

Alcohol (ethanol) is one of the most commonly abused drugs throughout the world. In the United States, about half of the adult population actively consumes alcoholic beverages and about 15 to 20 million people suffer from alcoholism (1). According to a 1990 survey by the US Department of Health and Human Services, alcohol is the third leading cause of preventable mortality, behind cigarette smoking and obesity, with about 100,000 deaths annually and an annual economic burden of more than $100 billion in the US (2,3).
Alcohol consumption can lead to a variety of abnormalities in the liver including steatosis, alcoholic hepatitis, and hepatic fibrosis, which typically precede the development of alcoholic cirrhosis, an end-stage liver disease most often requiring liver transplantation. Alcoholic cirrhosis is also a major risk factor for hepatocellular carcinoma that accounts for nearly 6% of all human cancers. Alcoholic liver disease affects more than 2 million people in the United States. According to a US surveillance report, liver cirrhosis was the 10th leading cause of mortality in 1997 accounting for approximately 25,000 deaths in that year (4). Another 10,000 died of liver cancer, which in the majority of the cases involves underlying cirrhosis.
GENDER
The most important factors in the pathogenesis of alcoholic liver disease are the amount and duration of alcohol consumed. It has also been consistently observed that the incidence of alcohol-induced liver injury is higher and progresses faster among women than among men with a similar history of alcohol abuse (5,6). In addition, women are at a higher risk of developing liver injury than men, even when factors such as body weight and amount of alcohol consumed are taken into consideration. The average threshold alcohol intake to produce liver injury is 40 g/day to 60 g /day (3 to 5 drinks) in men but only 20 g/day (<2 drinks) in women with a drink defined as 12 oz (354 mL) of beer, 5 oz (148 mL) of wine or 1.5 oz (44 mL) of 80-proof distilled spirits, each containing about 12g of alcohol (7,8). In a 12-year longitudinal study of more than 13,000 individuals in Denmark, a steep dose-dependent increase in relative risk of liver disease was found above a threshold of 14–27 drinks per week in men and 7–13 drinks per week in women (9). Although there was a dose-dependent increase in relative risk of developing liver disease for both men and women, women were at significantly higher risk at all levels of alcohol intake. Thus, a level of alcohol consumption considered safe in men is not necessarily safe in women.
For a given amount of alcohol consumed, women have higher blood alcohol concentrations than men. This is due, in part, to the fact that the distribution of alcohol occurs in a smaller water space in women because of their smaller body size and a higher proportion of fat (10). However, the major mechanism implicated is women's lower gastric alcohol dehydrogenase (ADH) activity compared with men under 50 years of age (10,11). ADH is the enzyme responsible for the metabolism of a substantial amount of alcohol before it enters systemic circulation (discussed below). Thus, lower ADH activity results in decreased breakdown of alcohol in the stomach and higher levels in the bloodstream. Alcohol abuse decreases gastric ADH activity even further (10). Consequently, a large proportion of ingested alcohol may reach the liver, thus contributing to the greater susceptibility of women to liver injury. Older women, however, have comparable or even higher gastric ADH activities, because gastric ADH decreases with age only in men and not in women (11).
ETHNICITY
Apart from gender differences in alcohol metabolism, there is evidence that ethnicity may influence drinking behavior and the risk of alcoholic liver injury. Genetic studies have shown that the liver enzymes ADH and acetaldehyde dehydrogenase (ALDH, the enzyme that catalyzes the conversion of acetaldehyde to acetate) are polymorphic in humans and display strikingly different catalytic properties among different racial populations. The ADH2*2 and ADH3*1 alleles, which encode enzymes that metabolize alcohol rapidly, are more frequent in Chinese and Japanese populations (12,13). ALDH exists in two major forms in the liver, ALDH1 and ALDH2, the latter showing high affinity, [low Michaelis constant (Km)] for acetaldehyde and is responsible for most acetaldehyde oxidation. The ALDH2 gene exists in two allelic forms: ALDH2*1 and the inactive ALDH2*2 form. The inactive isoenzyme is present in about 50% of the Chinese and Japanese population. The highly active ADH and the inactive ALDH alleles in these populations are associated with aversion to alcohol and protection against alcoholism because of acetaldehyde toxicity (14). Shortly after drinking alcohol, these individuals develop facial flushing, palpitation, hypotension, and nausea, which lead to alcohol avoidance and to a decreased risk of alcoholism and alcohol-related liver disease.
ALCOHOL METABOLISM AND LIVER INJURY
Two major enzyme systems are involved in the metabolism of alcohol in the liver: ADH and the microsomal ethanol-oxidizing system (MEOS) (Figure). ADH has a high affinity (low Km: 0.2–2 mM) for alcohol and accounts for essentially all alcohol metabolism when blood and tissue concentrations are low. However, when the concentrations are high, MEOS, with a lower affinity (high Km: 8–10 mM) for alcohol, can also contribute to alcohol metabolism.
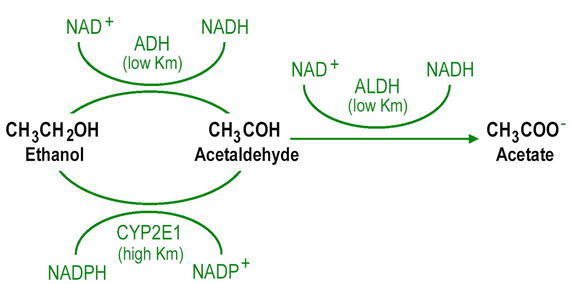

ADH converts alcohol to acetaldehyde by removing hydrogen. Then a second enzyme, aldehyde dehydrogenase in hepatic mitochondria, oxidizes acetaldehyde to acetate by removing additional hydrogen and adding oxygen. In peripheral tissues, acetate is converted to acetyl coenzymes A and subsequently to CO2 and water. Acetaldehyde is a potent toxic metabolite of alcohol and contributes to liver injury in several different ways (15). Acetaldehyde promotes cell death by depleting glutathione levels and inducing oxidative damage. Acetaldehyde binds to specific amino acid residues on structural and functional proteins of the cells to form acetaldehyde-protein adduct leading to cellular injury. For example, by binding to the highly reactive lysine residue on the cytoskeletal protein tubulin, acetaldehyde can impair microtubule assembly and function. Acetaldehyde-protein adducts also activate hepatic stellate cells to produce excess extracellular matrix protein leading to alcoholic fibrosis (16). These adducts have been detected by immunohistochemical staining of liver biopsies of individuals with early-stage liver disease (17,18). Oxidized nicotinamide-adenine dinucleotide (NAD+) is converted to reduced NAD (NADH) during the production of acetaldehyde, leading to a shift in the redox state of the hepatocytes which, in turn, results in the inhibition of fatty acid oxidation that favors steatosis and hyperlipidemia (15).
The other alcohol oxidation pathway MEOS also converts alcohol to acetaldehyde. The critical component that mediates this conversion is cytochrome P-450 (CYP) 2E1, which is induced predominantly in the hepatocytes by alcohol (19). CYP2E1 utilizes reduced niotinamide-adenine dinucleotide phosphate NADPH, which is converted to NADP+ by NADPH oxidation with the release of oxygen-derived free radicals as a by-product. Thus induction of CYP2E1 can result in greater rates of NADPH oxidation leading to increased production of reactive oxygen species (ROS). A number of in vitro studies have shown that induction of CYP2E1 by ethanol is associated with generation of ROS, lipid peroxidation, and mitochondrial damage (20). This is further supported by a number of animal studies showing a correlation between CYP2E1 induction and alcohol induced liver disease (20). However, the precise role of CYP2E1 from these studies remains to be understood in view of the evidence that alcoholic liver injury in experimental animals can be prevented despite the induction of CYP2E1 (21). CYP2E1 is also involved in the metabolism of several drugs including halogenated anesthetics, the nonsteroidal anti-inflammatory drug phenylbutazone, acetaminophen, and even vitamins such as retinol and its precursor β-carotene (22). Thus, induction of CYP2E1 by alcohol may result in the conversion of these substances to hepatotoxic metabolites that can further contribute to alcoholic liver injury.
Evidence from a number of studies indicates that Kupffer cells, the resident hepatic macrophages, play a pivotal role in ethanol-induced hepatotoxicity (20). Alcohol is known to increase gut permeability to Gram-negative bacterial endotoxins. Thus, activation of Kupffer cells by such endotoxins can result in the activation of NADPH oxidase, a major oxidant-generating enzyme leading to the release of ROS and the production of inflammatory cytokines and chemokines including IL-1, IL-6, TNF-alpha and IL-8, which have deleterious effects on hepatocytes (23). NADPH-oxidase-deficient mice treated with alcohol do not develop liver pathology in contrast to the alcohol-fed normal mice (24). The NADPH-oxidase-deficient mice did not show ethanol-induced increases in free radical production, TNF-alpha transcripts and activation of NF- kappa B, suggesting that NADPH-oxidase plays an important role in alcoholic liver injury. In addition, ROS produced via alcohol metabolism may itself stimulate the production and release of cytokines and chemokines.
Increased circulating levels of TNF-alpha, IL-6, and IL-8 have been shown in the sera of patients with alcoholic liver cirrhosis (20). Plasma TNF-alpha levels in patients with alcoholic hepatitis correlate with severity of disease and risk of mortality. TNF-alpha can induce cytotoxicity by activating the TNF receptor p55 (TNFR p55) or TNF receptor 1 (TNF-R1), and by initiating signaling through the death domain (25,26). Consistent with these mechanisms, TNF-R1 knock-out mice have been shown to be resistant to alcoholic liver injury (27), and, in another study, plasma levels of TNFR p55 have been shown to negatively correlate with total blood glutathione in patients with acute alcoholic hepatitis (28). The Kupffer cell- derived TNF-alpha can also induce hepatocytes to produce IL-8 and macrophage inflammatory protein-2 (MIP-2), the potent chemoattractants for neutrophils that facilitate the interaction between inflammatory leukocytes and target cells and exacerbate cytotoxicity (29,30).
Plasma levels of IL-6 are elevated in patients with alcoholic hepatitis and correlate with severity of liver disease (20). A recent study suggests that IL-6 in alcoholic liver disease is involved in mediating antiapoptotic signals to overcome alcohol-induced hepatic apoptosis (31). The study demonstrated that livers from chronically ethanol-fed IL-6 (−/−) mice but not IL-6 (+/+) mice showed significant apoptosis. The expression of antiapoptotic proteins Bcl-2 and Bcl-x (L) was also markedly elevated in the livers of ethanol-fed IL-6 (+/+) mice, suggesting that IL-6 prevents alcohol-induced apoptosis by induction of Bcl-2 and Bcl-x (L).
In a number of studies TGF-beta expression was elevated in patients with alcoholic liver disease (20). TGF-beta increases collagen gene expression and collagen synthesis in hepatic cells. Patients with active liver disease showed a 97% increase in type 1 collagen synthesis in liver biopsy specimens (32), suggesting that TGF-beta may be the key factor in the development of hepatic fibrosis in alcoholic liver disease.
SUMMARY AND CONCLUSIONS
Excessive and prolonged use of alcoholic beverages is a human behavior with serious social and medical consequences in a significant population of drinkers. Although the risk is lower among persons who drink less, there is an increased risk of progression to cirrhosis in females than in males ingesting the same relative amount of alcohol. The heptotoxic effects of alcohol result from its oxidation through the ADH and MEOS pathways, which produce acetaldehyde that is metabolized to acetate by ALDH. Past treatment efforts have been focused on the management of the complications of cirrhosis such as bleeding and ascites. However, a better understanding of alcohol metabolism, alcohol-induced metabolic disorders, and their sequelae should result in improved preventive and therapeutic approaches. Some of these approaches will target nutritional deficiencies, altered redox state, and inflammatory responses (22).

Dr. Prakash is the Head of the Laboratory of Molecular Oncology at Ochsner Clinic Foundation and Professor (conjoint) in the Department of Microbiology, Immunology, and Parasitology at the Louisiana State University Health Sciences Center in New Orleans. His main research interest is in the molecular mechanisms of HIV pathogenesis and alcohol effects on anti-HIV therapies.

Dr. Steve Nelson is the John H. Seabury Professor of Medicine in the Section of Pulmonary and Critical Care Medicine at the Louisiana State University Health Sciences Center in New Orleans. He is the Director of the National Institutes of Health funded Alcohol Research Center at LSUHSC. His main research interests are the pathophysiology of infections and the health consequences of alcohol abuse.
REFERENCES
- Stinson F. S., Yi Y-H., Grant B. F. Drinking in the U.S.: Main findings from the National Longitudinal Alcohol Epidemiologic Survey. 1997;((6)) In: U.S. Alcohol Epidemiologic Data References Manual NIH Publication no. 99-3519. 1st ed. Washington, DC: U.S. Government Printing Office. [Google Scholar]
- McGinnis J. M., Foege W. H. Actual causes of death in the United States. JAMA. 1993;270:2207–2212. [PubMed] [Google Scholar]
- Angell M., Kassirer J. P. Alcohol and other drugs – toward a more rational and consistent policy. N Engl J Med. 1994;331:537–539. doi: 10.1056/NEJM199408253310810. [DOI] [PubMed] [Google Scholar]
- Saadatmand F., Stinson F. S., Grant B. F. Surveillance Report #54: Liver Cirrhosis Mortality in the United States 1970–97. National Institute on Alcohol Abuse and Alcoholism, Division of Biometry and Epidemiology. 2000 Alcohol Epidemiologic Data System, Bethesda, MD. [Google Scholar]
- Morgan M. Y., Sherlock S. Sex-related differences among 100 patients with alcoholic liver disease. Br Med J. 1977;1:939–941. doi: 10.1136/bmj.1.6066.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S., Takezawa Y., Sato T. Alcoholic liver disease in women. Tohoku J Exp Med. 1979;129:351–355. doi: 10.1620/tjem.129.351. [DOI] [PubMed] [Google Scholar]
- Pequignot G., Tuyns A. J., Berta J. L. Ascitic cirrhosis in relation to alcohol consumption. Int J Epidemiol. 1978;7:113–120. doi: 10.1093/ije/7.2.113. [DOI] [PubMed] [Google Scholar]
- Congress on alcohol and health: Highlights from current research from the Secretary of Health and Human Services. 2000:429–30. 10th special report to U.S. U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health. National Institute on Alcohol Abuse and Alcoholism. NIH publication no. 00-1583. [Google Scholar]
- Becker U., Deis A., Sorensen T. I. Prediction of risk of liver disease by alcohol intake, sex, and age: a prospective population study. Hepatology. 1996;23:1025–1029. doi: 10.1002/hep.510230513. [DOI] [PubMed] [Google Scholar]
- Frezza M., di Padova C., Pozzato G. High blood alcohol levels in women. The role of decreased gastric alcohol dehydrogenase activity and first-pass metabolism. N Engl J Med. 1990;322:95–99. doi: 10.1056/NEJM199001113220205. [DOI] [PubMed] [Google Scholar]
- Seitz H. K., Egerer G., Simanowski U. A. Human gastric alcohol dehydrogenase activity: effect of age, sex, and alcoholism. Gut. 1993;34:1433–1437. doi: 10.1136/gut.34.10.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosron W. F., Li T. K. Genetic polymorphism of human liver alcohol and aldehyde dehydrogenases, and their relationship to alcohol metabolism and alcoholism. Hepatology. 1986;6:502–510. doi: 10.1002/hep.1840060330. [DOI] [PubMed] [Google Scholar]
- Chao Y. C., Liou S. R., Chung Y. Y. Polymorphism of alcohol and aldehyde dehydrogenase genes and alcoholic cirrhosis in Chinese patients. Hepatology. 1994;19:360–366. [PubMed] [Google Scholar]
- Shibuya A., Yoshida A. Genotypes of alcohol-metabolizing enzymes in Japanese with alcohol liver diseases: a strong association of the usual Caucasian-type aldehyde dehydrogenase gene (ALDH1(2)) with the disease. Am J Hum Genet. 1988;43:744–748. [PMC free article] [PubMed] [Google Scholar]
- Lieber C. S. Alcohol and the liver: 1994 update. Gastroenterology. 1994;106:1085–1105. doi: 10.1016/0016-5085(94)90772-2. [DOI] [PubMed] [Google Scholar]
- Friedman S. L. Stellate cell activation in alcoholic fibrosis—an overview. Alcohol Clin Exp Res. 1999;23:904–910. [PubMed] [Google Scholar]
- Niemela O., Juvonen T., Parkkila S. Immunohistochemical demonstration of acetaldehyde-modified epitopes in human liver after alcohol consumption. J Clin Invest. 1991;87:1367–1374. doi: 10.1172/JCI115141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstege A., Bedossa P., Poynard T. Acetaldehyde-modified epitopes in liver biopsy specimens of alcoholic and nonalcoholic patients: localization and association with progression of liver fibrosis. Hepatology. 1994;19:367–374. [PubMed] [Google Scholar]
- Ronis M. J., Huang J., Crouch J. Cytochrome P450 CYP 2E1 induction during chronic alcohol exposure occurs by a two-step mechanism associated with blood alcohol concentrations in rats. J Pharmacol Exp Ther. 1993;264:944–950. [PubMed] [Google Scholar]
- Prakash O., Mason A., Luftig R. B. Hepatitis C virus (HCV) and human immunodeficiency virus type 1 (hiv-1) infections in alcoholics. Front Biosci. 2002;7:e286–300. doi: 10.2741/A924. [DOI] [PubMed] [Google Scholar]
- Koop D. R., Klopfenstein B., Iimuro Y. Gadolinium chloride blocks alcohol-dependent liver toxicity in rats treated chronically with intragastric alcohol despite the induction of CYP2E1. Mol Pharmacol. 1997;51:944–950. doi: 10.1124/mol.51.6.944. [DOI] [PubMed] [Google Scholar]
- Lieber C. S. Alcoholic liver disease: new insights in pathogenesis lead to new treatments. J Hepatol. 2000;32((1 Suppl)):113–128. doi: 10.1016/s0168-8278(00)80420-1. [DOI] [PubMed] [Google Scholar]
- Bautista A. P. Free radicals, chemokines, and cell injury in HIV-1 and SIV infections and alcoholic hepatitis. Free Radic Biol Med. 2001;31:1527–1532. doi: 10.1016/s0891-5849(01)00745-6. [DOI] [PubMed] [Google Scholar]
- Kono H., Rusyn I., Yin M. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J Clin Invest. 2000;106:867–872. doi: 10.1172/JCI9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser A., Evan G. A. A license to kill. Cell. 1996;85:781–784. doi: 10.1016/s0092-8674(00)81005-3. [DOI] [PubMed] [Google Scholar]
- Tewari M., Dixit V. M. Recent advances in tumor necrosis factor and CD40 signaling. Curr Opin Genet Dev. 1996;6:39–44. doi: 10.1016/s0959-437x(96)90008-8. [DOI] [PubMed] [Google Scholar]
- Yin M., Wheeler M. D., Kono H. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology. 1999;117:942–952. doi: 10.1016/s0016-5085(99)70354-9. [DOI] [PubMed] [Google Scholar]
- Naveau S., Abella A., Raynard B. Tumor necrosis factor soluble receptor p55 and lipid peroxidation in patients with acute alcoholic hepatitis. Am J Gastroenterol. 2001;96:3361–3367. doi: 10.1111/j.1572-0241.2001.05338.x. [DOI] [PubMed] [Google Scholar]
- Thornton A. J., Strieter R. M., Lindley I. Cytokine-induced gene expression of a neutrophil chemotactic factor/IL-8 in human hepatocytes. J Immunol. 1990;144:2609–2613. [PubMed] [Google Scholar]
- Ohkubo K., Masumoto T., Horiike N. Induction of CINC (interleukin-8) production in rat liver by non-parenchymal cells. J Gastroenterol Hepatol. 1998;13:696–702. doi: 10.1111/j.1440-1746.1998.tb00716.x. [DOI] [PubMed] [Google Scholar]
- Hong F., Kim W. H., Tian Z. Elevated interleukin-6 during ethanol consumption acts as a potential endogenous protective cytokine against ethanol-induced apoptosis in the liver: involvement of induction of Bcl-2 and Bcl-x(L) proteins. Oncogene. 2002;21:32–43. doi: 10.1038/sj.onc.1205016. [DOI] [PubMed] [Google Scholar]
- Annoni G., Weiner F. R., Zern M. A. Increased transforming growth factor-beta 1 gene expression in human liver disease. J Hepatol. 1992;14:259–264. doi: 10.1016/0168-8278(92)90168-o. [DOI] [PubMed] [Google Scholar]