. In addition, there was recruitment of vinculin to I3C-induced focal adhesions, previously shown to increase the stability of these adhesions (69). Current studies are aimed at delineating the I3C induced cellular cascade that stimulates the dynamics of focal adhesion formation and thereby controls cell motility.
In conclusion, our study demonstrates that the RhoA/ROCK pathway is necessary for the I3C-regulated formation of stress fibers and focal adhesions in MDA-MB-231 breast cancer cells. RhoA signaling contributes to cancer primarily through effects on cell migration (43, 48, 52, 70), thus inhibition of this process by I3C limits metastatic capability of breast cancer cells, which in combination with the potent anti-proliferative effects of this indole (6–8, 13, 14), implicates I3C as an intriguing anti-cancer therapeutic agent for breast cancer.
Virtually nothing is known about the mechanism by which the anti-cancer phytochemical Indole-3-carbinol (I3C) limits the metastatic capacity of cancer cells. Our study demonstrates that I3C induces stress fibers and peripheral focal adhesions in a Rho kinase-dependent manner, and thereby activates a key cell signaling pathway that leads to an inhibition of motility of highly invasive human breast cancer cells.
Acknowledgements
We thank Kim Failor, Hui Cen, Nicola Rubenstein, Hanh Garcia, and Shyam Sundar for helpful discussions during the course of our study. Grant support: NIH Public Service grant CA102360 from the National Cancer Institute (G.L.F.). C.T.B. was awarded a postdoctoral fellowship 9FB-0149 from the California Breast Cancer Research Program.
Abbreviations
- FAK
focal adhesion kinase
- I3C
indole-3-carbinol
- MYPT1
myosin phosphatase
- ROCK
Rho kinase
References
- 1.Lopez-Otin C, Diamandis EP. Breast and prostate cancer: an analysis of common epidemiological, genetic, and biochemical features. Endocr Rev. 1998;19:365–396. doi: 10.1210/edrv.19.4.0337. [DOI] [PubMed] [Google Scholar]
- 2.Higdon JV, Delage B, Williams DE, Dashwood RH. Cruciferous vegetables and human cancer risk: epidemiologic evidence and mechanistic basis. Pharmacol Res. 2007;55:224–236. doi: 10.1016/j.phrs.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Terry P, Wolk A, Persson I, Magnusson C. Brassica vegetables and breast cancer risk. Jama. 2001;285:2975–2977. doi: 10.1001/jama.285.23.2975. [DOI] [PubMed] [Google Scholar]
- 4.van Poppel G, Verhoeven DT, Verhagen H, Goldbohm RA. Brassica vegetables and cancer prevention. Epidemiology and mechanisms. Adv Exp Med Biol. 1999;472:159–168. doi: 10.1007/978-1-4757-3230-6_14. [DOI] [PubMed] [Google Scholar]
- 5.Bresnick E, Birt DF, Wolterman K, Wheeler M, Markin RS. Reduction in mammary tumorigenesis in the rat by cabbage and cabbage residue. Carcinogenesis. 1990;11:1159–1163. doi: 10.1093/carcin/11.7.1159. [DOI] [PubMed] [Google Scholar]
- 6.Aggarwal BB, Ichikawa H. Molecular targets and anti-cancer potential of indole-3-carbinol and its derivatives. Cell Cycle. 2005;4:1201–1215. doi: 10.4161/cc.4.9.1993. [DOI] [PubMed] [Google Scholar]
- 7.Weng JR, Tsai CH, Kulp SK, Chen CS. Indole-3-carbinol as a chemopreventive and anti-cancer agent. Cancer Lett. 2008;262:153–163. doi: 10.1016/j.canlet.2008.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim YS, Milner JA. Targets for indole-3-carbinol in cancer prevention. Journal of Nutritional Biochemistry. 2005;16:65–73. doi: 10.1016/j.jnutbio.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 9.Sharma S, Stutzman JD, Kelloff GJ, Steele VE. Screening of potential chemopreventive agents using biochemical markers of carcinogenesis. Cancer Res. 1994;54:5848–5855. [PubMed] [Google Scholar]
- 10.Bradlow HL, Michnovicz J, Telang NT, Osborne MP. Effects of dietary indole-3-carbinol on estradiol metabolism and spontaneous mammary tumors in mice. Carcinogenesis. 1991;12:1571–1574. doi: 10.1093/carcin/12.9.1571. [DOI] [PubMed] [Google Scholar]
- 11.Wattenberg LW, Loub WD. Inhibition of polycyclic aromatic hydrocarbon-induced neoplasia by naturally occurring indoles. Cancer Res. 1978;38:1410–1413. [PubMed] [Google Scholar]
- 12.Grubbs CJ, Steele VE, Casebolt T, Juliana MM, Eto I, Whitaker LM, Dragnev KH, Kelloff GJ, Lubet RL. Chemoprevention of chemically-induced mammary carcinogenesis by indole-3-carbinol. Anticancer Res. 1995;15:709–716. [PubMed] [Google Scholar]
- 13.Firestone GL, Bjeldanes LF. Indole-3-Carbinol (I3C) and 3-3'diindolylmethane (DIM) anti-proliferative signaling pathways control cell cycle gene transcription in human breast cancer cells by regulating promoter-Sp1 tanscription factor interactions. J. Nutrition. 2003;133:2448S–2455S. doi: 10.1093/jn/133.7.2448S. [DOI] [PubMed] [Google Scholar]
- 14.Cover CM, Hsieh SJ, Tran SH, Hallden G, Kim GS, Bjeldanes LF, Firestone GL. Indole-3-carbinol inhibits the expression of cyclin-dependent kinase-6 and induces a G1 cell cycle arrest of human breast cancer cells independent of estrogen receptor signaling. J Biol Chem. 1998;273:3838–3847. doi: 10.1074/jbc.273.7.3838. [DOI] [PubMed] [Google Scholar]
- 15.Cover CM, Hsieh SJ, Cram EJ, Hong C, Riby JE, Bjeldanes LF, Firestone GL. Indole-3-carbinol and tamoxifen cooperate to arrest the cell cycle of MCF-7 human breast cancer cells. Cancer Res. 1999;59:1244–1251. [PubMed] [Google Scholar]
- 16.Cram EJ, Liu BD, Bjeldanes LF, Firestone GL. Indole-3-carbinol inhibits CDK6 expression in human MCF-7 breast cancer cells by disrupting Sp1 transcription factor interactions with a composite element in the CDK6 gene promoter. J Biol Chem. 2001;276:22332–22340. doi: 10.1074/jbc.M010539200. [DOI] [PubMed] [Google Scholar]
- 17.Chen DZ, Qi M, Auborn KJ, Carter TH. Indole-3-carbinol and diindolylmethane induce apoptosis of human cervical cancer cells and in murine HPV16-transgenic preneoplastic cervical epithelium. J Nutr. 2001;131:3294–3302. doi: 10.1093/jn/131.12.3294. [DOI] [PubMed] [Google Scholar]
- 18.Chinni SR, Li Y, Upadhyay S, Koppolu PK, Sarkar FH. Indole-3-carbinol (I3C) induced cell growth inhibition, G1 cell cycle arrest and apoptosis in prostate cancer cells. Oncogene. 2001;20:2927–2936. doi: 10.1038/sj.onc.1204365. [DOI] [PubMed] [Google Scholar]
- 19.Zhang J, Hsu JC, Kinseth MA, Bjeldanes LF, Firestone GL. Indole-3-carbinol (I3C) induces a G1 cell cycle arrest of human LNCaP prostate cancer cells and inhibits expression of prostate specific antigen. Cancer. 2003;98:2511–2520. doi: 10.1002/cncr.11844. [DOI] [PubMed] [Google Scholar]
- 20.Garcia HH, Brar GA, Nguyen DH, Bjeldanes LF, Firestone GL. Indole-3-carbinol (I3C) inhibits cyclin-dependent kinase-2 function in human breast cancer cells by regulating the size distribution, associated cyclin E forms, and subcellular localization of the CDK2 protein complex. J Biol Chem. 2005;280:8756–8764. doi: 10.1074/jbc.M407957200. [DOI] [PubMed] [Google Scholar]
- 21.Ge X, Fares FA, Yannai S. Induction of apoptosis in MCF-7 cells by indol-3-carbinol is independent of p53 and bax. Anticancer Res. 1999;19:3199–3203. [PubMed] [Google Scholar]
- 22.Howells LM, Gallacher-Horley B, Houghton CE, Manson MM, Hudson EA. Indole-3-carbinol inhibits protein kinase B/Akt and induces apoptosis in the human breast tumor cell line MDA MB468 but not in the nontumorigenic HBL100 line. Mol Cancer Ther. 2002;1:1161–1172. [PubMed] [Google Scholar]
- 23.Hsu JC, Dev A, Wing A, Brew CT, Bjeldanes LF, Firestone GL. Indole-3-carbinol mediated cell cycle arrest of LNCaP human prostate cancer cells requires the induced production of activated p53 tumor suppressor protein. Biochem Pharmacol. 2006;72:1714–1723. doi: 10.1016/j.bcp.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 24.Bradlow HL, Michnovicz JJ, Halper M, Miller DG, Wong GY, Osborne MP. Long-term responses of women to indole-3-carbinol or a high fiber diet. Cancer Epidemiol Biomarkers Prev. 1994;3:591–595. [PubMed] [Google Scholar]
- 25.Sundar SN, Kerekatte V, Equinozio CN, Doan VB, Bjeldanes LF, Firestone GL. Indole-3-carbinol selectively uncouples expression and activity of estrogen receptor subtypes in human breast cancer cells. Mol Endocrinol. 2006;20:3070–3082. doi: 10.1210/me.2005-0263. [DOI] [PubMed] [Google Scholar]
- 26.Wang TT, Milner MJ, Milner JA, Kim YS. Estrogen receptor alpha as a target for indole-3-carbinol. J Nutr Biochem. 2006;17:659–664. doi: 10.1016/j.jnutbio.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 27.Brew CT, Aronchik I, Hsu JC, Sheen JH, Dickson RB, Bjeldanes LF, Firestone GL. Indole-3-carbinol activates the ATM signaling pathway independent of DNA damage to stabilize p53 and induce G1 arrest of human mammary epithelial cells. Int J Cancer. 2006;118:857–868. doi: 10.1002/ijc.21445. [DOI] [PubMed] [Google Scholar]
- 28.Meng Q, Goldberg ID, Rosen EM, Fan S. Inhibitory effects of Indole-3-carbinol on invasion and migration in human breast cancer cells. Breast Cancer Res Treat. 2000;63:147–152. doi: 10.1023/a:1006495824158. [DOI] [PubMed] [Google Scholar]
- 29.Rahman KM, Sarkar FH, Banerjee S, Wang Z, Liao DJ, Hong X, Sarkar NH. Therapeutic intervention of experimental breast cancer bone metastasis by indole-3-carbinol in SCID-human mouse model. Mol Cancer Ther. 2006;5:2747–2756. doi: 10.1158/1535-7163.MCT-06-0221. [DOI] [PubMed] [Google Scholar]
- 30.Meng Q, Qi M, Chen DZ, Yuan R, Goldberg ID, Rosen EM, Auborn K, Fan S. Suppression of breast cancer invasion and migration by indole-3- carbinol: associated with up-regulation of BRCA1 and E-cadherin/catenin complexes. J Mol Med. 2000;78:155–165. doi: 10.1007/s001090000088. [DOI] [PubMed] [Google Scholar]
- 31.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 32.Chrzanowska-Wodnicka M, Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol. 1996;133:1403–1415. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wozniak MA, Modzelewska K, Kwong L, Keely PJ. Focal adhesion regulation of cell behavior. Biochim Biophys Acta. 2004;1692:103–119. doi: 10.1016/j.bbamcr.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 34.Humphries JD, Wang P, Streuli C, Geiger B, Humphries MJ, Ballestrem C. Vinculin controls focal adhesion formation by direct interactions with talin and actin. J Cell Biol. 2007;179:1043–1057. doi: 10.1083/jcb.200703036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burridge K, Chrzanowska-Wodnicka M. Focal adhesions, contractility, and signaling. Annu Rev Cell Dev Biol. 1996;12:463–518. doi: 10.1146/annurev.cellbio.12.1.463. [DOI] [PubMed] [Google Scholar]
- 36.Parsons JT, Martin KH, Slack JK, Taylor JM, Weed SA. Focal adhesion kinase: a regulator of focal adhesion dynamics and cell movement. Oncogene. 2000;19:5606–5613. doi: 10.1038/sj.onc.1203877. [DOI] [PubMed] [Google Scholar]
- 37.Hsia DA, Mitra SK, Hauck CR, Streblow DN, Nelson JA, Ilic D, Huang S, Li E, Nemerow GR, Leng J, Spencer KS, Cheresh DA, Schlaepfer DD. Differential regulation of cell motility and invasion by FAK. J Cell Biol. 2003;160:753–767. doi: 10.1083/jcb.200212114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- 39.Critchley DR. Focal adhesions - the cytoskeletal connection. Curr Opin Cell Biol. 2000;12:133–139. doi: 10.1016/s0955-0674(99)00067-8. [DOI] [PubMed] [Google Scholar]
- 40.Sastry SK, Burridge K. Focal adhesions: a nexus for intracellular signaling and cytoskeletal dynamics. Exp Cell Res. 2000;261:25–36. doi: 10.1006/excr.2000.5043. [DOI] [PubMed] [Google Scholar]
- 41.Webb DJ, Brown CM, Horwitz AF. Illuminating adhesion complexes in migrating cells: moving toward a bright future. Curr Opin Cell Biol. 2003;15:614–620. doi: 10.1016/s0955-0674(03)00105-4. [DOI] [PubMed] [Google Scholar]
- 42.Ridley AJ. Rho GTPases and cell migration. J Cell Sci. 2001;114:2713–2722. doi: 10.1242/jcs.114.15.2713. [DOI] [PubMed] [Google Scholar]
- 43.Evers EE, Zondag GC, Malliri A, Price LS, ten Klooster JP, van der Kammen RA, Collard JG. Rho family proteins in cell adhesion and cell migration. Eur J Cancer. 2000;36:1269–1274. doi: 10.1016/s0959-8049(00)00091-5. [DOI] [PubMed] [Google Scholar]
- 44.Wennerberg K, Der CJ. Rho-family GTPases: it's not only Rac and Rho (and I like it) J Cell Sci. 2004;117:1301–1312. doi: 10.1242/jcs.01118. [DOI] [PubMed] [Google Scholar]
- 45.Matsui T, Amano M, Yamamoto T, Chihara K, Nakafuku M, Ito M, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. Embo J. 1996;15:2208–2216. [PMC free article] [PubMed] [Google Scholar]
- 46.Pellegrin S, Mellor H. Actin stress fibres. J Cell Sci. 2007;120:3491–3499. doi: 10.1242/jcs.018473. [DOI] [PubMed] [Google Scholar]
- 47.Baugher PJ, Krishnamoorthy L, Price JE, Dharmawardhane SF. Rac1 and Rac3 isoform activation is involved in the invasive and metastatic phenotype of human breast cancer cells. Breast Cancer Res. 2005;7:R965–R974. doi: 10.1186/bcr1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sequeira L, Dubyk CW, Riesenberger TA, Cooper CR, van Golen KL. Rho GTPases in PC-3 prostate cancer cell morphology, invasion and tumor cell diapedesis. Clin Exp Metastasis. 2008;25:569–579. doi: 10.1007/s10585-008-9173-3. [DOI] [PubMed] [Google Scholar]
- 49.Fritz G, Just I, Kaina B. Rho GTPases are over-expressed in human tumors. Int J Cancer. 1999;81:682–687. doi: 10.1002/(sici)1097-0215(19990531)81:5<682::aid-ijc2>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 50.McHardy LM, Sinotte R, Troussard A, Sheldon C, Church J, Williams DE, Andersen RJ, Dedhar S, Roberge M, Roskelley CD. The tumor invasion inhibitor dihydromotuporamine C activates RHO, remodels stress fibers and focal adhesions, and stimulates sodium-proton exchange. Cancer Res. 2004;64:1468–1474. doi: 10.1158/0008-5472.can-03-2733. [DOI] [PubMed] [Google Scholar]
- 51.Barker TH, Grenett HE, MacEwen MW, Tilden SG, Fuller GM, Settleman J, Woods A, Murphy-Ullrich J, Hagood JS. Thy-1 regulates fibroblast focal adhesions, cytoskeletal organization and migration through modulation of p190 RhoGAP and Rho GTPase activity. Exp Cell Res. 2004;295:488–496. doi: 10.1016/j.yexcr.2004.01.026. [DOI] [PubMed] [Google Scholar]
- 52.Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004;18:862–876. doi: 10.1101/gad.1185504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pille JY, Denoyelle C, Varet J, Bertrand JR, Soria J, Opolon P, Lu H, Pritchard LL, Vannier JP, Malvy C, Soria C, Li H. Anti-RhoA and anti-RhoC siRNAs inhibit the proliferation and invasiveness of MDA-MB-231 breast cancer cells in vitro and in vivo. Mol Ther. 2005;11:267–274. doi: 10.1016/j.ymthe.2004.08.029. [DOI] [PubMed] [Google Scholar]
- 54.Thompson EW, Paik S, Brunner N, Sommers CL, Zugmaier G, Clarke R, Shima TB, Torri J, Donahue S, Lippman ME, et al. Association of increased basement membrane invasiveness with absence of estrogen receptor and expression of vimentin in human breast cancer cell lines. J Cell Physiol. 1992;150:534–544. doi: 10.1002/jcp.1041500314. [DOI] [PubMed] [Google Scholar]
- 55.Denoyelle C, Vasse M, Korner M, Mishal Z, Ganne F, Vannier JP, Soria J, Soria C. Cerivastatin, an inhibitor of HMG-CoA reductase, inhibits the signaling pathways involved in the invasiveness and metastatic properties of highly invasive breast cancer cell lines: an in vitro study. Carcinogenesis. 2001;22:1139–1148. doi: 10.1093/carcin/22.8.1139. [DOI] [PubMed] [Google Scholar]
- 56.McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell. 2004;6:483–495. doi: 10.1016/s1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- 57.Cox EA, Sastry SK, Huttenlocher A. Integrin-mediated adhesion regulates cell polarity and membrane protrusion through the Rho family of GTPases. Mol Biol Cell. 2001;12:265–277. doi: 10.1091/mbc.12.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 59.Riento K, Guasch RM, Garg R, Jin B, Ridley AJ. RhoE binds to ROCK I and inhibits downstream signaling. Mol Cell Biol. 2003;23:4219–4229. doi: 10.1128/MCB.23.12.4219-4229.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Narumiya S, Ishizaki T, Uehata M. Use and properties of ROCK-specific inhibitor Y-27632. Methods Enzymol. 2000;325:273–284. doi: 10.1016/s0076-6879(00)25449-9. [DOI] [PubMed] [Google Scholar]
- 61.Amano M, Fukata Y, Shimokawa H, Kaibuchi K. Purification and in vitro activity of Rho-associated kinase. Methods Enzymol. 2000;325:149–155. doi: 10.1016/s0076-6879(00)25439-6. [DOI] [PubMed] [Google Scholar]
- 62.Goi T, Rusanescu G, Urano T, Feig LA. Ral-specific guanine nucleotide exchange factor activity opposes other Ras effectors in PC12 cells by inhibiting neurite outgrowth. Mol Cell Biol. 1999;19:1731–1741. doi: 10.1128/mcb.19.3.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Allen WE, Zicha D, Ridley AJ, Jones GE. A role for Cdc42 in macrophage chemotaxis. J Cell Biol. 1998;141:1147–1157. doi: 10.1083/jcb.141.5.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jaffe AB, Hall A. Rho GTPases in transformation and metastasis. Adv Cancer Res. 2002;84:57–80. doi: 10.1016/s0065-230x(02)84003-9. [DOI] [PubMed] [Google Scholar]
- 65.Arthur WT, Burridge K. RhoA inactivation by p190RhoGAP regulates cell spreading and migration by promoting membrane protrusion and polarity. Mol Biol Cell. 2001;12:2711–2720. doi: 10.1091/mbc.12.9.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vial E, Sahai E, Marshall CJ. ERK-MAPK signaling coordinately regulates activity of Rac1 and RhoA for tumor cell motility. Cancer Cell. 2003;4:67–79. doi: 10.1016/s1535-6108(03)00162-4. [DOI] [PubMed] [Google Scholar]
- 67.Denoyelle C, Albanese P, Uzan G, Hong L, Vannier JP, Soria J, Soria C. Molecular mechanism of the anti-cancer activity of cerivastatin, an inhibitor of HMG-CoA reductase, on aggressive human breast cancer cells. Cell Signal. 2003;15:327–338. doi: 10.1016/s0898-6568(02)00124-9. [DOI] [PubMed] [Google Scholar]
- 68.Sahai E, Olson MF, Marshall CJ. Cross-talk between Ras and Rho signalling pathways in transformation favours proliferation and increased motility. Embo J. 2001;20:755–766. doi: 10.1093/emboj/20.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crowley E, Horwitz AF. Tyrosine phosphorylation and cytoskeletal tension regulate the release of fibroblast adhesions. J Cell Biol. 1995;131:525–537. doi: 10.1083/jcb.131.2.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kusama T, Mukai M, Tatsuta M, Nakamura H, Inoue M. Inhibition of transendothelial migration and invasion of human breast cancer cells by preventing geranylgeranylation of Rho. Int J Oncol. 2006;29:217–223. [PubMed] [Google Scholar]