

Abstract
We surveyed gene–gene interactions (epistasis) in human body mass index (BMI) in four European populations (n<1200) via exhaustive pair-wise genome scans where interactions were computed as F ratios by testing a linear regression model fitting two single-nucleotide polymorphisms (SNPs) with interactions against the one without. Before the association tests, BMI was corrected for sex and age, normalised and adjusted for relatedness. Neither single SNPs nor SNP interactions were genome-wide significant in either cohort based on the consensus threshold (P=5.0E−08) and a Bonferroni corrected threshold (P=1.1E−12), respectively. Next we compared sub genome-wide significant SNP interactions (P<5.0E−08) across cohorts to identify common epistatic signals, where SNPs were annotated to genes to test for gene ontology (GO) enrichment. Among the epistatic genes contributing to the commonly enriched GO terms, 19 were shared across study cohorts of which 15 are previously published genome-wide association loci, including CDH13 (cadherin 13) associated with height and SORCS2 (sortilin-related VPS10 domain containing receptor 2) associated with circulating insulin-like growth factor 1 and binding protein 3. Interactions between the 19 shared epistatic genes and those involving BMI candidate loci (P<5.0E−08) were tested across cohorts and found eight replicated at the SNP level (P<0.05) in at least one cohort, which were further tested and showed limited replication in a separate European population (n>5000). We conclude that genome-wide analysis of epistasis in multiple populations is an effective approach to provide new insights into the genetic regulation of BMI but requires additional efforts to confirm the findings.
Keywords: body mass index, BMI, gene interaction, epistasis, pair-wise genome scan
Introduction
Body mass index (BMI) is the most commonly used anthropometric method to define human obesity. BMI is a complex trait affected by many environmental (eg, diet, age, physical activity) and genetic factors, with heritability estimates that vary from 40–80% in twin studies, 20–50% in family studies and 20–60% in adoption studies.1 Recent genome-wide association (GWA) studies have successfully identified numerous single-nucleotide polymorphisms (SNPs) that are robustly associated with obesity related traits, including BMI.2, 3, 4 They shed light on the biological basis of obesity and suggest a role for neuronal influences on the regulation of appetite and/or energy balance. However, the identified genetic variants jointly explained only a small proportion of the trait variation and thus had limited predictive value for obesity risk.5 For example, in a recent meta-analysis (249 796 individuals) 32 identified and replicated SNPs together explained only 1.45% of the inter-individual variation in BMI where the strongest SNP accounted for just 0.34% of the variance.3 The 32 BMI SNPs map to 32 different genes that are referred to as BMI loci hereafter.
Gene–gene interactions (epistasis) are thought to be potential sources of the unexplained genetic variation,6, 7, 8 but they remain largely unexplored in the GWA studies conducted for BMI so far. A major hurdle for analysing epistasis in GWA studies was the lack of fast methods to enumerate billions of interaction tests in a full pair-wise genome scan to map different types of epistasis (eg, with or without main effects) while keeping false-positive rates under control.9, 10 Another hurdle for studying epistasis is the relatively small sample size in many existing GWA cohorts that may limit the power of detection and replication of epistasis signals unless the epistatic effects to be detected are large.11, 12 It was showed in simulation that more than 4000 case–control pairs were needed to achieve 80% power of detection of epistasis with an odds ratio of 3.0 in complex diseases.13 For quantitative traits, sample sizes need to be substantially (eg, 45%) larger than case–control phenotypes to achieve a similar power.14
With the advances in computing technologies, the major hurdle is gradually easing and full pair-wise genome scans are beginning to be applied to GWA populations individually.15, 16 Meta-analysis of epistasis as applied in GWA studies3 could be a good way to overcome the sample size hurdle but requires new methods to accommodate imputed SNP genotype data. Various approaches in search space reduction (ie, less stringent significance thresholds as result of the much reduced number of tests) can be applied to improve the power of detection of epistasis in individual GWA populations.11 Testing interactions involving genome-wide significant loci (of marginal effects) with a threshold corrected for the actual number of tests has been suggested10, 17, 18 and applied successfully in recent studies.16, 19, 20, 21 Another approach is to select SNPs based on existing biological knowledge (eg, protein–protein interactions) and test interactions among them only.22, 23 However, cautions should be taken when making the SNP selection12 because biological knowledge may not be directly related to the trait studied and any biases in the pre-identified loci could lead to false-positive epistatic signals.
Here we demonstrate a different approach to exploit the value of genome-wide analysis of epistasis using multiple populations. First we performed full pair-wise genome scans for epistasis in BMI in four GWA populations to which we had direct access: the Scottish ORCADES,24 the CROATIA-Vis25 and CROATIA-Korcula,26 and the Italian MICROS27 study cohorts. Each of these cohorts has a relatively small sample size and is sampled from distinct European regions with widely differing lifestyles and diets. Second, we identified common and potentially important gene–gene interactions using the epistasis signals uncovered in each cohort and their gene ontology (GO) enrichment across populations. In addition, we also identified a set of interactions involving the BMI loci (as prior knowledge) in different cohorts. Third, we tested the identified interactions in each cohort for replication and then the replicated signals in the Northern Finland Birth Cohort 1966 (NFBC1966).28 We aim to address the question whether epistasis analysis is of value for the dissection of the genetic regulation of BMI in these study cohorts.
Materials and methods
Study cohorts and ethics statement
The four study cohorts have been described in detail elsewhere.24, 25, 26, 27, 29 Briefly, the Scottish ORCADES cohort was recruited from a subgroup of 10 islands of the archipelago of Orkney. This study was approved by the NHS Orkney Research Ethics Committee and the North of Scotland REC. The CROATIA-Vis and CROATIA-Korcula cohorts were recruited from the island of Vis and the island of Korcula, respectively. Both studies were approved by the Ethical Committee of the Medical School, University of Zagreb and the Multi-Centre Research Ethics Committee for Scotland. The Italian MICROS cohort was recruited from villages in an isolated highland area of the South Tyrol. The study was approved by the ethical committee of the Autonomous Province of Bolzano. All participants gave written informed consent and were measured for a number of traits, including weight and height from which BMI values were calculated.
DNA samples were genotyped with Illumina Infinium HumanHap300v1/v2 (for CROATIA-Vis by the Wellcome Trust Clinical Facility in Einburgh, UK) or HumanCNV370v1 SNP bead microarrays (for CROATIA-Korcula, ORCADES and MICROS by the Helmholtz Zentrum Munchen in Munich, Germany) and analysed using the BeadStudio software (Illumina). Quality control of the genotype data was performed for each cohort using the R/GenABEL package (Version 1.6-7)30 based on a common set of criteria: individual call rate at 97%, SNP call rate at 95%, P-value for deviation from Hardy–Weinberg equilibrium at 1.0e-10, minor allele frequency at 2%. The NFBC1966 data were provided by the database of Genotype and Phenotype (dbGaP) via specific Data Use Certification and used as the replication cohort. NFBC1966 includes nearly all individuals born in 1966 in the two northernmost Finnish provinces that were genotyped with HumanCNV370v1 SNP bead microarrays28 and was put through the same quality control procedure as above. The summary information of each cohort after quality control and excluding individuals without BMI or age records or with extremely high BMI (ie, BMI>50 kg/m2) is given in Table 1.
Table 1. Summary information of uncorrected BMI (kg/m2)a.
| Vis | Korcula | ORCADES | MICROS | NFBC1966 | |
|---|---|---|---|---|---|
| N | 901 | 880 | 695 | 1177 | 5071 |
| Male/female ratio | 0.74 | 0.56 | 0.87 | 0.78 | 1.0 |
| Age range (years) | 18–91 | 18–98 | 18–92 | 18–88 | 31 |
| Number of SNPs | 300265 | 307712 | 309202 | 293913 | 323697 |
| BMI median | 27.10 | 27.70 | 27.08 | 24.89 | 24.00 |
| BMI mean±SD | 27.30±4.19 | 27.93±4.05 | 27.76±4.86 | 25.51±4.57 | 24.70±4.24 |
| BMI heritability | 0.356 | 0.399 | 0.514 | 0.450 | 0. 216 |
| GWA lambda | 1.001 | 0.999 | 0.997 | 0.996 | 1.005 |
Vis and Korcula represent CROATIA-Vis and CROATIA-Korcula, respectively.
Statistical analysis
The raw BMI data in each of the four study cohorts were corrected for age and sex and normalised using the rntransform function that is implemented in the GenABEL package performing quantile normalisation of residuals from a generalised linear model analysis. The normalised BMI residuals were then analysed using a linear mixed model to correct for polygenic effects due to relatedness using the polygenic function in the GenABEL package and the resultant environmental residuals (ie, pgresidualY in GenABEL) were used as the trait to test for association.31 The polygenic heritability was estimated at the mixed-model step. Following the original GWA study,28 in the NFBC1966 cohort individuals with pregnancy and/or self reported BMI measures were excluded, and the raw BMI values were corrected for the SexOCPG factor (recoded according to gender, status of taking oral contraception and pregnancy) and then normalised and corrected for relatedness as above.
A single-SNP based GWA scan was performed in each population using a score test method (based on the additive model) implemented in the mmscore function in the GenABEL package. The consensus GWA threshold of 7.3 (−log10(5.0E−08)) was applied to identify GWA significant SNPs.32 We also performed a full pair-wise genome scan using the regression models described below. Considering a pair of SNPs denoted as SNP1 and SNP2, the following genetic models were used to detect epistasis where genotypes of each SNP (ie, homozygote of the minor allele, homozygote of the major allele and heterozygote) were fitted as fixed factors:
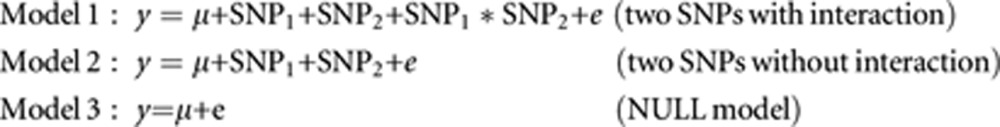 |
where y is the trait of interest, μ is the model constant, SNP1 (or SNP2) is a fixed factor with three levels (genotype classes), SNP1*SNP2 is the interaction term, e is the random error term. The F ratio test of Model 1 against Model 3 evaluates the whole pair effect, including interaction (ie, Fpair, 8 degrees of freedom). The F ratio test of Model 1 against Model 2 evaluates the interaction between the two SNPs (ie, Fint, 4 degrees of freedom). P-values were calculated based on the F distribution with relevant degrees of freedom and transformed to the −log10 scale (ie, −log10 Ppair for the Fpair test, −log10 Pint for the Fint test). We were concerned mainly about the Fint tests in this study.
Genome-wide significant thresholds (all in the −log10 scale) were derived based on Bonferroni correction for multiple tests, that is, the 5% nominal P value corrected by the number of tests performed. Considering 300 000 SNPs, a full pair-wise genome scan perform 4.5E+10 association tests and thus the 5% genome-wide threshold is 11.95 (ie, −log10(0.05/4.5E+10)). After each pair-wise genome scan, results were evaluated using the predefined threshold to identify genome-wide significant interaction signals. Each SNP in the results was annotated to the nearest gene within a window of 20 kilobases flanking the SNP (based on the physical distance to either the start or end of transcription of a gene; the distance is considered as zero if the SNP is within a gene).
A GO enrichment analysis was conducted for each study cohort using the running mode of ‘Two unranked lists of genes' in Gorilla33 based on the standard Hyper Geometric statistics, where the annotated epistatic genes were used as the target with the full list of human genes as the background. For simplicity, we chose to use the same −log10 P value as the consensus GWA threshold (ie, −log10 Pint >7.3) to select SNP pairs of each cohort and used their gene annotations as the input for the GO enrichment analysis. The GO terms enriched (P<1.0E−03) were compared across study cohorts to identify firstly common GO terms and then their member genes shared by the cohorts. The shared epistatic genes were examined further for biological functions via literature mining and their associated interactions in the retained results of each cohort to identify potentially important interactions for replication tests. The BMI loci involved SNP pairs (−log10 Pint >7.3) in each study cohort were also identified as potentially important interaction signals for replication tests.
Genome-wide significant SNP pairs and those identified as potentially important interactions were tested for replication across the four study cohorts. The replicated SNP pairs were further tested for replication in the NFBC1966 cohort. Each replication test was done at both the SNP and region levels. At the SNP level, each replicated SNP was exactly the same as the corresponding epistatic SNP and thus the 5% nominal significance threshold (ie, −log10(0.05)=1.30) was used because only one replication test was needed. At the region level, interactions between each of 10 adjacent SNPs (ie, five upstream and five downstream) of the first epistatic SNP and each of those of the second were tested, to accommodate the situation where multiple SNPs may tag a same mutant of a gene. Permutation was used to derive significance thresholds for replication of each epistatic pair at the region level, where phenotypes were permuted and the highest −log10 Pint value of 121 (ie, 11 × 11) interaction tests was recorded in each of 1000 iterations. The replicated SNP pairs were fitted together into the full model to calculate the proportion of phenotypic variance explained in each study cohort.
Results
The mean BMI was similar across the CROATIA-Vis, CROATIA-Korcula and ORCADES cohorts but lower in MICROS (Table 1). The polygenic heritability estimates varied from 0.356 (CROATIA-Vis) to 0.514 (ORCADES). Conventional GWA scans found no genome-wide significant SNPs in any single cohort. The inflation factor lambda (computed by regression of observed association P-values against the expected) of each GWA scan was very close to 1 (Table 1), suggesting the family relatedness in each cohort was well accounted for. Only 8 out of the 32 BMI SNPs previously identified3 were genotyped in the four study cohorts and none of these showed a strong association with BMI (Supplementary Table S1).
Full pair-wise genome scans found no SNP pairs that passed the genome-wide threshold (−log10 Pint=11.95) in any of the four study cohorts (Figure 1). Considering interaction signals with −log10 Pint >7.3, MICROS had the least number of SNP pairs and consequently the least number of annotated genes, whereas the remaining three cohorts had relatively similar numbers of SNP pairs and annotated genes (Table 2). Five out of the 32 BMI loci (but not the BMI SNPs) were involved in 7 epistatic pairs in CROATIA-Vis: FTO, KCTD15, LRP1B, NEGR1 and PRKD1. Similarly, three BMI loci (NEGR1, NRXN3 and PRKD1) were involved in CROATIA-Korcula, two (FTO and MTCH2) in ORCADES and two (FTO and LRP1B) in MICROS.
Figure 1.

Pair-wise epistatic signals in each study cohort. (a) Pairwise epistatic signals in CROATIA-Vis. (b) Pairwise epistatic signals in CROATIA-Korcula. (c) Pairwise epistatic signals in ORCADES. (d) Pairwise epistatic signals in MICROS.
Table 2. Number of SNP pairs and genes annotated at −log10 Pint>7.3 in each study cohorta.
| Cohort | SNP pair | Gene annotated | SNP pair with BMI loci | BMI loci |
|---|---|---|---|---|
| Vis | 1365 | 1011 | 7 | 5 |
| Korcula | 1237 | 971 | 5 | 3 |
| ORCADES | 1335 | 890 | 4 | 2 |
| MICROS | 785 | 639 | 3 | 2 |
Vis and Korcula represent CROATIA-Vis and CROATIA-Korcula, respectively.
GO terms enriched by epistatic genes (−log10 Pint >7.3) in each cohort were compared (Supplementary Table S2) and identified 9 common in all four cohorts (Table 3) that might indicate common regulation mechanisms (eg, GO:0008038 – neuron recognition). Among the epistatic genes that enriched the 9 GO terms, we found 19 epistatic genes shared by the four cohorts of which 15 are previously published GWA loci (mostly not genome-wide significant) associating with various phenotypes34 (Supplementary Table S3). Most of the 19 shared epistatic genes interacted with one another despite their interactions being relatively weak (−log10 Pint<7.3, Supplementary Table S4) in general, including CDH13 (cadherin 13) associated with height35 and SORCS2 (sortilin-related VPS10 domain containing receptor 2) associated with circulating insulin-like growth factor 1 and insulin-like growth factor-binding protein-3, which are important for anthropometric traits and risk of cancer and cardiovascular disease.36
Table 3. Common GO terms enriched by epistatic genes in CROATIA-Vis (Vis), CROATIA-Korcula (Korcula), ORCADES and MICROSa.
| GO term | Description | Vis | Korcula | ORCADES | MICROS |
|---|---|---|---|---|---|
| GO:0008038 | Neuron recognition | 2.07E−06 | 1.78E−05 | 8.38E−07 | 1.13E−04 |
| GO:0022610 | Biological adhesion | 2.49E−06 | 3.16E−05 | 3.72E−05 | 3.59E−05 |
| GO:0007155 | Cell adhesion | 2.49E−06 | 3.16E−05 | 3.72E−05 | 3.59E−05 |
| GO:0044459 | Plasma membrane part | 1.51E−09 | 5.88E−08 | 6.06E−13 | 1.54E−05 |
| GO:0042995 | Cell projection | 1.16E−07 | 6.42E−06 | 1.63E−07 | 1.81E−07 |
| GO:0043005 | Neuron projection | 1.23E−05 | 7.05E−06 | 2.56E−06 | 1.59E−07 |
| GO:0044425 | Membrane part | 2.82E−05 | 1.43E−05 | 2.00E−11 | 3.25E−06 |
| GO:0044456 | Synapse part | 4.15E−05 | 6.97E−04 | 7.50E−05 | 4.07E−04 |
| GO:0005886 | Plasma membrane | 6.02E−04 | 1.85E−06 | 4.29E−07 | 6.28E−06 |
The process, function and component GO terms are in the first, second and third panels, respectively.
We further tested replication of the SNP pairs involving either BMI loci (19, Table 2) or two shared epistatic genes across the study cohorts (50, Supplementary Table S4). Despite none of the 69 SNP pairs being genome-wide significant, eight of them had a replication in one or more cohorts at the SNP level (ie, −log10 Pint>1.30; Table 4). The best replicated pairs at the SNP level were rs2202167 (NRXN3) rs11150880 (−log10 Pint was 8.19, 1.68 and 1.43 in CROATIA-Korcula, CROATIA-Vis and ORCADES, respectively) and rs1474056 (MTCH2) - rs7250947 (PLIN4) (−log10 Pint was 8.08 in ORCADES and 2.44 in CROATIA-Korcula). The rs11150880 SNP is near the RPH3AL gene, which is known to have a key role in insulin secretion by pancreatic cells.37 The PLIN4 gene may be important for intracellular and neutral lipid storage droplets.38 The eight replicated SNP pairs together explained the phenotypic variance of BMI by 4, 4, 2 and 0.5% in CROATIA-Vis, CROATIA-Korcula, ORCADES and MICROS, respectively. By testing replication at the region level, we found the pair of rs9858278 (NAALADL2) - rs7198915 (CDH13) replicated in CROATIA-Vis, CROATIA-Korcula and MICROS (exceed the 5% thresholds, Table 4 and Supplementary Table S5). Further testing the nine replicated SNP pairs in the NFBC1966 cohort found none replicated at either the SNP or region levels. However, seven out of the nine pairs had −log10 Pint>2, of which three exceed the 20% thresholds (Table 4 and Supplementary Table S5).
Table 4. Replicated interactions involving either the BMI loci or two shared epistatic genes across cohortsa.
| SNP1 | Chr1 | Gene1 | SNP2 | Chr2 | Gene2 | Vis | Korcula | ORCADES | MICROS | NFBC1966 |
|---|---|---|---|---|---|---|---|---|---|---|
| rs953104 | 1 | ATP1A4 | rs8008553 | 14 | PRKD1b | 8.46c (5.53d) | 0.27 (2.84) | 0.19 (1.59) | 1.34c (2.13) | 0.12 (2.11) |
| rs10256700 | 7 | CNTNAP2 | rs10833498 | 11 | NELL1 | 0.03 (1.94) | 6.05c (5.04d) | 0.02 (1.66) | 1.35c (2.27) | 0.42 (2.68f) |
| rs587791 | 9 | PTPRD | rs1408219 | 13 | GPC6 | 5.05c (3.71d) | 0.45 (1.43) | 0.06 (2.31) | 1.41c (2.60) | 0.38 (2.05) |
| rs3847291 | 9 | PTPRD | rs1554941 | 21 | DSCAM | 2.21c (2.91e) | 0.59 (2.07) | 5.03c (3.01e) | 0.38 (1.46) | 0.77 (2.03) |
| rs1474056 | 11 | MTCH2b | rs7250947 | 19 | PLIN4 | 0.19 (1.56) | 2.44c (1.62) | 8.07c (7.84d) | 0.36 (2.60) | 0.25 (1.24) |
| rs3783297 | 14 | PRKD1b | rs7146371 | 14 | (−) | 0.85 (1.86) | 7.52c (6.77d) | 0.48 (2.13) | 1.67c (1.76) | 0.07 (2.90f) |
| rs2202167 | 14 | NRXN3b | rs11150880 | 17 | (−) | 1.68c (2.51) | 8.19c (5.98d) | 1.43c (2.96) | 0.29 (1.54) | 0.10 (2.25) |
| rs11071868 | 15 | MEGF11 | rs2665272 | 16 | FTOb | 7.97c (3.81d) | 1.38c (2.30) | 0.31 (1.73) | 0.03 (2.39) | 0.09 (2.80f) |
| rs9858278 | 3 | NAALADL2 | rs7198915 | 16 | CDH13 | 4.24c (3.48d) | 0.01 (3.17d) | 0.43 (2.08) | 0.93 (3.16d) | 0.02 (1.20) |
SNP1 (SNP2): the first (second) SNP name; chr1 (chr2): the chromosome where SNP1 (SNP2) locates; gene1 (gene2): symbol of the gene annotated by SNP1 (SNP2); −log10Pint value at the SNP (region) level; Vis: CROATIA-Vis; Korcula: CROATIA-Korcula; (–): no gene annotated; 5%, 10% and 20% thresholds derived for replication at the region level in Supplementary Table S5.
One of the BMI loci.
−log10Pint>1.30 at the SNP level.
Significant at the 5% threshold.
Significant at the 10% threshold.
Significant at the 20% threshold.
Discussion
Gene–gene interactions have been suggested as sources of the hidden genetic variations in GWA studies,6, 7 but the extent of their role in this regard has yet to be demonstrated. One big challenge is that the sample sizes of many GWA data sets are relatively small (eg, less than 4000 individuals) and hence the power to detect epistasis could be low.8, 13 Therefore studying epistasis in a single GWA population is unlikely to be fruitful. This is certainly true in our case where exhaustive genome scans in the four study cohorts found no genome-wide significant epistasis associated with BMI. We suggest to tackle the challenge by looking for common (thus potentially important) gene–gene interactions from sub genome-wide significant epistatic signals (−log10 Pint>7.3) in multiple GWA populations. We showed that GO enrichment analysis could be used to identify common GO terms (ie gene function groups) enriched by the epistatic signals in the four study cohorts from which 19 shared epistatic genes were identified. Most of the 19 shared epistatic genes are previously identified GWA loci associating with phenotypes other than BMI and interacted with one another. Their interactions were considered potentially important because they belong to one or multiple commonly enriched GO terms. Interactions involving at least one of the 32 BMI loci with −log10 Pint>7.3 were also considered potentially important assuming the BMI loci are likely interactive.
Being aware of possible noises in those potentially important interactions, we used replication to identify the most reliable epistatic signals across the study cohorts. Eight epistatic pairs involving either the BMI loci or two shared epistatic genes showed replication at the SNP level in at least one cohort (Table 4). The eight epistatic pairs together could indeed explain a considerable proportion of the BMI variation in each individual cohort. Nevertheless, caution is recommended in light of the potential overestimation of the effects due to the ‘winner's curse'.39 Besides, none of the eight epistatic pairs were replicated in all of the four study cohorts, or in the replication cohort NFBC1966. Further replication tests in other populations and/or functional assays are useful to confirm whether they are true signals.
Statistical replication has been used as the golden rule to prevent reporting false positives in GWA studies. This however appears to be far more difficult for epistatic signals than for single SNP signals for reasons, including power, minor allele frequency change, and linkage disequilibrium between epistatic SNP and mutant for both loci.16 The moderate −log10 Pint values of the epistatic pairs tested for replication suggest that the linkage disequilibrium between epistatic SNPs and mutants is not high so replication of these pairs will be difficult. Furthermore, different environments may cause different phenotype distributions in the discovery and replication cohorts. The lack of replication in the NFBC1966 cohort could be due to two important environmental factors of BMI: age40 (ie, 31 vs a range between 18 and 90 in the study cohorts) and diets.29
The approach based on common gene–gene interactions in multiple GWA populations is an effective solution to the issue of limited power of detection of epistasis. It is just a partial solution though because some ignored interactions may be important as well. Comparison of sub genome-wide significant epistatic signals across multiple populations can be made at either the SNP, or gene or pathway level and seem more fruitful at the gene or pathway level than the SNP level. The approach may become more useful if better annotation methods (considering only GWA signals without interactions)41 can be adapted to epistasis. For example, not all epistatic SNPs were annotated to genes in the study and hence did not contribute to the enrichment analysis. The approach will likely remain important even once new tools for meta-analysis of epistasis in GWA data sets become available to increase power for detection of epistasis.
Acknowledgments
We are grateful to Dr Bill Hill (MRC Human Genetics Unit) for his excellent computing support. We thank all the staff and participants in the ORCADES, CROATIA-Vis, CROATIA-Korcula and MICROS studies, in particular those who contributed to data and sample collection, genotyping, trait measurement and data analysis. We acknowledge the data access to the STAMPEED study (Northern Finland Birth Cohort 1966, phs000276.v1.p1) granted by dbGaP (http://www.ncbi.nlm.nih.gov/gap). We acknowledge the financial support provided by the MRC Core Fund. BBSRC funded WHW, AG and CSH's time in part (grant reference: BB/H024484/1).
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary Material
References
- Vimaleswaran KS, Loos RJ. Progress in the genetics of common obesity and type 2 diabetes. Expert Rev Mol Med. 2010;12:e7. doi: 10.1017/S1462399410001389. [DOI] [PubMed] [Google Scholar]
- Heid IM, Jackson AU, Randall JC, et al. Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat Genet. 2010;42:949–960. doi: 10.1038/ng.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speliotes EK, Willer CJ, Berndt SI, et al. Association analyses of 249 796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer CJ, Speliotes EK, Loos RJ, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Zhao JH, Luan J, et al. Cumulative effects and predictive value of common obesity-susceptibility variants identified by genome-wide association studies. Am J Clin Nutr. 2010;91:184–190. doi: 10.3945/ajcn.2009.28403. [DOI] [PubMed] [Google Scholar]
- Eichler EE, Flint J, Gibson G, et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11:446–450. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordovas JM, Robertson R, Cleirigh EN. Gene-gene and gene-environment interactions defining lipid-related traits. Curr Opin Lipidol. 2011;22:129–136. doi: 10.1097/MOL.0b013e32834477a9. [DOI] [PubMed] [Google Scholar]
- Schupbach T, Xenarios I, Bergmann S, et al. FastEpistasis: a high performance computing solution for quantitative trait epistasis. Bioinformatics. 2010;26:1468–1469. doi: 10.1093/bioinformatics/btq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei WH, Knott S, Haley CS, et al. Controlling false positives in the mapping of epistatic QTL. Heredity. 2010;104:401–409. doi: 10.1038/hdy.2009.129. [DOI] [PubMed] [Google Scholar]
- Cordell HJ. Detecting gene-gene interactions that underlie human diseases. Nat Rev Genet. 2009;10:392–404. doi: 10.1038/nrg2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips PC. Epistasis – the essential role of gene interactions in the structure and evolution of genetic systems. Nat Rev Genet. 2008;9:855–867. doi: 10.1038/nrg2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauderman WJ. Sample size requirements for association studies of gene-gene interaction. Am J Epidemiol. 2002;155:478–484. doi: 10.1093/aje/155.5.478. [DOI] [PubMed] [Google Scholar]
- Yang J, Wray NR, Visscher PM. Comparing apples and oranges: equating the power of case-control and quantitative trait association studies. Genet Epidemiol. 2010;34:254–257. doi: 10.1002/gepi.20456. [DOI] [PubMed] [Google Scholar]
- Ma L, Yang J, Runesha HB, et al. Genome-wide association analysis of total cholesterol and high-density lipoprotein cholesterol levels using the Framingham heart study data. BMC Med Genet. 2010;11:55. doi: 10.1186/1471-2350-11-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Hemani G, Hicks AA, et al. Characterisation of genome-wide association epistasis signals for serum uric acid in human population isolates. PLoS One. 2011;6:e23836. doi: 10.1371/journal.pone.0023836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooperberg C, Leblanc M. Increasing the power of identifying gene x gene interactions in genome-wide association studies. Genet Epidemiol. 2008;32:255–263. doi: 10.1002/gepi.20300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam AC, Powell J, Wei WH, et al. A combined strategy for quantitative trait loci detection by genome-wide association. BMC Proc. 2009;3 (Suppl 1:S6. doi: 10.1186/1753-6561-3-s1-s6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DM, Spencer CCA, Pointon JJ, et al. Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility. Nat Genet. 2011;43:761–767. doi: 10.1038/ng.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Xu H, Chen S, et al. Genome-wide interaction-based association analysis identified multiple new susceptibility loci for common diseases. PLoS Genet. 2011;7:e1001338. doi: 10.1371/journal.pgen.1001338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strange A, Capon F, Spencer CC, et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet. 2010;42:985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akula N, Baranova A, Seto D, et al. A network-based approach to prioritize results from genome-wide association studies. PLoS One. 2011;6:e24220. doi: 10.1371/journal.pone.0024220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emily M, Mailund T, Hein J, et al. Using biological networks to search for interacting loci in genome-wide association studies. Eur J Hum Genet. 2009;17:1231–1240. doi: 10.1038/ejhg.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuillan R, Leutenegger AL, Abdel-Rahman R, et al. Runs of homozygosity in European populations. Am J Hum Genet. 2008;83:359–372. doi: 10.1016/j.ajhg.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitart V, Biloglav Z, Hayward C, et al. 3000 years of solitude: extreme differentiation in the island isolates of Dalmatia, Croatia. Eur J Hum Genet. 2006;14:478–487. doi: 10.1038/sj.ejhg.5201589. [DOI] [PubMed] [Google Scholar]
- Polasek O, Marusic A, Rotim K, et al. Genome-wide association study of anthropometric traits in Korcula Island, Croatia. Croat Med J. 2009;50:7–16. doi: 10.3325/cmj.2009.50.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattaro C, Marroni F, Riegler A, et al. The genetic study of three population microisolates in South Tyrol (MICROS): study design and epidemiological perspectives. BMC Med Genet. 2007;8:29. doi: 10.1186/1471-2350-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatti C, Service SK, Hartikainen AL, et al. Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nat Genet. 2009;41:35–46. doi: 10.1038/ng.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igl W, Johansson A, Wilson JF, et al. Modeling of environmental effects in genome-wide association studies identifies SLC2A2 and HP as novel loci influencing serum cholesterol levels. PLoS Genet. 2010;6:e1000798. doi: 10.1371/journal.pgen.1000798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aulchenko YS, Ripke S, Isaacs A, et al. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23:1294–1296. doi: 10.1093/bioinformatics/btm108. [DOI] [PubMed] [Google Scholar]
- Aulchenko YS, de Koning DJ, Haley C. Genomewide rapid association using mixed model and regression: a fast and simple method for genomewide pedigree-based quantitative trait loci association analysis. Genetics. 2007;177:577–585. doi: 10.1534/genetics.107.075614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MI, Abecasis GR, Cardon LR, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, et al. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Kamatani Y, Takahashi A, et al. A genome-wide association study in 19 633 Japanese subjects identified LHX3-QSOX2 and IGF1 as adult height loci. Hum Mol Genet. 2010;19:2303–2312. doi: 10.1093/hmg/ddq091. [DOI] [PubMed] [Google Scholar]
- Kaplan RC, Petersen AK, Chen MH, et al. A genome-wide association study identifies novel loci associated with circulating IGF-I and IGFBP-3. Hum Mol Genet. 2011;20:1241–1251. doi: 10.1093/hmg/ddq560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmugam C, Katkoori VR, Jhala NC, et al. Immunohistochemical expression of rabphilin-3A-like (Noc2) in normal and tumor tissues of human endocrine pancreas. Biotech Histochem. 2009;84:39–45. doi: 10.1080/10520290902738878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel AR, Brasaemle DL, McAndrews-Hill M, et al. Adoption of PERILIPIN as a unifying nomenclature for the mammalian PAT-family of intracellular lipid storage droplet proteins. J Lipid Res. 2010;51:468–471. doi: 10.1194/jlr.R000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goring HH, Terwilliger JD, Blangero J. Large upward bias in estimation of locus-specific effects from genomewide scans. Am J Hum Genet. 2001;69:1357–1369. doi: 10.1086/324471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy R, Wills AK, Wong A, et al. Life course variations in the associations between FTO and MC4R gene variants and body size. Hum Mol Genet. 2010;19:545–552. doi: 10.1093/hmg/ddp504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MX, Gui HS, Kwan JS, et al. GATES: a rapid and powerful gene-based association test using extended Simes procedure. Am J Hum Genet. 2011;88:283–293. doi: 10.1016/j.ajhg.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.