

Abstract
In this work we return to the problem of the determination of ligand–receptor binding stoichiometry and binding constants. In many cases the ligand is a fluorescent dye which has low fluorescence quantum yield in free state but forms highly fluorescent complex with target receptor. That is why many researchers use dye fluorescence for determination of its binding parameters with receptor, but they leave out of account that fluorescence intensity is proportional to the part of the light absorbed by the solution rather than to the concentration of bound dye. We showed how ligand–receptor binding parameters can be determined by spectrophotometry of the solutions prepared by equilibrium microdialysis. We determined the binding parameters of ANS – human serum albumin (HSA) and ANS – bovine serum albumin (BSA) interaction, absorption spectra, concentration and molar extinction coefficient, as well as fluorescence quantum yield of the bound dye. It was found that HSA and BSA have two binding modes with significantly different affinity to ANS. Correct determination of the binding parameters of ligand–receptor interaction is important for fundamental investigations and practical aspects of molecule medicine and pharmaceutics. The data obtained for albumins are important in connection with their role as drugs transporters.
Introduction
Correct determination of ligand-receptor binding stoichiometry and binding constants is very important both for fundamental investigations and practical aspects of molecule medicine, pharmaceutics and at the development of biosensor systems of high social significance [1], [2], [3], [4], [5], [6]. The determination of binding parameters significantly enriches tried-and-true method based on fluorescence of extrinsic dyes which is widely used in molecular and cellular biology for investigation of protein’s folding, structural changes induced by different agents, interaction with each other, aggregation, amyloid fibril formation etc. In recent years it is more and more extensively used for practical diagnostics due to significant advances in molecular medicine. In all cases the knowledge of binding parameters is difficult to overestimate. The use of fluorescent dyes is based on their ability to form a highly fluorescent complex with target receptor [7], [8]. That is why it seems natural that many scientists used fluorescence for characterization dye–receptor interaction. In almost all of the studies focused on this problem, the binding constants and binding stoichiometry were evaluated on the basis of the dependence of dye fluorescence intensity on its or receptor concentration (see e.g. [1], [3], [9], [10], [11], [12], [13], [14], [15]). These works were based on the assumption that the fluorescence intensity as a function of dye concentration reaches a plateau when all of the binding sites are occupied [3], [16]. In this work, we demonstrate that the dependence of fluorescence intensity on optical density of solution is always the curve with saturation for any fluorophore (even without receptor). We show that fluorescence intensity depends not only on optical density of fluorophore but on total optical density of solution and proposed how it can be corrected on this value. And finally one must not forget that fluorescence intensity depends also on fluorophore molar extinction coefficient and its quantum yield which are not equal for free and bound dye. We for the first time determined these values for 1-anilino-8-naphthalene sulfonate (ANS) bound to proteins. In this work we showed how problems that cannot be solved by fluorescence can be solved by spectrophotometric determination of the concentration of bound dye if sample and reference solutions are prepared by equilibrium microdialysis. The knowledge of the concentration of bound dye gives an opportunity of correct determination of binding stoichiometry and binding constants. Furthermore, only the use of equilibrium microdialysis for preparation reference solution appropriate for the sample solution allows to determine absorption spectrum of bound dye and its molar extinction coefficient. Finally, fluorescence intensity of the same solutions can be used for determination quantum yield of bound dye and even fluorescence quantum yields of the dye bound to sites of different binding modes. All said above we illustrated by ANS–serum albumins interaction.
The interaction of ANS with proteins was recognized almost 60 years ago [17]. The dye has a low fluorescence quantum yield in polar environments, which is greatly enhanced on interaction with many proteins. ANS was used as a probe for biological membranes studies [18], for the analysis of conformational changes, folding–unfolding processes in proteins and molten globule test [19]. In the work [20] we make an assumption that ANS binds to the aggregates of proteins in molten globule state rather than to the hydrophobic clusters on the surface of a protein in the molten globule state. In several works the possibility of charge interaction of ANS with proteins is mentioned [20], [21], [22]. In the work [23] it was shown that ANS binds to a solvent exposed region and take part in formation of a hydrophobic regions (clusters, environment), though in its environment positively charged arginine residue was found, also. As a hydrophobic probe it has been frequently used to study the binding properties of hydrophobic species of albumins [24], [25]. Serum albumins are the most predominant proteins in blood plasma. The extensive study of these proteins is connected with their unique ability to bind and transfer a wide variety of different substances such as metal ions, metabolites, fatty acids, others biologically active species including drugs, etc. [26], [27].
Materials and Methods
1. Reagents
ANS from Serva (Germany), quinine sulfate (QS) from AnaSpec (Fremont, California, USA), HSA from (Ronkonkoma, NY, USA) and BSA from Koch-Light Lab Ltd (Coinbrook, Bucks., U.K.) were used without afterpurification. HSA was dissolved in 20 mM phosphate buffer (pH 6.3). BSA was dissolved in 10 mM PBS buffer (pH 7.4) [3]. Fluorescence measurements were performed with homemade spectrofluorimeter [28] and spectrofluorimeter Cary Varian Eclipse (Mulgrave, Victoria, Australia). The solution of QS in 0.5 M H2SO4, whose fluorescence and absorption spectra are similar to that of ANS, was taken as a reference for determining the fluorescence quantum yield of ANS bound to proteins.
2. Fluorescence and Spectrophotometric Measurements
Fluorescence of ANS and QS was excited at 365 nm and recorded at 470 nm. The spectral slits width was 5 nm in all experiments. Change of spectral slits width did not influence on the experimental results.
For determination the fluorescence quantum yield of the dye bound to receptor the correction coefficients κ(λ em) for QS and for the ANS – BSA and ANS – HSA solutions were determined:
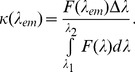 |
(1) |
Here F(λ
em) is the fluorescence intensity at the fixed wavelength of registration and 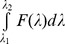 is the total fluorescence intensity. These coefficients were used for calculation total fluorescence intensity of ANS-BSA and ANS-HSA solutions (FANS) and for QS solution (FQS). The fluorescence quantum yield of QS was taken as 0.546 (AnaSpec Catalog # 80040, 2010).
is the total fluorescence intensity. These coefficients were used for calculation total fluorescence intensity of ANS-BSA and ANS-HSA solutions (FANS) and for QS solution (FQS). The fluorescence quantum yield of QS was taken as 0.546 (AnaSpec Catalog # 80040, 2010).
Absorption spectra were recorded by spectrophotometer Hitachi U-3900H (Tokyo, Japan).
3. Equilibrium Microdialysis
Equilibrium microdialysis was performed with a Harvard Apparatus/Amika (Holliston, Massachusetts, USA) device, which consists of two chambers (500 µL each) separated by membrane (MWCO 10,000) impermeable to particles larger than 10,000 Da. Equilibrium microdialysis implies allocation of two interacting agents, a ligand and receptor, in two chambers (#2 and #1, respectively) divided by a membrane permeable to the ligand and impermeable to the receptor. After that the microdialysis device are put on a rocking-bar in a thermostatted box for 48 hours. All experiments were performed at 23°C.
In the test experiments, we put a solution of ANS of concentration C
0 in chamber # 2 and the solvent in chamber # 1. After 24 h of dialysis, the absorption spectra of the samples from chambers # 1 and # 2 coincide (D
#1(λ) = D
#2(λ) =  D
0(λ)). This means that 24 h is enough time to allow the dye to equilibrate between chambers # 1 and # 2 and that ANS does not interact with membrane or chambers walls.
D
0(λ)). This means that 24 h is enough time to allow the dye to equilibrate between chambers # 1 and # 2 and that ANS does not interact with membrane or chambers walls.
Results and Discussion
The interaction of any ligand to receptor is characterized by several binding parameters: the number of binding modes, stoichiometry and binding constants of each binding mode. In the case when ligand binding sites on receptor are identical and independent of each other, the binding constant of the ligand to receptor (Kb) is determined by the ratio of the ligand – receptor complex concentration, i.e. concentration of bound ligand (Cb), to the product of the concentration of free binding sites of receptor (nCp – Cb) and concentration of free ligand (Cf):
| (2) |
where Cp is the receptor concentration and n is the number of ligand binding sites on the receptor. Consequently, nCp is the concentration of binding sites. The value of the binding constant, Kb, and the number of ligand binding sites on the receptor, n, can be determined on the basis of the experimental dependence of Cb on Cf.
The only assumption made in our work was the independence of binding sites. When processing in the frame of one mode failed to satisfy the experimental data we proposed the existence of two binding modes. The determined binding parameters of ANS-HSA and ANS-BSA interaction fit well this model. It should be noted that this model is an approximation and more binding modes can exist. The use of any more complicated model, e.g. Adair model [3], [29] which determines the sequential binding of ligands to acceptor, needs special justification.
1. Results that can be Obtained on the Basis of Equilibrium Microdialysis
The concentration of bound component can be determined by absorption spectrophotometry if the concentration the dye in reference solution equals to its concentration in sample solution. These solutions can be prepared by equilibrium microdialysis. Surprisingly, this approach, which is inherently designed for determining dye–receptor binding stoichiometry, has rarely been used, and only in a truncated form. The proposed approach permits to determine not only the concentration of bound and free dye but also the absorption spectra and molar extinction coefficients of dye bound to protein. We showed that fluorescence quantum yield of the dye bound to the sites of the different binding modes can be determined on the basis of the fluorescence intensity of the solutions prepared by equilibrium microdialysis.
1.1. Determination of the dependence of Cb on Cf
Equilibrium microdialysis implies allocation of two interacting agents, a ligand and receptor, in two chambers (#2 and #1, respectively). In our case the solution of protein with an initial concentration Cp, was placed in chamber #1, and the solution of ANS in the same buffer with an initial concentration C0 was placed in chamber #2. After equilibration, the concentrations of free dye in chambers #1 and #2 become equal (Cf), while the total ANS concentration in chamber #1 is greater than that in chamber #2 by the concentration of the bound dye (Cb), thus:
| (3) |
The initial concentration of ANS in chamber #2 (C0) and the dye concentration in this chamber after equilibration (Cf), which equals the concentration of free dye in chamber #1, can be determined by absorption spectrophotometry. The concentration of the dye bound to protein can be determined on the basis of eq. 3.
1.2. Determination of binding parameters: the number of binding modes, stoichiometry and binding constants
Equilibrium microdialysis was done for a large number of different initial ANS concentrations (C0) with subsequent spectrophotometric determination of the free (Cf) and on the basis of eq. 3 bound dye concentration (Cb). Traditionally these data are presented as Scatchard plot:
| (4) |
which allows graphical determination of binding parameters. The nonlinear character of Scatchard plots for ANS bound to HSA and BSA (Figure 1) indicate on the existence of more that one binding mode. If the binding sites are independent of each other, 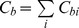 and equation similar to eq. 2 can be written for each mode:
and equation similar to eq. 2 can be written for each mode:
Figure 1. Scatchard plots for ANS interaction with BSA and HSA.

Experimental data (circles) and best fit curve with binding constants (Kbi) and number of binding sites (ni) given on the panels.
| (5) |
Then the dependences of Cb on Cf will be as follows:
| (6) |
This model gives adequate description of the experimental data for ANS binding to HSA and BSA at i = 2. The values of Kbi and ni were found as the best fit of eq. 6 using GraphPad Prism 5. The values of Kbi and ni are given in Figure 1 and in Table 1. The ANS binding constants of the sites with higher affinity is about 70 and 300 times greater than that of the sites with lower affinity for BSA and HSA, respectively. The binding constant of the mode with largest affinity for HSA is more than four times greater than that for BSA. The number of binding sites with high affinity are 4 and 6 for HSA and BSA, respectively.
Table 1. Characteristics of ANS – BSA and ANS – HSA interaction.
| protein | mode | λmax, nm | εi, max 10−3, M−1cm−1 | εi, 365 10−3, M−1cm−1 | Kbi 10−4, M−1 | n i | qi |
| BSA | 1 | 374.5±0.5 | 5.7±0.2 | 5.4±0.2 | 13.8±0.5 | 5.6±0.8 | 0.45±0.01 |
| 2 | 376.0±0.5 | 5.2±0.3 | 4.8±0.4 | 0.2±0.2 | 12.0±1.0 | 0.08±0.03 | |
| HSA | 1 | 373.5±0.5 | 5.5±0.1 | 5.3±0.1 | 61.2±0.7 | 4.0±0.1 | 0.42±0.01 |
| 2 | 374.0±0.5 | 5.6±0.2 | 5.3±0.2 | 0.2±0.1 | 10.0±0.2 | 0.07±0.01 |
1.3. Absorption spectrum of ANS bound to HSA and BSA
At equilibrium, the absorption spectrum of the solution in chamber #2 represents the absorption spectrum of free ANS at a concentration of Cf (Df (λ)), and the absorption spectrum of the solution in chamber #1 represents the superposition of the absorption spectra of free ANS (concentration Cf ) and ANS bound to protein at a concentration of Cb (Db(λ)). Thus, in chambers #1 and #2, we have a sample and optimal reference solutions for determination of the absorption spectrum of ANS bound to proteins. The analysis of these spectra (Figure 2) shows that the absorption spectrum of ANS bound to HSA and BSA are significantly red-shifted in comparison to that of free ANS. It is evident that the absorptioin spectra of ANS bound to HSA and BSA are complex. Apparently, it is determined by overlapping of two spectral bands.
Figure 2. Absorption spectra of ANS bound to BSA and HSA.

Curves 1 and 2 represent absorption spectra of ANS in chamber #2 (free ANS in concentration Cf) and in chamber #1 (superposition of absorption spectra of free ANS in concentration Cf, ANS bound to proteins in concentration Cb) after reaching equilibrium. Curve 3 is the absorption spectra of ANS bound to proteins. Curve 4 is the absorption spectra of free dye in a concentration equal to that of bound dye.
1.4. Molar extinction coefficient of ANS bound to HSA and BSA
In the case of one binding mode, the measured absorption spectrum can easily be presented in the units of the molar extinction coefficient (Lambert-Beer law). If there are two binding modes, then the concentrations of dye bound to the sites of each mode can be calculated on the basis of the Kd1, Kd2, n 1 and n 2 values:
| (7) |
Figure 3a shows the decomposition of 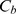 into two components,
into two components, 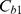 and
and 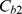 , for ANS-HSA and ANS-BSA interaction. Taking into account that.
, for ANS-HSA and ANS-BSA interaction. Taking into account that.
| (8) |
the values of 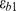 (λ) and
(λ) and 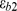 (λ) can be determined using the known values of
(λ) can be determined using the known values of  ,
, 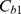 and
and 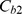 by multiple linear regression (e.g., using Graph Pad Prism 5). Figure 3b shows the relation between Db (at λ = λex = 365 nm) and Cb1 and Cb2 values that result in values of
by multiple linear regression (e.g., using Graph Pad Prism 5). Figure 3b shows the relation between Db (at λ = λex = 365 nm) and Cb1 and Cb2 values that result in values of 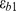 and
and 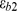 for ANS bound to HSA and BSA. Similarly, the values of
for ANS bound to HSA and BSA. Similarly, the values of 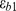 and
and 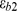 can be determined at the other wavelengths. Figure 3c shows the absorption spectra of the dye bound to the sites of each of the two modes of protein in units of the molar extinction coefficient. These data show that ANS binding to proteins is accompanied not only by the change of the position of its absorption spectra but by the change of its molar extinction coefficient, also.
can be determined at the other wavelengths. Figure 3c shows the absorption spectra of the dye bound to the sites of each of the two modes of protein in units of the molar extinction coefficient. These data show that ANS binding to proteins is accompanied not only by the change of the position of its absorption spectra but by the change of its molar extinction coefficient, also.
Figure 3. Determination of the molar extinction coefficient of ANS bound to BSA and HSA.

(a) Concentration of ANS bound to proteins (Cb) as a superposition of the concentrations of the dye bound to the sites of mode 1 (Cb1) and mode 2 (Cb2). (b) The dependence 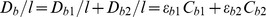 on Cb1, Cb2. In the panel experimental data, best-fit curve and the values of molar extinction coefficients εb1 and εb2 obtained by multiple nonlinear regression are presented. (c) Absorption spectra of ANS bound to the sites of mode 1 and mode 2 in the units of the molar extinction coefficient.
on Cb1, Cb2. In the panel experimental data, best-fit curve and the values of molar extinction coefficients εb1 and εb2 obtained by multiple nonlinear regression are presented. (c) Absorption spectra of ANS bound to the sites of mode 1 and mode 2 in the units of the molar extinction coefficient.
2. Can Equilibrium Microdialysis be Replaced by Fluorescence?
Frequently, the external dyes are used as a common tool to study the receptor’s structure. For example, fluorescent dye ANS is usually used for the study of the process of protein’s folding, detection partially folded proteins state known as molten globule and examining their properties [19], [20], [30], [31], and fluorescent dye thioflavin T is used for detection and study of amyloid fibrils [32], [33], [34], [35], [36]. These dyes do not fluoresce in the free state, more exactly have very low fluorescence quantum yield in free state, and form highly fluorescent complexes while binding to receptor. That is why it seems natural to use fluorescence of bound dyes for determination of their binding parameters. Many researchers being not specialists in fluorescence assumed that fluorescence intensity as a function of dye concentration reaches a plateau when all of the binding sites are occupied. In reality, the dependence of fluorescence intensity on dye concentration is the curve with saturation for any (even unbound) fluorophore and it is linear only in the limit when dye concentration tend to zero (Figure 4). Furthermore, the researchers did not take into account that fluorescence intensity depends not only on bound fluorophore concentration but also on its fluorescence quantum yield and molar extinction coefficient which usually differ from that of free dye.
Figure 4. The dependences of fluorescence intensity of quinine sulfate (QS) on its optical density.
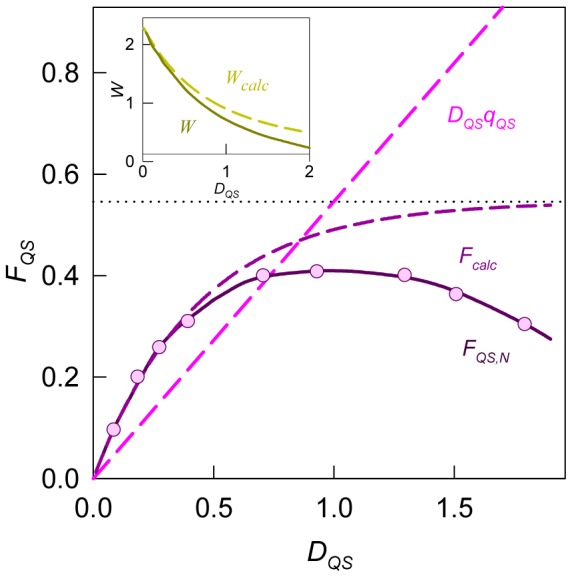
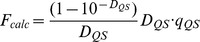 are calculated values and
are calculated values and 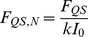 are experimentally recorded and normalized values of fluorescence intensity of QS. Dots are the limiting value of
are experimentally recorded and normalized values of fluorescence intensity of QS. Dots are the limiting value of 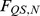 at
at 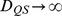 . The dependences of calculated values
. The dependences of calculated values 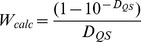 and experimentally determined values
and experimentally determined values 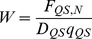 on total optical density are given in the Insert. Strait line is the dependence of
on total optical density are given in the Insert. Strait line is the dependence of 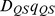 on
on 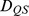 (
(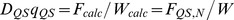 ).
).
2.1. Dependence of the fluorescence intensity on the optical density of the fluorescence substance and on the total optical density of the solution
Any ligand–receptor solution contains ligand molecules both free and bound to receptor. So the solution of ANS in the presence of serum albumins is a two-component system in which one component (free dye unbound to protein) absorbs the excitation light (its optical density is Df) but does not fluoresce, while the other component (dye bound to protein) absorbs the excitation light (its optical density is Db) and fluoresces with a quantum yield qb. In this case the fluorescence intensity of solution FANS, excited by light with fluorescence intensity I0 can be presented as follows:
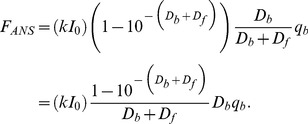 |
(9) |
Here k is the proportionality coefficient. If protein has several binding sites of ANS which are independent of each other but can be divided into several groups (binding modes) which differ by the value of binding constants (K bi), binding stoichiometry (ni) or by the properties of bound dye (absorption spectra D bi(λ), molar extinction coefficients (εi), fluorescent quantum yields (q bi)) then eq. 9 will be as follows:
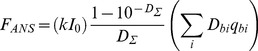 |
(10) |
Here 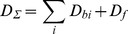 is total optical density of solution. The fluorescence intensity of a single-component solution can be presented in the same manner. In particular, for the fluorescent dye quinine sulfate (QS), which was used in this work as a standard substance with known quantum yield, the fluorescence intensity is:
is total optical density of solution. The fluorescence intensity of a single-component solution can be presented in the same manner. In particular, for the fluorescent dye quinine sulfate (QS), which was used in this work as a standard substance with known quantum yield, the fluorescence intensity is:
| (11) |
Thus in the equations 9–11 for fluorescence intensity three factors can be marked out:
the factor kI0 depends only on the device used in the experiment. It is determined by the intensity of the excitation light, the wavelength of excitation and registration of fluorescence, the spectral slits width of the monochromators in the excitation and registration pathways, sensitivity of photodetector and signal amplification.
the factor
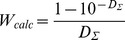 is determined by total optical density (DΣ) of solution (at λ = λex) and does not depend on the contribution in absorption of fluorescent and non-fluorescent components of solution. The calculated dependence of this factor on the optical density of QS solution (DQS = DΣ) is given in Figure 4, Insert.
is determined by total optical density (DΣ) of solution (at λ = λex) and does not depend on the contribution in absorption of fluorescent and non-fluorescent components of solution. The calculated dependence of this factor on the optical density of QS solution (DQS = DΣ) is given in Figure 4, Insert.
The experimental dependence of fluorescence intensity on the optical density of the fluorescent substance can differ from the calculated one (Figure 4). The fact that the recorded fluorescence intensity, after reaching some value of optical density, begins to decrease with an increase in the content of the fluorescent substance is a general property of such dependences and does not indicate the existence of self-quenching or dye aggregation, as it has been frequently suggested (see e.g. [37], [38], [39]). In reality, this effect is determined by an increased absorption of excitation light by the solution layers adjacent to the front wall of the spectrofluorimeter cell on the increase in the total optical density of the solution. The detection system of the spectrofluorimeter “sees” only the central part of the cell, which is reached by a respectively smaller amount of excitation light. Because of this effect, the recorded fluorescence intensity begins to diminish after the optical density reaches a certain value. The effects discussed above depend on the particular instrument used in the experiment and must be taken into account. This can be performed by replacing Wcalc with an experimentally determined value W. The measurement of QS fluorescence intensity at different optical densities gives an opportunity to determine the dependence of W on total optical density (Figure 4, Insert) and to choose the normalization coefficient kI 0 in order to correct the dependence of fluorescence intensity on the total optical density of solution and to normalize it in the units of the product of optical density and fluorescence quantum yield.
In fact the value of kI 0 must be chosen so that W → W calc at D QS → 0 and normalized and corrected fluorescence intensity of QS (FQS, 0) would equal to:
| (12) |
the third factor is the product of optical density and quantum yield of the fluorescent component in the case of one binding mode or the sum of the products in the case of several binding modes. Only this factor is an informative element which bears information about the properties of fluorescent component (or fluorescent components) in solution. In this case eqs 9 and 10 for normalized and corrected fluorescence intensities (FANS, 0) will be as follows:
| (13) |
or
| (14) |
2.2. Determination of the fluorescence quantum yield of the dye bound to proteins
If all binding sites of ligand to receptor are identical and independent of each other then fluorescence quantum yield is determined as a slope of the dependence 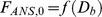 (see eq. 13). In the case of two binding modes, a relation similar to eq. (14) is valid. To determine the values of qb1 and qb2, it is necessary to know the set of three related values
(see eq. 13). In the case of two binding modes, a relation similar to eq. (14) is valid. To determine the values of qb1 and qb2, it is necessary to know the set of three related values  ,
, 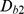 and
and 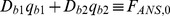 , that correspond to one microdialysis experiment. The dependences of the values of
, that correspond to one microdialysis experiment. The dependences of the values of 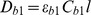 and
and 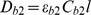 on Db can be determined on the basis of the values of the molar extinction coefficients and concentrations of ANS molecules bound to the sites of two binding modes for the samples obtained after microdialysis (Figure 5a). The dependence of the values
on Db can be determined on the basis of the values of the molar extinction coefficients and concentrations of ANS molecules bound to the sites of two binding modes for the samples obtained after microdialysis (Figure 5a). The dependence of the values 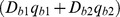 on Db1 and Db2 can be determined from eq. 14. On the basis of these data, the fluorescence quantum yields of the dye bound to the sites of each binding mode can be determined using multiple linear regression (Figure 5b, Table 1).
on Db1 and Db2 can be determined from eq. 14. On the basis of these data, the fluorescence quantum yields of the dye bound to the sites of each binding mode can be determined using multiple linear regression (Figure 5b, Table 1).
Figure 5. Determination of the fluorescence quantum yield of ANS bound to BSA and HSA.

(a) Optical density of ANS bound to proteins as a superposition of optical densities of the dye bound to the sites of mode 1 (Db1) and mode 2 (Db2). (b) 3D dependence of 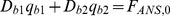 on Db1 and Db2. Experimental data, best-fit curve and the values of qb1 and qb2, determined by multiple non-linear regression are presented.
on Db1 and Db2. Experimental data, best-fit curve and the values of qb1 and qb2, determined by multiple non-linear regression are presented.
Conclusion
In this work two significantly different binding modes were determined for ANS binding to serum albumins. The ANS binding constant of the sites with higher affinity is about 70 and 300 times greater than that with lower affinity for BSA and HSA, respectively. Fluorescence quantum yields of ANS bound with higher affinity are about 0.45 and 0.42 and that with lower affinity are about 0.08 and 0.07 for BSA and HSA. It could be thought that interaction with low affinity has nonspecific character [40] but even lower quantum yield is 20 times greater than that of free ANS in buffer solution (0.0032 [41]). This can be interpreted as the existence of several hydrophobic pockets on the surface of serum albumins which bind ANS with high affinity and numerous binding sites on the surface of proteins, perhaps, formed by hydrophobic clusters and/or charge groups which bind ANS with low affinity. The possibility of both types of ANS interaction with proteins can be found in literature [3], [4], [17], [23], [42], [43], [44], [45]. Since the first work of Weber, ANS was assigned to hydrophobic probes [17]. At the same time ANS molecule being negatively charged strongly binds to cationc groups of polyamino acids. [22] The existence of negative or positive charge groups on the surface of protein or in its hydrophobic pockets can prevent or promote its binding to protein [30], [44], [46]. ANS ability to bind hydrophobic clusters on the surface of the protein in the molten globule state make it one of the main probes for the appearance of such state [19], [45]. According to our views, ANS binds to the hydrophobic pockets in aggregates formed by proteins in molten globule state rather than with hydrophobic clusters on the surface of protein in molten globule state [20]. ANS can intercalate in the hydrophobic pockets of multidomain and/or multimeric proteins [31]. Aggregation of such proteins in the molten globule state results in the formation of new hydrophobic pockets between protein molecules that leads to the increase in number of bound ANS molecules [20], [31].
Though, there are numerous works on the investigation of ANS binding to serum albumins, yet there are no consensus about the number of binding modes, binding sites and values of binding constants [3], [12], [13], [24], [25], [42], [44], [47], [48], [49]. This is caused by significant difficulties of the binding parameters determination. Togashi and Ryder pointed that the broad range of the values of the reported binding parameters is caused by the use of different assumption and models of dye – protein interaction, the absence of the method for estimating the free ligand concentration and overlooking the true dependence of fluorescence intensity on total optical density of solution (overlooking of the inner filter effect) [3].
In our work we for the first time proposed correct estimation of the influence of the total optical density of fluorescent dye solution (free and bound dye) on its fluorescence. We showed that total fluorescence intensity is determined by the factor that is a function of total optical density of the solution only and does not depend on the relative contributions of the optical densities of the fluorescent and non-fluorescent components, and by the other factor that is a product of the optical density and the quantum yield of the fluorescent component. But even corrected fluorescence intensity is determined not only by the concentration of fluorescence dye, but also by its molar extinction coefficient and fluorescence quantum yield of bound dye that are not known and usually differ from that of free dye. For correct determination of the concentration of bound and free dye we proposed to use equilibrium microdialysis. The use of equilibrium microdialysis gave us the opportunity to determine not only the values of binding stoichiometry and binding constants but also for the first time to register absorption spectrum of bound dye and to determine molar extinction coefficient and fluorescence quantum yield of ANS bound to sites of different binding mode.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported in part by the Program “Molecular and Cell Biology” of the Russian Academy of Sciences and Dmitry Zimin’s Russian Charitable Foundation “Dynasty” (A.I.S.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Groenning M. 2009. Binding mode of Thioflavin T and other molecular probes in the context of amyloid fibrils-current status. J Chem Biol: DOI 10.1007/s12154-2009-0027–12155 [DOI] [PMC free article] [PubMed]
- 2.Oravcova J, Bohs B, Lindner W. Drug-protein binding sites. New trends in analytical and experimental methodology. J Chromatogr B Biomed Appl. 1996;677:1–28. doi: 10.1016/0378-4347(95)00425-4. [DOI] [PubMed] [Google Scholar]
- 3.Togashi DM, Ryder AG. A fluorescence analysis of ANS bound to bovine serum albumin: binding properties revisited by using energy transfer. J Fluoresc. 2008;18:519–526. doi: 10.1007/s10895-007-0294-x. [DOI] [PubMed] [Google Scholar]
- 4.Togashi DM, Ryder AG. Assessing protein-surface interactions with a series of multi-labeled BSA using fluorescence lifetime microscopy and Forster Energy Resonance Transfer. Biophys Chem. 2010;152:55–64. doi: 10.1016/j.bpc.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 5.Jain TK, Varshney M, Maitra A. Structural Studies of Aerosol OT Reverse Micellar Aggregates by FT-IR Spectroscopy. J Phys Chem. 1989;93:7409–7416. [Google Scholar]
- 6.Ivy MA, Gallagher LT, Ellington AD, Anslyn EV. Exploration of plasticizer and plastic explosive detection and differentiation with serum albumin cross-reactive arrays. Chemical Science. 2012;3:1773–1779. [Google Scholar]
- 7.Hawe A, Sutter M, Jiskoot W. Extrinsic fluorescent dyes as tools for protein characterization. Pharm Res. 2008;25:1487–1499. doi: 10.1007/s11095-007-9516-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.LeVine H, 3rd. Thioflavine T interaction with amyloid-sheet structures. Amyloid: Int J Exp Clin Invest. 1995;2:1–6. [Google Scholar]
- 9.Groenning M, Norrman M, Flink JM, van de Weert M, Bukrinsky JT, et al. Binding mode of Thioflavin T in insulin amyloid fibrils. J Struct Biol. 2007;159:483–497. doi: 10.1016/j.jsb.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 10.Panjehshahin MR, Bowmer CJ, Yates MS. A pitfall in the use of double-reciprocal plots to estimate the intrinsic molar fluorescence of ligands bound to albumin. Biochem Pharmacol. 1989;38:155–159. doi: 10.1016/0006-2952(89)90162-7. [DOI] [PubMed] [Google Scholar]
- 11.Sen P, Fatima S, Ahmad B, Khan RH. Interactions of thioflavin T with serum albumins: spectroscopic analyses. Spectrochim Acta A Mol Biomol Spectrosc. 2009;74:94–99. doi: 10.1016/j.saa.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 12.Bagatolli LA, Kivatinitz SC, Fidelio GD. Interaction of small ligands with human serum albumin IIIA subdomain. How to determine the affinity constant using an easy steady state fluorescent method. J Pharm Sci. 1996;85:1131–1132. doi: 10.1021/js960142k. [DOI] [PubMed] [Google Scholar]
- 13.Naik DV, Paul WL, Schulman SG. Fluorometric determination of drug-protein association constants: binding of pamaquine by bovine serum albumin. J Pharm Sci. 1975;64:1677–1680. doi: 10.1002/jps.2600641020. [DOI] [PubMed] [Google Scholar]
- 14.Kubarych CJ, Adams MM, Anslyn EV. Serum Albumins as Differential Receptors for the Discrimination of Fatty Acids and Oils. Organic Letters. 2010;12:4780–4783. doi: 10.1021/ol101906g. [DOI] [PubMed] [Google Scholar]
- 15.Adams MM, Anslyn EV. Differential Sensing Using Proteins: Exploiting the Cross-Reactivity of Serum Albumin To Pattern Individual Terpenes and Terpenes in Perfume. Journal of the American Chemical Society. 2009;131:17068–17069. doi: 10.1021/ja908319m. [DOI] [PubMed] [Google Scholar]
- 16.Morimoto K, Kawabata K, Kunii S, Hamano K, Saito T, et al. Characterization of type I collagen fibril formation using thioflavin T fluorescent dye. J Biochem. 2009;145:677–684. doi: 10.1093/jb/mvp025. [DOI] [PubMed] [Google Scholar]
- 17.Weber G. Polarization of the fluorescence of macromolecules. II. Fluorescent conjugates of ovalbumin and bovine serum albumin. Biochem J. 1952;51:155–167. doi: 10.1042/bj0510155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Slavik J. Anilinonaphthalene sulfonate as a probe of membrane composition and function. Biochim Biophys Acta. 1982;694:1–25. doi: 10.1016/0304-4157(82)90012-0. [DOI] [PubMed] [Google Scholar]
- 19.Semisotnov GV, Rodionova NA, Razgulyaev OI, Uversky VN, Gripas AF, et al. Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers. 1991;31:119–128. doi: 10.1002/bip.360310111. [DOI] [PubMed] [Google Scholar]
- 20.Povarova OI, Kuznetsova IM, Turoverov KK. Differences in the pathways of proteins unfolding induced by urea and guanidine hydrochloride: molten globule state and aggregates. PLoS One. 2010;5:e15035. doi: 10.1371/journal.pone.0015035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anufrieva EV, Nekrasova TN, Sheveleva TV, Krakovyak MG. [Structure and structural transformations of macromolecules water-soluble polymers and luminescence of magnesium salt of 8-anilinenaphthalene-1-sulfonic acid.] Vysokomol Soed 36 449–456. 1994.
- 22.Gasymov OK, Glasgow BJ. ANS fluorescence: potential to augment the identification of the external binding sites of proteins. Biochim Biophys Acta. 2007;1774:403–411. doi: 10.1016/j.bbapap.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schonbrunn E, Eschenburg S, Luger K, Kabsch W, Amrhein N. Structural basis for the interaction of the fluorescence probe 8-anilino-1-naphthalene sulfonate (ANS) with the antibiotic target MurA. Proc Natl Acad Sci U S A. 2000;97:6345–6349. doi: 10.1073/pnas.120120397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cardamone M, Puri NK. Spectrofluorimetric assessment of the surface hydrophobicity of proteins. Biochem J 282 (Pt. 1992;2):589–593. doi: 10.1042/bj2820589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takehara K, Yuki K, Shirasawa M, Yamasaki S, Yamada S. Binding properties of hydrophobic molecules to human serum albumin studied by fluorescence titration. Anal Sci. 2009;25:115–120. doi: 10.2116/analsci.25.115. [DOI] [PubMed] [Google Scholar]
- 26.Sudlow G, Birkett DJ, Wade DN. The characterization of two specific drug binding sites on human serum albumin. Mol Pharmacol. 1975;11:824–832. [PubMed] [Google Scholar]
- 27.Peters T. All about albumin: biochemistry, genetics, and medical application New York: Academic Press. 432 p. 1996.
- 28.Turoverov KK, Biktashev AG, Dorofeiuk AV, Kuznetsova IM. [A complex of apparatus and programs for the measurement of spectral, polarization and kinetic characteristics of fluorescence in solution]. Tsitologiia. 1998;40:806–817. [PubMed] [Google Scholar]
- 29.Konkoli Z. Safe uses of Hill’s model: an exact comparison with the Adair-Klotz model. Theor Biol Med Model. 2011;8:1–17. doi: 10.1186/1742-4682-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Collini M, D’Alfonso L, Baldini G. New insight on beta-lactoglobulin binding sites by 1-anilinonaphthalene-8-sulfonate fluorescence decay. Protein Sci. 2000;9:1968–1974. doi: 10.1110/ps.9.10.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fan YX, Zhou JM, Kihara H, Tsou CL. Unfolding and refolding of dimeric creatine kinase equilibrium and kinetic studies. Protein Sci. 1998;7:2631–2641. doi: 10.1002/pro.5560071217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naiki H, Higuchi K, Hosokawa M, Takeda T. Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavin T1. Anal Biochem. 1989;177:244–249. doi: 10.1016/0003-2697(89)90046-8. [DOI] [PubMed] [Google Scholar]
- 33.LeVine H, 3rd. Thioflavine T interaction with synthetic Alzheimer’s disease beta-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 1993;2:404–410. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Voropay ES, Samtsov MP, Kaplevsky KN, Maskevich AA, Stepuro VI, et al. Spectral properties of Thioflavin T and its complexes with amyloid fibrils. J Appl Spectrosc. 2003;70:868–874. [Google Scholar]
- 35.Sulatskaya AI, Kuznetsova IM, Turoverov KK. Interaction of tеhioflavin T with amyloid fibril: fluorescence quantum yield of bound dye.. J Phys Chem B. 2012;116:2538–2544. doi: 10.1021/jp2083055. [DOI] [PubMed] [Google Scholar]
- 36.Sulatskaya AI, Kuznetsova IM, Turoverov KK. The interaction of tеhioflavin T with amyloid fibril: stoichiometry and affinity of dye binding, absorption spectra of bound dye.. J Phys Chem B. 2011;115:11519–11524. doi: 10.1021/jp207118x. [DOI] [PubMed] [Google Scholar]
- 37.Dzwolak W, Pecul M. Chiral bias of amyloid fibrils revealed by the twisted conformation of Thioflavin T: an induced circular dichroism/DFT study. FEBS Lett. 2005;579:6601–6603. doi: 10.1016/j.febslet.2005.10.048. [DOI] [PubMed] [Google Scholar]
- 38.Uversky VN, Winter S, Lober G. Use of fluorescence decay times of 8-ANS-protein complexes to study the conformational transitions in proteins which unfold through the molten globule state. Biophys Chem. 1996;60:79–88. doi: 10.1016/0301-4622(96)00009-9. [DOI] [PubMed] [Google Scholar]
- 39.Uversky VN, Winter S, Lober G. Self-association of 8-anilino-1-naphthalene-sulfonate molecules: spectroscopic characterization and application to the investigation of protein folding. Biochim Biophys Acta. 1998;1388:133–142. doi: 10.1016/s0167-4838(98)00173-3. [DOI] [PubMed] [Google Scholar]
- 40.Kurganov BI. Analysis of negative cooperativity for glutamate dehydrogenase. Biophys Chem. 2000;87:185–199. doi: 10.1016/s0301-4622(00)00193-9. [DOI] [PubMed] [Google Scholar]
- 41.Lee AG, Rogers J, Wilton DC, Ghiggino KP, Phillips D. Spectroscopic resolution of drug binding sites in biological membranes. FEBS Lett. 1978;94:171–174. doi: 10.1016/0014-5793(78)80930-2. [DOI] [PubMed] [Google Scholar]
- 42.Weber G, Young LB. Fragmentation of Bovine Serum Albumin by Pepsin. I. the Origin of the Acid Expansion of the Albumin Molecule. J Biol Chem. 1964;239:1415–1423. [PubMed] [Google Scholar]
- 43.Stryer L. The interaction of a naphthalene dye with apomyoglobin and apohemoglobin. A fluorescent probe of non-polar binding sites. J Mol Biol. 1965;13:482–495. doi: 10.1016/s0022-2836(65)80111-5. [DOI] [PubMed] [Google Scholar]
- 44.Matulis D, Lovrien R. 1-Anilino-8-naphthalene sulfonate anion-protein binding depends primarily on ion pair formation. Biophys J. 1998;74:422–429. doi: 10.1016/S0006-3495(98)77799-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ptitsyn OB. Molten globule and protein folding. Adv Protein Chem. 1995;47:83–229. doi: 10.1016/s0065-3233(08)60546-x. [DOI] [PubMed] [Google Scholar]
- 46.Bismuto E, Gratton E, Lamb DC. Dynamics of ANS binding to tuna apomyoglobin measured with fluorescence correlation spectroscopy. Biophys J. 2001;81:3510–3521. doi: 10.1016/S0006-3495(01)75982-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Georgiou ME, Georgiou CA, Koupparis MA. Automated flow injection gradient technique for binding studies of micromolecules to proteins using potentiometric sensors: application to bovine serum albumin with anilinonaphthalenesulfonate probe and drugs. Anal Chem. 1999;71:2541–2550. doi: 10.1021/ac981019b. [DOI] [PubMed] [Google Scholar]
- 48.Hazra P, Chakrabarty D, Chakraborty A, Sarkar N. Probing protein-surfactant interaction by steady state and time-resolved fluorescence spectroscopy. Biochem Biophys Res Commun. 2004;314:543–549. doi: 10.1016/j.bbrc.2003.12.118. [DOI] [PubMed] [Google Scholar]
- 49.Suarez Varela A, Sandez Macho MI, Minones J. Spectrofluorimetric study of the binding of 1-anilinonaphthalene-8-sulfonate to bovine serum albumin. J Pharm Sci. 1992;81:842–844. doi: 10.1002/jps.2600810825. [DOI] [PubMed] [Google Scholar]