

Abstract
There is some overlap in the biological activities of cell-penetrating peptides (CPPs) and antimicrobial peptides (AMPs). We compared nine AMPs, seven CPPs, and a fusion peptide with regard to their ability to cluster anionic lipids in a mixture mimicking the cytoplasmic membrane of Gram-negative bacteria, as measured by differential scanning calorimetry. We also studied their bacteriostatic effect on several bacterial strains, and examined their conformational changes upon membrane binding using circular dichroism. A remarkable correlation was found between the net positive charge of the peptides and their capacity to induce anionic lipid clustering, which was independent of their secondary structure. Among the peptides studied, six AMPs and four CPPs were found to have strong anionic lipid clustering activity. These peptides also had bacteriostatic activity against several strains (particularly Gram-negative Escherichia coli) that are sensitive to lipid clustering agents. AMPs and CPPs that did not cluster anionic lipids were not toxic to E. coli. As shown previously for several types of AMPs, anionic lipid clustering likely contributes to the mechanism of antibacterial action of highly cationic CPPs. The same mechanism could explain the escape of CPPs from intracellular endosomes that are enriched with anionic lipids.
Introduction
It has recently become evident that there is functional overlap in the properties of antimicrobial peptides (AMPs) and cell-penetrating peptides (CPPs) (1–3). Both classes of peptides exhibit a wide variety of sequences, lengths, hydrophobicities, and structures, and can operate via several different mechanisms. They are generally polycationic and amphiphilic, and hence have a high affinity for negatively charged membranes. Some CPPs can permeabilize membranes and exhibit antimicrobial activity, whereas some AMPs are toxic without membrane permeabilization. In addition, peptides that act as AMPs can be bacteriostatic against one class of bacteria while being bactericidal to others (4,5).
The preferential interaction of these peptides with negatively charged lipids could result in the lateral segregation of such lipids from zwitterionic ones, leading to the formation of membrane domains. Such clustering of anionic lipids could contribute to the toxicity against bacteria (6–8). Anionic lipid clustering has not yet been studied with CPPs. Differential scanning calorimetry (DSC) is a suitable tool for characterizing such changes in membrane properties. Phase separation by lipid clustering is different from that which produces peptide-rich and -poor domains with distinct transitions. The phenomenon of clustering of anionic lipids away from zwitterionic ones was used to predict the specificity of several antimicrobial agents against different bacterial species (9). It was shown that a mixture of 1-palmitoyl-2-oleoyl-phosphatidylethanolamine (POPE) and tetraoleoyl cardiolipin (TOCL) in a 75:25 ratio, which mimics the lipid composition of Gram-negative bacteria, is a simple system that can be used to probe the clustering of anionic lipid by peptides and proteins (10) at ambient temperatures. The gel-to-liquid crystalline phase transition temperature of pure TOCL is <0°C. If an agent causes anionic lipid clustering by segregating TOCL, the phase transition temperature of the other component in the lipid mixture will increase, bringing it closer to that of pure POPE, i.e., ∼24°C. This effect persists over several heating and cooling cycles.
Candidates for inducing lipid clustering are Arg and Lys residues. The Arg side chains not only contribute to the net cationic charge, they also help to stabilize membrane interactions and peptide insertion (11). Recently, the contribution of individual Arg residues to the overall antimicrobial activity was dissected with fragments of LL-37 (12). The role of Arg in promoting anionic lipid clustering and bacterial species selectivity was reported in a study of three homologous, small, Arg-rich AMPs (10). Arg has also been the subject of extensive study in CPPs (13). In general, CPPs contain more Arg compared with AMPs. Because of their bidentate structure, Arg residues generate a negative Gaussian curvature (i.e., negative curvature along two perpendicular principal directions), whereas Lys generates a negative curvature along one direction only (2,14). Some of the most effective CPPs for delivering cargo are the Arg-rich ones, such as HIV-TAT and penetratin (15,16). For this reason, Arg residues in membranes have received much attention (2,15,17,18). Both Arg and Lys residues have high pK values, so they remain protonated at physiological pH, thereby creating an energetic barrier for partitioning into the membrane. Nevertheless, the guanidinium moiety in Arg can form two hydrogen bonds with the phosphate group of phospholipids, thus allowing for easier membrane penetration (19). Recent computer simulations indicated that the translocation of an Arg through the membrane involves the formation of a water-filled defect that keeps the charged group hydrated (20). Most of the energy penalty for inserting Arg into membranes is paid for by insertion of the first Arg residue; the remaining ones can use the same water-filled defect without much further cost. Because Arg induces Gaussian curvature, whereas Lys forms monodentate hydrogen bonds and generates only negative curvature (2), Lys-rich AMPs are more lytic and damaging to membranes compared with Arg-rich CPPs.
Because polycationic peptides bind strongly to anionic membranes, one might expect CPPs to be able to cluster anionic lipids away from zwitterionic ones, as shown previously for AMPs (6–9,21,22). Therefore, in this work we compared anionic lipid clustering in the presence of several different representative polycationic peptides (Table 1), all of which were previously characterized (23–37). To compare the relative effectiveness of Arg versus Lys residues in modulating anionic lipid clustering, we modified some of the peptides by replacing all of their Lys residues with Arg.
Table 1.
Peptides and their sequence and primary function
| Peptide | Sequence | Function |
|---|---|---|
| Gramicidin S | cyclo(PVOLfPVOLf)∗ | AMP |
| MSI-103 | KIAGKIAKIAGKIAKIAGKIA-amide | AMP |
| MSI-103-Arg | RIAGRIARIAGRIARIAGRIA-amide | AMP |
| PGLa | GMASKAGAIAGKIAKVALKAL-amide | AMP |
| PGLa-Arg | GMASRAGAIAGRIARVALRAL-amide | AMP |
| Magainin 2 | GIGKFLHSAKKFGKAFVGEIMNS | AMP |
| Magainin 2-Arg | GIGRFLHSARRFGRAFVGEIMNS | AMP |
| KIGAKI | KIGAKIKIGAKIKIGAKI-amide | AMP |
| BP100 | KKLFKKILKYL-amide | AMP |
| MAP | KLALKLALKALKAALKLA-amide | CPP |
| MAP-Arg | RLALRLALRALRAALRLA-amide | CPP |
| Penetratin | RQIKIWFQNRRMKWKK | CPP |
| SAP | VRLPPPVRLPPPVRLPPP | CPP |
| PEP1 | KETWWETWWTEWSQPKKKRKV | CPP |
| Transportan | GWTLNSAGYLLGKINLKALAALAKKIL | CPP |
| HIV-TAT | GRKKRRQRRRPPQ | CPP |
| FP23 | AVGIGALFLGFLGAAGSTMGARS-amide | FP |
All cationic side chains are shown in bold, and the N-terminus contributes an extra charge.
D-amino acids are presented in lower case; O stands for ornithine.
Experimental Methods
Materials
All lipids used in this study were obtained from Avanti Polar Lipids (Alabaster, AL). Chemicals for peptide synthesis were obtained from Iris Biotech GmbH (Marktedwitz, Germany), and solvents for synthesis and purification were purchased from Acros Organics (Geel, Belgium) or Biosolve (Valkenswaard, Netherlands). All of the peptides listed in Table 1 were synthesized (as C-terminal amides, with the exception of magainin 2, penetratin, SAP, PEP1, transportan, and HIV-TAT) by Fmoc solid-phase peptide synthesis on a Syro II multiple peptide synthesizer (Syro II; MultiSyntech, Witten, Germany). They were purified by preparative high-performance liquid chromatography (HPLC) with water/acetonitrile gradients containing 5 mM HCl as the ion pairing agent as described previously (32,38,39). The identity was confirmed by analytical HPLC (Agilent Technologies, Waldbronn, Germany) coupled to a mass spectrometer (μ-TOF, Bruker, Bremen, Germany). The purity was >95%.
Differential scanning calorimetry
Measurements were performed with a NANO II differential scanning calorimeter (Calorimeter Sciences, Linden, UT) as previously described (9). Lipid films were prepared with POPE/TOCL (75:25). After hydration, the samples were brought to 0°C and scanned from 0 to 35°C with several cycles of heating and cooling. The scan rate was 1°/min. Degassed buffer was placed in the reference cell, and each cell volume was 0.34 mL. The buffer was 20 mM PIPES, 140 mM NaCl, 1 mM EDTA, pH 7.4. The total lipid concentration in the calorimeter cell was 2.5 mg/mL. Calorimeter data were analyzed with the use of a program provided by Microcal and plotted with Origin 7.0.
Determination of minimal inhibitory concentration
The minimal inhibitory concentrations (MICs) of the peptides were determined in sterile 96-well plates. Peptide stock solutions (1.024 mg/mL) were prepared in water and successively diluted in series (1:1 v/v) using 50 μl Müller-Hinton (MH) broth to obtain the desired peptide concentration in each well in 50 μl total volume. To each well, an aliquot of 50 μl of a bacterial suspension containing 106 colony-forming units/mL in MH media was added to obtain a final volume of 100 μl. Antibacterial activities are expressed as the MIC and correspond to the lowest concentration of peptide, in micromolar units, at which no bacterial growth was detected after overnight incubation at 37°C. The MIC was determined against Escherichia coli DSM 1103, Pseudomonas aeruginosa DSM 1117, Staphylococcus aureus DSM 1104, and Staphylococcus epidermidis DSM 1798.
Circular dichroism spectroscopy
Circular dichroism (CD) spectroscopy was performed with a Jasco J-815 spectropolarimeter (Jasco, Groß-Umstadt, Germany). The spectra were recorded in thermostated quartz cuvettes maintained at 30°C, from 260 to 185 nm in 0.1 nm intervals. CD spectra of the peptides were measured in both the presence and absence of small unilamellar vesicles (SUVs) composed of POPE/TOCL (75:25) at a lipid/peptide (L/P) molar ratio of 20 or 100. Three repeat scans were recorded at a scan rate of 20 nm/min, with a response time of 8 s with 1 nm bandwidth. The resulting spectra were averaged and corrected for the baseline spectra corresponding to peptide-free lipid suspension. The resulting spectra were smoothed with the use of Jasco Spectra Analysis software. At high peptide concentration, we observed the precipitation of vesicles in a few cases, which caused significant light scattering. This effect, together with other CD artifacts, led to absorption flattening, i.e., to reduced signal intensity toward low wavelengths (vide infra).
Results
DSC
Using DSC, we evaluated the phase behavior of a mixture of POPE/TOCL (75:25) in a suspension of multilamellar vesicles, in the presence and absence of peptides, at an L/P molar ratio of 20. The DSC curves are shown in Fig. 1, A and B, and the shift in the transition temperature of the higher melting component is shown in Table 2. At a lower L/P ratio, we observed precipitation and/or clarification of the lipid mixture, whereas at higher ratios the thermotropic effects were less pronounced. Hence, we report the results only for the mixtures with L/P = 20.
Figure 1.

DSC of the lipid mixture POPE/TOCL (75:25) in the absence and presence of peptides at L/P = 20. Positive values correspond to heating, and negative values correspond to cooling. The first cycle of heating and cooling from 0°C to 25°C is in the lipid alone, followed by two cycles of heating and cooling in the presence of peptide. All scans were performed at a scan rate of 1°/min. (A) Clustering by AMPs. (B) Clustering by CPPs.
Table 2.
Physical properties of the peptides and the shifts caused in the transition temperature of the binary mixture POPE/TOCL (75:25)
| Net charge | Charge density∗ | Mean hydrophobicity (Hφ) | Anionic lipid clustering Δλ (°C)† | |
|---|---|---|---|---|
| Gramicidin S | 2 | 0.20 (10 aa) | not available‡ | 0.0 |
| MSI-103 | 7 | 0.33 (21 aa) | −1.0 (0.185) | 5.0 |
| MSI-103-Arg | 7 | 0.33 (21 aa) | −1.02 (−0.580) | 6.0 |
| PGLa | 5 | 0.24 (21 aa) | −0.63 (0.279) | 3.7 |
| PGLa-Arg | 5 | 0.24 (21 aa) | −0.65 (−0.082) | 2.8 |
| Magainin 2 | 3.5 | 0.15 (23 aa) | −0.58 (0.173) | 0.9 |
| Magainin 2-Arg | 3.5 | 0.15 (23 aa) | −0.60 (−0.006) | 3.2 |
| KIGAKI | 7 | 0.39 (18 aa) | −0.98 (0.163)§ | 6.2 |
| BP100 | 6 | 0.55 (11 aa) | 0.07 (−1.490) | 1.7 |
| MAP | 6 | 0.33 (18 aa) | 0.65 (0.202) | 4.4 |
| MAP-Arg | 6 | 0.33 (18 aa) | 0.62 (−0.084) | 4.7 |
| Penetratin | 7 | 0.44 (16 aa) | −2.33 (−0.552) | 5.4 |
| SAP | 3 | 0.17 (18 aa) | 0.53 (−0.090) | 0.6 |
| PEP1 | 3 | 0.14 (21 aa) | −2.55 (−0.390) | 0.9 |
| Transportan | 4 | 0.15 (27 aa) | 0.84 (0.320) | 3.5 |
| HIV-TAT | 8 | 0.62 (14 aa) | −7.29 (−1.474) | 2.0 |
| FP23 | 2 | 0.10 (23 aa) | 0.81 (0.514) | 0.0 |
Charge density = net charge per number of residues.
Δλ = maximum temperature shift (in °C) for L/P = 20 from the DSC transition of the pure lipid.
Amphiphilic cyclic β-sheet. Hydrophobicity could not be calculated by the same programs used for the other peptides because of the presence of Orn.
Overall, the net positive charge of the peptide was the major contributing element to the clustering effect. The cyclic gramicidin S and the fusogenic peptide FP23 (corresponding to the N-terminal 23 residues of the gp41 protein of HIV-1), with only two charges each, did not have any effect on clustering. A clear correlation with net positive charge is seen for the series of homologous peptides MSI-103, PGLa, KIGAKI, and MAP (Fig. 1 A). The substitution of Arg by Lys did not have any effect on anionic lipid clustering. The only exception was magainin 2-Arg, which became more active in segregating anionic lipids. Peptides that are too hydrophilic or too short to penetrate deeply into the membrane (e.g., BP100 and HIV-TAT) have only a marginal or no effect (Fig. 1, A and B). Peptides that are very hydrophobic cannot cluster anionic lipids if there are not enough cationic charges present (or if they are preaggregated, as is the case with SAP), but they are good at clustering provided that sufficient cationic charges are present (as with transportan; Fig. 1 B). Of note, the lipid-clustering experiments did not distinguish between the functionalities of AMP and CPP.
Approximately four cationic charges are required to produce a clustering effect in peptides that are capable of forming an amphipathic helix. Magainin 2 is at the borderline, with 3.5 charges, which may explain its sensitivity to Arg-Lys substitution. Recent calculations performed with a coarse-grained physical model that takes into account peptide amphiphilicity and the presence of anionic lipids showed that a peptide net cationic charge of ≥4 was necessary for the bacteria selectivity of AMPs (40).
Minimal inhibitory concentration
Even though the MICs for some of the peptides studied here have already been reported in the literature, we redetermined them under identical conditions so that the values would be comparable. The MIC values of the peptides were determined against two Gram-negative strains (E. coli and P. aeruginosa) and two Gram-positive strains (S. aureus and S. epidermidis). The MIC values shown in Table 3 have been converted to micromolar units to account for the different lengths of the peptides. As previously noted, Gram-positive strains that are devoid of an outer membrane are more susceptible to amphiphilic membrane-active peptides as compared with Gram-negative strains. All of the investigated AMPs show moderate to high antimicrobial activity, as previously noted in the literature (28,31,32,36). Similar antimicrobial activities are also observed for several CPPs, as previously reported, e.g., for MAP (41) and penetratin (42,43). CPPs in general have less activity than AMPs, i.e., they have higher MIC values. However, some CPPs that can fold into amphipathic α-helical structure (here, MAP, MAP-Arg, penetratin, and transportan) do show significant antimicrobial action. As expected, the fusogenic peptide FP23 did not show any antimicrobial activity against any of the strains, in similarity to the other less toxic CPPs (SAP, PEP1, and HIV-TAT).
Table 3.
Minimal inhibitory concentration (μM)
| Peptides | E. coli (G−) | P. aeruginosa (G−) | S. aureus (G+) | S. epidermidis (G+) |
|---|---|---|---|---|
| Gramicidin S | 53 | >211 | 1 | 2 |
| MSI-103 | 3 | 28 | 3 | 2 |
| MSI-103-Arg | 6 | 26 | 3 | 2 |
| PGLa | 30 | 119 | 15 | 7 |
| PGLa-Arg | 7 | 113 | 4 | 2 |
| Magainin 2 | 99 | >99 | 99 | 99 |
| Magainin 2-Arg | 47 | 95 | 95 | 24 |
| KIGAKI | 8 | 8 | 4 | 8 |
| BP100 | 10 | 20 | 5 | 5 |
| MAP | 31 | >122 | 8 | 4 |
| MAP-Arg | 57 | >115 | 29 | 4 |
| Penetratin | 51 | 51 | 51 | 13 |
| SAP | >122 | >122 | >122 | >122 |
| PEP1 | >87 | >87 | >87 | >87 |
| Transportan | 43 | >86 | 43 | 5 |
| HIV-TAT | >128 | 128 | 128 | >128 |
| FP23 | >117 | >117 | >117 | >117 |
The known lipid membrane compositions of the bacterial strains used in this work (10,44) are shown in Table 4. The POPE/TOCL (75:25) mixture was previously found to mimic the composition of Gram-negative bacteria very well (6,10). Most Gram-negative bacteria are rich in PE, although there are a few species in which PE is not the major lipid (e.g., Caulobacter crescentus). In contrast, it is common for Gram-positive bacteria to have little zwitterionic lipid. A frequently used example is S. aureus. It should be borne in mind, however, that the membrane composition of different Gram-positive bacterial species normally varies widely and is subject to changes according to environmental conditions. In addition, some Gram-positive bacteria contain a variety of glycolipids in different proportions (7,10). Furthermore, in certain strains, the phosphatidylglycerol (PG) and cardiolipin (CL) can be acylated with amino acids, producing cationic lipids that can repel cationic antimicrobial agents, leading to resistance (7,10,45). Thus, most Gram-negative bacteria will be susceptible to agents that induce anionic lipid clustering. However, we must obtain more detailed knowledge about the lipid composition of Gram-positive bacteria before we can predict their susceptibility to anionic lipid clustering agents.
Table 4.
Lipid composition of the cytoplasmic membrane
| Bacteria | PE | CL | PG |
|---|---|---|---|
| E. coli (G−) | 80 | 5 | 15 |
| P. aeruginosa (G−) | 60 | 11 | 21 |
| S. aureus (G+) | 0 | 19 | 57 |
| S. epidermidis (G+) | 0 | 1 | 90 |
CD spectroscopy
CD spectroscopy was used to monitor the conformational changes of all the investigated peptides (AMPs, CPPs, and FP23) upon addition of SUVs composed of POPE/TOCL (75:25). The CD experiments had to be done with SUVs to avoid scattering artifacts, even though DSC was carried out with MLVs. First, the peptide spectra were recorded in 10 mM phosphate buffer (pH = 7) in the absence of lipids (data not shown). Most of the peptides show a completely disordered conformation (random coil) with a characteristic negative band around 196 nm, or they are largely disordered (PEP1, transportan, and MAP) with a minimum around 201–202 nm. The only exceptional spectra are seen for gramicidin S, because of its cyclic structure, and SAP, which is known to be preaggregated in aqueous solution (46).
In the presence of lipids, amphiphilic peptides tend to acquire their preferred secondary structure. In our CD experiments, the addition of SUVs composed of POPE/TOCL (75:25), caused severe precipitation at an L/P ratio of 20 in the case of MSI-103, MSI-103-Arg, MAP, and BP100. Precipitation decreased the amount of material (lipids and peptides), and in some cases led to the appearance of weak, random, coil line shapes due to residual unbound peptide in solution. Furthermore, it gave rise to light scattering and other CD artifacts such as absorption flattening (47,48). Therefore, the CD experiments shown in Fig. 2 were performed at L/P = 100 instead of L/P = 20 as in the DSC experiments with MLVs, for which the CD data are not shown. Aside from the artifacts, we observed no obvious conformational differences for the different L/P ratios.
Figure 2.
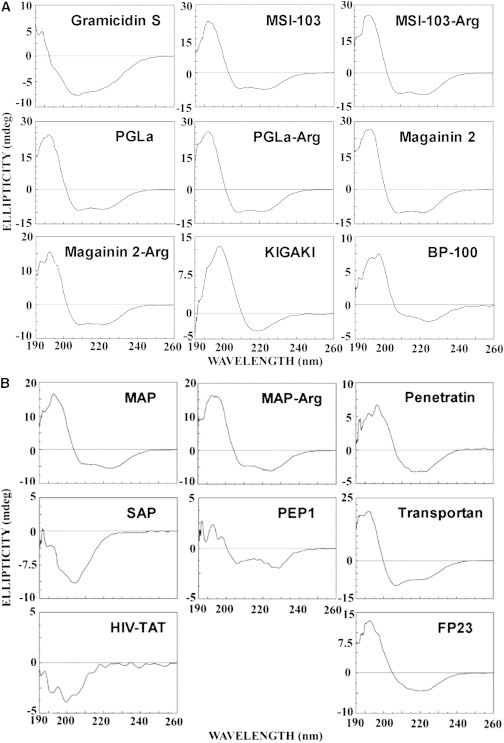
CD spectra measured at 30°C of the investigated peptides in the presence of SUVs composed of POPE/TOCL (75:25) at an L/P molar ratio of 100. (A) CD spectra of the AMPs (gramicidin S, MSI-103, MSI-103-Arg, PGLa, PGLa-Arg, magainin 2, magainin 2-Arg, KIGAKI, and BP100). (B) CD spectra of the CPPs (MAP, MAP-Arg, penetratin, SAP, PEP1, transportan, HIV-TAT) and the fusion peptide (FP23).
Only the designer-made β-stranded KIGAKI (Fig. 2 A) and the fusogenic FP23 (Fig. 2 B) showed a pronounced structural transition from disordered to β-sheets, as characterized by a positive band around 203–205 nm and a broad minimum between 215 and 219 nm. Most other peptides showed a transition from disordered to largely α-helical, as characterized by a positive band around 190 nm and two negative bands around 208 and 220 nm. This includes the AMPs PGLa, PGLa-Arg, magainin 2, magainin 2-Arg, MSI-103, and MSI-103-Arg (Fig. 2 A), and the CPPs MAP, MAP-Arg, and transportan (Fig. 2 B). We note that none of the Lys-to-Arg substitutions caused any conformational change in the α-helical PGLa, magainin 2, MSI-103, or MAP. PEP1 and penetratin showed a mixture of α-helical and β-pleated folded states, as characterized by the appearance of a negative band at 217–220 nm (Fig. 2 B). Gramicidin S (Fig. 2 A), SAP (Fig. 2 B), and HIV-TAT (Fig. 2 B) did not show any change in secondary structure upon addition of lipids. This observation can be attributed to the cyclic conformation of gramicidin S, the preaggregation of SAP, and the complete lack of secondary structure of the Arg-rich peptide HIV-TAT.
Discussion
In this study, we focused on the possible role of anionic lipid clustering in the antimicrobial action of CPPs. It was recently suggested that a common feature among CPPs is their ability to induce negative Gaussian curvature in lipid bilayers, and that this enables the internalization of peptides into cells (15) as well as cytoskeletal remodeling (2). Previously, much of the research on cell-penetrating mechanisms was focused on distinguishing between direct entry and endocytic pathways. A new physicochemical focus on a common aspect that can account for all pathways would advance our understanding of these agents. However, because CPPs have several different entry pathways, and different CPPs favor different mechanisms, it is unlikely that changes in a single membrane property can explain all variations in CPP behavior. One of the arguments given in a previous study is that similar trends are seen for CPPs and AMPs when one plots the ratio of the number of Arg residues over the total number of charges, i.e., NArg/(NLys + NArg), versus the mean hydrophobicity (see Fig. 2 G in Mishra et al. (2)). We evaluated whether the peptides used in this work fall on these curves, but we found significant deviations. For example, a CPP or AMP with a positive mean hydrophobicity, or one close to zero, would be predicted by those curves to have a low value for the fraction of Arg residues. Some peptides fit this generalization, but others, such as PGLa-Arg, Mag 2-Arg, MAP-Arg, and SAP, do not. In addition, the fusogenic peptide FP23 has the highest hydrophobicity but carries only one Arg and no Lys.
Negative Gaussian curvature should result in the promotion of inverted micelles or bicontinuous cubic phases, as has been observed with the TAT peptide in dimyristoylphosphatidylcholine (DMPC), and in the eukaryotic mimic phosphatidylcholine (PC)/phosphatidylethanolamine (PE)/phosphatidylserine (PS) (23,49). Many other peptides that are not known to be CPPs also promote negative Gaussian curvature, as evidenced by the formation of bicontinuous cubic phases. These include the influenza fusion peptide (50) (which facilitates the bending of membranes (51,52), even though it has no activity as either a CPP or an AMP), the antimicrobial defensins in PC/PE/PS (14), alamethicin in DEPE (53), gramicidin S in microbial lipid extracts (54), lactoferricin derivatives in an E. coli lipid extract (55), and protegrin-1 (PG-1) and PGLa in POPE (56). Hence, it is likely that the promotion of negative Gaussian curvature per se is not a requirement for the function of a CPP, nor would all agents that promote this curvature implicitly function as CPPs. In addition, the promotion of Gaussian curvature is dependent on the lipid composition of the membrane. Although the ability to promote membrane curvature is not a required property of CPPs or AMPs, this does not mean that it cannot play a role for some peptides.
Our purpose in this study was not to find an alternative mechanism to the promotion of negative Gaussian curvature, but rather to determine whether there are other factors that contribute to the pronounced antimicrobial activity shown by some CPPs. One such factor is lipid clustering. This phenomenon was monitored here by alterations in the DSC profile upon addition of the peptide. The effect is readily observed in a binary mixture of POPE containing 20–30 mol % TOCL, which is initially well mixed. Note that comparable mixtures containing PG tend to phase separate, which leads to DSC profiles with multiple peaks and thus makes them unsuitable for studying the effects of peptides. As we previously noted (8,10), the peptide-induced segregation of CL from PE is not caused by preferential binding of certain peptides to PE. Any polycationic peptide would bind more strongly to an anionic lipid than to a zwitterionic one. This is demonstrated by the fact that such peptides always shift the phase transition of the PE component toward that of the pure lipid, regardless of whether the anionic lipid is the higher or lower melting component in the mixture.
By this DSC criterion, it is clear that anionic lipid clustering is promoted by several AMPs and CPPs (Fig. 1, Table 2), although this phenomenon is not required for a peptide to behave as a CPP, nor is a specific secondary structure required. Remarkably, we find an excellent correlation of the transition temperature shift Δλ (obtained experimentally by DSC) with the net positive charge of the peptides, as illustrated in Fig. 3. It is seen that the AMP gramicidin S and the fusogenic FP23, with only two positive charges each, cannot cluster anionic lipids. The CPPs SAP and PEP1, with only three positive charges each, have a weak clustering effect. The linear correlation continues with the α-helical MSI-103, PGLa, transportan, KIGAKI, PEP1, and penetratin. The only exceptions are BP100 and HIV-TAT, both of which are very hydrophilic and very short in length, which may lead to a larger entropic penalty. Peptides that were studied as a pair, and carry either Lys or Arg as the basic residues, generally have similar activities. The only deviation is seen for magainin 2, which has much more lipid clustering activity in the Arg form compared with the natural Lys form (Fig. 3). These results suggest that if the Arg and Lys peptides differ in lipid clustering activity, the Arg form should be more potent. This relationship was previously observed and ascribed to differences in the hydrogen-bonding capabilities of these two different side chains (2). We find that this newly described linear correlation also applies to other previously studied AMPs, but to ensure that all of the data refer to the same experimental conditions, they are not included in Fig. 3.
Figure 3.
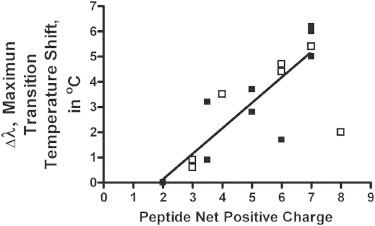
Maximum temperature shift of the main transition obtained by DSC in the presence of peptide (Fig. 1, A and B, Table 2), with respect to the lipid mixture in the absence of peptide, Δλ, plotted as a function of the peptide's net positive charge. The short peptides BP100 and HIV-TAT fall out of line. The linear relationship holds for the AMPs (solid squares) as well as for the CPPs (open squares).
Note that an excellent correlation is obtained only when the net charges of the peptides (i.e., the effective number of cationic charges added to the lipid mixture) are considered. When only the number of cationic side chains is counted, and the compensating effect of anionic residues and the C-terminus is ignored (Table 2), we do not obtain such a good correlation. A poor correlation is also obtained when we consider the charge density per amino acid residue (Table 2), suggesting that the anionic lipids are simply not influenced by the presence of any uncharged regions of the peptides, whether they are long or short. We did not find any correlation between the hydrophobicity of the peptides and their lipid-clustering ability (Table 2). It should be pointed out, however, that the mean hydrophobicities of the peptide pairs studied with either Lys or Arg as the basic amino acid (Table 2) did not differ significantly when calculated with the consensus scale used in the program HydroMCalc (57), but they differed significantly when calculated with the consensus scale of Eisenberg (58). Because these peptide pairs generally have similar biological properties, this suggests that other factors are also required to explain the observed activities in addition to promoting negative Gaussian curvature.
It was previously recognized that many CPPs have AMP-like activity (1,3). Furthermore, members of both classes are able to fuse vesicles composed of lipids mimicking eukaryotic membranes or containing a late-endosome-specific anionic lipid (34,59). Here, we show that the antimicrobial activity of CPPs could result from a simple contribution made by anionic lipid clustering. This phenomenon is expected to be more important for Gram-negative bacteria than for most Gram-positive bacteria, because the cytoplasmic membrane of Gram-negative species contains a sizeable amount of zwitterionic lipids from which anionic lipids can segregate. The Gram-negative bacterium E. coli in particular was reported to be a good model because of its susceptibility to lipid clustering by AMPs (10). In this work, we found that the lipids of this bacterium exhibit the same susceptibility to clustering when exposed to the presence of certain CPPs.
In Gram-positive bacteria, with the predominance of anionic lipids in the membrane and zwitterionic lipids mostly absent, one would expect that other membrane-perturbing mechanisms, such as pore formation, would predominate, leading to a bacteriostatic/bactericidal action in the presence of AMPs or CPPs. With regard to CPPs, we found only two that are poor in anionic lipid clustering: SAP and PEP1. These peptides also have the weakest antimicrobial activity of all the CPPs tested (Table 3). In several species of bacteria that do not have much uncharged or zwitterionic lipid in their cytoplasmic membranes (e.g., S. aureus and S. epidermidis), they were also effective as bacteriostatic agents. It is conceivable that preferential interaction with one of the anionic lipids can lead to clustering in mixtures of anionic lipids, as we previously found (60). This phenomenon, however, did not correlate well with the measured MIC values in Gram-positive bacteria.
In mammalian cells, almost all of the anionic lipids are located in the cytoplasmic leaflet of the cell membrane. Nevertheless, there are several nonphospholipid anionic species exposed on the outer surface of eukaryotic cells, including the protein nucleolin, and proteoglycans such as heparin sulfate, that have been suggested to make initial contact with CPPs (61). However, it was recently shown for penetratin that binding to the cell surface does not correlate well with the internalization efficiency of the peptide (62). It was also reported that several different CPPs can induce the clustering of glycosaminoglycans (63). At higher peptide concentrations, there is an accumulation of the peptide on glycosaminoglycans, and the activation of endocytosis results in peptide internalization (62). Hence, binding of CPPs to anionic lipid components on mammalian cell membranes is not generally expected to be required for internalization. It is also known that processes such as apoptosis or the initiation of blood coagulation will promote the appearance of anionic PS on the outside of cells, and that this is accelerated by oxidized lipids via flip-flop (64). Apparently, CPPs by themselves do not promote the flip-flop of lipid molecules (65). However, the function of CPPs in eukaryotic cells is to penetrate the cell membrane, which would be facilitated by the presence of Arg residues or aromatic ones, or increased hydrophobicity. Hence, during their penetration of the membrane, CPPs would access the cytoplasmic leaflet of the cell that is rich in anionic lipid.
Of note, the anionic lipid bis(monoacylglycero)phosphate (BMP) is known to be enriched in late endosomes, and was postulated to be involved in escape of Arg-rich peptides (59). A leaky fusion mechanism has been suggested for the escape of HIV-TAT after endocytosis. Even though this particular CPP showed an unusually low lipid clustering capacity in our study on POPE/TOCL mixtures, the concept of leaky fusion would be fully supported by any process that can induce lipid phase segregation. For example, early studies attributed the fusogenic effect of Ca2+ ions against anionic lipid vesicles to the formation of gel phase domains (66). This mechanism need not even require the formation of gel phase domains, but most likely could also occur to some extent at any boundary or defect-rich line between two distinct membrane domains. The possibility of endosomal escape by lipid clustering should therefore also be considered as a possible mechanism in membranes that contain BMP as the anionic agent.
In summary, when a biologically active polycationic peptide approaches a bacterial plasma membrane, it encounters the lipid headgroups, gravitating toward the negatively charge ones and promoting clustering. We have demonstrated here that clustering of anionic lipids that are typically found in bacterial membranes can contribute to the antimicrobial activity of CPPs, as was previously shown for AMPs. The major factor in clustering is the net charge of the peptide, regardless of its primary or secondary structure. In interactions with mammalian cells, peptide-induced lipid clustering could result from an encounter of a CPP with anionic lipids in the inner leaflet of the plasma membrane, or, more likely, after endocytosis in a late endosome that is enriched in anionic lipid BMP.
Acknowledgments
The authors thank Andrea Eisele and Kerstin Scheubeck for their technical support.
This research was partly supported by the Canadian Institute of Health Research (grant MOP 86608), the DFG Center for Functional Nanostructures (TP E1.2), and the HGF BioInterfaces Program.
References
- 1.Bobone S., Piazzon A., Stella L. The thin line between cell-penetrating and antimicrobial peptides: the case of Pep-1 and Pep-1-K. J. Pept. Sci. 2011;17:335–341. doi: 10.1002/psc.1340. [DOI] [PubMed] [Google Scholar]
- 2.Mishra A., Lai G.H., Wong G.C. Translocation of HIV TAT peptide and analogues induced by multiplexed membrane and cytoskeletal interactions. Proc. Natl. Acad. Sci. USA. 2011;108:16883–16888. doi: 10.1073/pnas.1108795108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Splith K., Neundorf I. Antimicrobial peptides with cell-penetrating peptide properties and vice versa. Eur. Biophys. J. 2011;40:387–397. doi: 10.1007/s00249-011-0682-7. [DOI] [PubMed] [Google Scholar]
- 4.Samy R.P., Stiles B.G., Chow V.T. Antimicrobial proteins from snake venoms: direct bacterial damage and activation of innate immunity against Staphylococcus aureus skin infection. Curr. Med. Chem. 2011;18:5104–5113. doi: 10.2174/092986711797636108. [DOI] [PubMed] [Google Scholar]
- 5.Spindler E.C., Hale J.D., Gill R.T. Deciphering the mode of action of the synthetic antimicrobial peptide Bac8c. Antimicrob. Agents Chemother. 2011;55:1706–1716. doi: 10.1128/AAC.01053-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Epand R.M., Epand R.F. Lipid domains in bacterial membranes and the action of antimicrobial agents. Biochim. Biophys. Acta. 2009;1788:289–294. doi: 10.1016/j.bbamem.2008.08.023. [DOI] [PubMed] [Google Scholar]
- 7.Epand R.M., Epand R.F. Biophysical analysis of membrane-targeting antimicrobial peptides: membrane properties and the design of peptides specifically targeting Gram-negative bacteria. In: Wang G., editor. Antimicrobial Peptides: Discovery, Design and Novel Therapeutic Strategies. CABI; Wallingford, UK: 2010. pp. 116–127. [Google Scholar]
- 8.Epand R.M., Epand R.F. Bacterial membrane lipids in the action of antimicrobial agents. J. Pept. Sci. 2011;17:298–305. doi: 10.1002/psc.1319. [DOI] [PubMed] [Google Scholar]
- 9.Epand R.M., Rotem S., Epand R.F. Bacterial membranes as predictors of antimicrobial potency. J. Am. Chem. Soc. 2008;130:14346–14352. doi: 10.1021/ja8062327. [DOI] [PubMed] [Google Scholar]
- 10.Epand R.M., Epand R.F., Shai Y. Lipid clustering by three homologous arginine-rich antimicrobial peptides is insensitive to amino acid arrangement and induced secondary structure. Biochim. Biophys. Acta. 2010;1798:1272–1280. doi: 10.1016/j.bbamem.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 11.Schow E.V., Freites J.A., Tobias D.J. Arginine in membranes: the connection between molecular dynamics simulations and translocon-mediated insertion experiments. J. Membr. Biol. 2011;239:35–48. doi: 10.1007/s00232-010-9330-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang G., Epand R.F., Epand R.M. Decoding the functional roles of cationic side chains of the major antimicrobial region of human cathelicidin LL-37. Antimicrob. Agents Chemother. 2012;56:845–856. doi: 10.1128/AAC.05637-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Esbjörner E.K., Lincoln P., Nordén B. Counterion-mediated membrane penetration: cationic cell-penetrating peptides overcome Born energy barrier by ion-pairing with phospholipids. Biochim. Biophys. Acta. 2007;1768:1550–1558. doi: 10.1016/j.bbamem.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 14.Schmidt N.W., Mishra A., Wong G.C. Criterion for amino acid composition of defensins and antimicrobial peptides based on geometry of membrane destabilization. J. Am. Chem. Soc. 2011;133:6720–6727. doi: 10.1021/ja200079a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt N., Mishra A., Wong G.C. Arginine-rich cell-penetrating peptides. FEBS Lett. 2010;584:1806–1813. doi: 10.1016/j.febslet.2009.11.046. [DOI] [PubMed] [Google Scholar]
- 16.Su Y., Waring A.J., Hong M. Membrane-bound dynamic structure of an arginine-rich cell-penetrating peptide, the protein transduction domain of HIV TAT, from solid-state NMR. Biochemistry. 2010;49:6009–6020. doi: 10.1021/bi100642n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hristova K., Wimley W.C. A look at arginine in membranes. J. Membr. Biol. 2011;239:49–56. doi: 10.1007/s00232-010-9323-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoo J., Cui Q. Does arginine remain protonated in the lipid membrane? Insights from microscopic pKa calculations. Biophys. J. 2008;94:L61–L63. doi: 10.1529/biophysj.107.122945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deol S.S., Domene C., Sansom M.S. Anionic phospholipid interactions with the potassium channel KcsA: simulation studies. Biophys. J. 2006;90:822–830. doi: 10.1529/biophysj.105.071407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MacCallum J.L., Bennett W.F., Tieleman D.P. Transfer of arginine into lipid bilayers is nonadditive. Biophys. J. 2011;101:110–117. doi: 10.1016/j.bpj.2011.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Epand R.F., Wang G., Epand R.M. Lipid segregation explains selective toxicity of a series of fragments derived from the human cathelicidin LL-37. Antimicrob. Agents Chemother. 2009;53:3705–3714. doi: 10.1128/AAC.00321-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Epand R.M., Epand R.F. Domains in bacterial membranes and the action of antimicrobial agents. Mol. Biosyst. 2009;5:580–587. doi: 10.1039/b900278m. [DOI] [PubMed] [Google Scholar]
- 23.Afonin S., Frey A., Ulrich A.S. The cell-penetrating peptide TAT(48-60) induces a non-lamellar phase in DMPC membranes. ChemPhysChem. 2006;7:2134–2142. doi: 10.1002/cphc.200600306. [DOI] [PubMed] [Google Scholar]
- 24.Afonin S., Grage S.L., Ulrich A.S. Temperature-dependent transmembrane insertion of the amphiphilic peptide PGLa in lipid bilayers observed by solid state 19F NMR spectroscopy. J. Am. Chem. Soc. 2008;130:16512–16514. doi: 10.1021/ja803156d. [DOI] [PubMed] [Google Scholar]
- 25.Bürck J., Roth S., Ulrich A.S. Conformation and membrane orientation of amphiphilic helical peptides by oriented circular dichroism. Biophys. J. 2008;95:3872–3881. doi: 10.1529/biophysj.108.136085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grasnick D., Sternberg U., Ulrich A.S. Irregular structure of the HIV fusion peptide in membranes demonstrated by solid-state NMR and MD simulations. Eur. Biophys. J. 2011;40:529–543. doi: 10.1007/s00249-011-0676-5. [DOI] [PubMed] [Google Scholar]
- 27.Ieronimo M., Afonin S., Ulrich A.S. 19F NMR analysis of the antimicrobial peptide PGLa bound to native cell membranes from bacterial protoplasts and human erythrocytes. J. Am. Chem. Soc. 2010;132:8822–8824. doi: 10.1021/ja101608z. [DOI] [PubMed] [Google Scholar]
- 28.Strandberg E., Kanithasen N., Ulrich A.S. Solid-state NMR analysis comparing the designer-made antibiotic MSI-103 with its parent peptide PGLa in lipid bilayers. Biochemistry. 2008;47:2601–2616. doi: 10.1021/bi701944r. [DOI] [PubMed] [Google Scholar]
- 29.Strandberg E., Tremouilhac P., Ulrich A.S. Synergistic transmembrane insertion of the heterodimeric PGLa/magainin 2 complex studied by solid-state NMR. Biochim. Biophys. Acta. 2009;1788:1667–1679. doi: 10.1016/j.bbamem.2008.12.018. [DOI] [PubMed] [Google Scholar]
- 30.Strandberg E., Wadhwani P., Ulrich A.S. Solid-state NMR analysis of the PGLa peptide orientation in DMPC bilayers: structural fidelity of 2H-labels versus high sensitivity of 19F-NMR. Biophys. J. 2006;90:1676–1686. doi: 10.1529/biophysj.105.073858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tremouilhac P., Strandberg E., Ulrich A.S. Synergistic transmembrane alignment of the antimicrobial heterodimer PGLa/magainin. J. Biol. Chem. 2006;281:32089–32094. doi: 10.1074/jbc.M604759200. [DOI] [PubMed] [Google Scholar]
- 32.Wadhwani P., Afonin S., Ulrich A.S. Optimized protocol for synthesis of cyclic gramicidin S: starting amino acid is key to high yield. J. Org. Chem. 2006;71:55–61. doi: 10.1021/jo051519m. [DOI] [PubMed] [Google Scholar]
- 33.Wadhwani P., Bürck J., Ulrich A.S. Using a sterically restrictive amino acid as a 19F NMR label to monitor and to control peptide aggregation in membranes. J. Am. Chem. Soc. 2008;130:16515–16517. doi: 10.1021/ja804928q. [DOI] [PubMed] [Google Scholar]
- 34.Wadhwani P., Reichert J., Ulrich A.S. Antimicrobial and cell-penetrating peptides induce lipid vesicle fusion by folding and aggregation. Eur. Biophys. J. 2012;41:177–187. doi: 10.1007/s00249-011-0771-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alves I.D., Jiao C.Y., Sagan S. Cell biology meets biophysics to unveil the different mechanisms of penetratin internalization in cells. Biochim. Biophys. Acta. 2010;1798:2231–2239. doi: 10.1016/j.bbamem.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 36.Blazyk J., Wiegand R., Kari U.P. A novel linear amphipathic β-sheet cationic antimicrobial peptide with enhanced selectivity for bacterial lipids. J. Biol. Chem. 2001;276:27899–27906. doi: 10.1074/jbc.M102865200. [DOI] [PubMed] [Google Scholar]
- 37.Koch K., Afonin S., Ulrich A.S. Solid-state (19)F-NMR of peptides in native membranes. Top. Curr. Chem. 2012;306:89–118. doi: 10.1007/128_2011_162. [DOI] [PubMed] [Google Scholar]
- 38.Afonin S., Glaser R.W., Ulrich A.S. 4-fluorophenylglycine as a label for 19F NMR structure analysis of membrane-associated peptides. ChemBioChem. 2003;4:1151–1163. doi: 10.1002/cbic.200300568. [DOI] [PubMed] [Google Scholar]
- 39.Fields G.B., Noble R.L. Solid phase peptide synthesis utilizing 9-fluorenylmethoxycarbonyl amino acids. Int. J. Pept. Protein Res. 1990;35:161–214. doi: 10.1111/j.1399-3011.1990.tb00939.x. [DOI] [PubMed] [Google Scholar]
- 40.Taheri-Araghi S., Ha B.Y. Cationic antimicrobial peptides: a physical basis for their selective membrane-disrupting activity. Soft Matter. 2010;6:1933–1940. [Google Scholar]
- 41.Dathe M., Schümann M., Bienert M. Peptide helicity and membrane surface charge modulate the balance of electrostatic and hydrophobic interactions with lipid bilayers and biological membranes. Biochemistry. 1996;35:12612–12622. doi: 10.1021/bi960835f. [DOI] [PubMed] [Google Scholar]
- 42.Garibotto F.M., Garro A.D., Enriz R.D. Penetratin analogues acting as antifungal agents. Eur. J. Med. Chem. 2011;46:370–377. doi: 10.1016/j.ejmech.2010.10.025. [DOI] [PubMed] [Google Scholar]
- 43.Zhu W.L., Shin S.Y. Antimicrobial and cytolytic activities and plausible mode of bactericidal action of the cell penetrating peptide penetratin and its lys-linked two-stranded peptide. Chem. Biol. Drug Des. 2009;73:209–215. doi: 10.1111/j.1747-0285.2008.00769.x. [DOI] [PubMed] [Google Scholar]
- 44.Epand R.F., Pollard J.E., Epand R.M. Depolarization, bacterial membrane composition, and the antimicrobial action of ceragenins. Antimicrob. Agents Chemother. 2010;54:3708–3713. doi: 10.1128/AAC.00380-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peschel A., Jack R.W., van Strijp J.A. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with l-lysine. J. Exp. Med. 2001;193:1067–1076. doi: 10.1084/jem.193.9.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pujals S., Giralt E. Proline-rich, amphipathic cell-penetrating peptides. Adv. Drug Deliv. Rev. 2008;60:473–484. doi: 10.1016/j.addr.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 47.Wallace B.A., Mao D. Circular dichroism analyses of membrane proteins: an examination of differential light scattering and absorption flattening effects in large membrane vesicles and membrane sheets. Anal. Biochem. 1984;142:317–328. doi: 10.1016/0003-2697(84)90471-8. [DOI] [PubMed] [Google Scholar]
- 48.Wallace B.A., Teeters C.L. Differential absorption flattening optical effects are significant in the circular dichroism spectra of large membrane fragments. Biochemistry. 1987;26:65–70. doi: 10.1021/bi00375a010. [DOI] [PubMed] [Google Scholar]
- 49.Mishra A., Gordon V.D., Wong G.C. HIV TAT forms pores in membranes by inducing saddle-splay curvature: potential role of bidentate hydrogen bonding. Angew. Chem. Int. Ed. Engl. 2008;47:2986–2989. doi: 10.1002/anie.200704444. [DOI] [PubMed] [Google Scholar]
- 50.Colotto A., Epand R.M. Structural study of the relationship between the rate of membrane fusion and the ability of the fusion peptide of influenza virus to perturb bilayers. Biochemistry. 1997;36:7644–7651. doi: 10.1021/bi970382u. [DOI] [PubMed] [Google Scholar]
- 51.Gabrys C.M., Yang R., Weliky D.P. Nuclear magnetic resonance evidence for retention of a lamellar membrane phase with curvature in the presence of large quantities of the HIV fusion peptide. Biochim. Biophys. Acta. 2010;1798:194–201. doi: 10.1016/j.bbamem.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tristram-Nagle S., Chan R., Nagle J.F. HIV fusion peptide penetrates, disorders, and softens T-cell membrane mimics. J. Mol. Biol. 2010;402:139–153. doi: 10.1016/j.jmb.2010.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keller S.L., Gruner S.M., Gawrisch K. Small concentrations of alamethicin induce a cubic phase in bulk phosphatidylethanolamine mixtures. Biochim. Biophys. Acta. 1996;1278:241–246. doi: 10.1016/0005-2736(95)00229-4. [DOI] [PubMed] [Google Scholar]
- 54.Staudegger E., Prenner E.J., Lohner K. X-ray studies on the interaction of the antimicrobial peptide gramicidin S with microbial lipid extracts: evidence for cubic phase formation. Biochim. Biophys. Acta. 2000;1468:213–230. doi: 10.1016/s0005-2736(00)00260-1. [DOI] [PubMed] [Google Scholar]
- 55.Zweytick D., Tumer S., Lohner K. Membrane curvature stress and antibacterial activity of lactoferricin derivatives. Biochem. Biophys. Res. Commun. 2008;369:395–400. doi: 10.1016/j.bbrc.2008.01.176. [DOI] [PubMed] [Google Scholar]
- 56.Hickel A., Danner-Pongratz S., Pabst G. Influence of antimicrobial peptides on the formation of nonlamellar lipid mesophases. Biochim. Biophys. Acta. 2008;1778:2325–2333. doi: 10.1016/j.bbamem.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 57.Tossi, A., L. Sandri, and L. Giangaspero. 2002. Mean hydrophobicities were calculated with the program HydroMCalc. Proc. 27th European Peptide Symp., Sorrento. 416–417.
- 58.Sweet R.M., Eisenberg D. Correlation of sequence hydrophobicities measures similarity in three-dimensional protein structure. J. Mol. Biol. 1983;171:479–488. doi: 10.1016/0022-2836(83)90041-4. [DOI] [PubMed] [Google Scholar]
- 59.Yang S.T., Zaitseva E., Melikov K. Cell-penetrating peptide induces leaky fusion of liposomes containing late endosome-specific anionic lipid. Biophys. J. 2010;99:2525–2533. doi: 10.1016/j.bpj.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Epand R.F., Maloy L., Epand R.M. Amphipathic helical cationic antimicrobial peptides promote rapid formation of crystalline states in the presence of phosphatidylglycerol: lipid clustering in anionic membranes. Biophys. J. 2010;98:2564–2573. doi: 10.1016/j.bpj.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gonçalves E., Kitas E., Seelig J. Binding of oligoarginine to membrane lipids and heparan sulfate: structural and thermodynamic characterization of a cell-penetrating peptide. Biochemistry. 2005;44:2692–2702. doi: 10.1021/bi048046i. [DOI] [PubMed] [Google Scholar]
- 62.Alves I.D., Bechara C., Sagan S. Relationships between membrane binding, affinity and cell internalization efficacy of a cell-penetrating peptide: penetratin as a case study. PLoS ONE. 2011;6:e24096. doi: 10.1371/journal.pone.0024096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ziegler A., Seelig J. Binding and clustering of glycosaminoglycans: a common property of mono- and multivalent cell-penetrating compounds. Biophys. J. 2008;94:2142–2149. doi: 10.1529/biophysj.107.113472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Volinsky R., Cwiklik L., Kinnunen P.K. Oxidized phosphatidylcholines facilitate phospholipid flip-flop in liposomes. Biophys. J. 2011;101:1376–1384. doi: 10.1016/j.bpj.2011.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Henriques S.T., Castanho M.A. Consequences of nonlytic membrane perturbation to the translocation of the cell penetrating peptide pep-1 in lipidic vesicles. Biochemistry. 2004;43:9716–9724. doi: 10.1021/bi036325k. [DOI] [PubMed] [Google Scholar]
- 66.Hay J.C. Calcium: a fundamental regulator of intracellular membrane fusion? EMBO Rep. 2007;8:236–240. doi: 10.1038/sj.embor.7400921. [DOI] [PMC free article] [PubMed] [Google Scholar]