

Abstract
Early afterdepolarizations (EADs) are linked to both triggered arrhythmias and reentrant arrhythmias by causing premature ventricular complexes (PVCs), focal excitations, or heterogeneous tissue substrates for reentry formation. However, a critical number of cells that synchronously exhibit EADs are needed to result in arrhythmia triggers and substrates in tissue. In this study, we use mathematical modeling and computer simulations to investigate EAD synchronization and arrhythmia induction in tissue models with random cell-to-cell variations. Our major observations are as follows. Random cell-to-cell variations in action potential duration without EAD presence do not cause large dispersion of refractoriness in well-coupled tissue. In the presence of phase-2 EADs, the cells may synchronously exhibit the same number of EADs or no EADs with a very small dispersion of refractoriness, or synchronize regionally to result in large dispersion of refractoriness. In the presence of phase-3 EADs, regional synchronization leads to propagating EADs, forming PVCs in tissue. Interestingly, even though the uncoupled cells exhibit either no EAD or only a single EAD, when these cells are coupled to form a tissue, more than one PVC can occur. When the PVCs occur at different locations and time, multifocal arrhythmias are triggered, with the foci shifting in space and time in an irregular manner. The focal arrhythmias either spontaneously terminate or degenerate into reentrant arrhythmias due to heterogeneities and spatiotemporal chaotic dynamics of the foci.
Introduction
Early afterdepolarizations (EADs) are secondary depolarizations of an action potential, which are associated with arrhythmogenesis in many cardiac diseases, such as long-QT syndrome (1–7) and heart failure (8,9). EADs have historically been linked to triggered arrhythmias maintained by focal excitations (10,11), but exactly how they trigger focal arrhythmias in cardiac tissue is not well understood. Later studies (2–4,12,13) have also proposed that EADs may cause reentrant arrhythmias by forming premature ventricular complexes (PVCs), or by regionally lengthening the action potential duration (APD) to increase dispersion of refractoriness, forming reentry substrates. However, EADs tend to occur irregularly from beat to beat in a given cell (8,14–16) due to dynamical chaos (17,18) or random ion channel fluctuations (19,20). They may occur randomly from cell to cell in a given beat due to random cell-to-cell variations (21,22), such as variations in ion channel conductance or kinetics, cell size and geometry, etc. How the irregularly occurring EADs synchronize in tissue to reach a critical size to form PVCs or reentry substrates remains incompletely understood.
In previous studies (17,18,23), we showed that irregularly occurring EADs could be explained by dynamical chaos. Due to the chaotic behavior of EADs, regional synchronization of EADs occurs in homogeneous tissue, with regions exhibiting EADs bordering to regions without EADs. This forms both PVCs and regionally lengthened APD, which gives rise to both triggers and substrates of arrhythmias, including reentrant and multiple shifting focal arrhythmias. However, real tissue is not homogeneous, but has random cell-to-cell fluctuations in cell parameters, such as the number of ion channels and regional heterogeneities. Because the properties of the cells in heterogeneous tissue are intrinsically different from each other, the way that EADs synchronize differs from that in the homogeneous tissue.
The essential difference is that in homogeneous tissue, because the cells are identical, synchronization means to maintain the cells in the same phase by gap junction coupling. The gap junction current is almost negligible after synchronization is achieved. In heterogeneous tissue, however, some cells are more prone to EADs than others. The ones more prone to EADs tend to promote EADs in the ones without EADs, and the ones without EADs tend to suppress EADs. Synchronization of EADs requires large gap junction currents to smooth out the intrinsic difference between cells. Therefore, understanding the roles of cellular heterogeneities and their interactions with dynamical instabilities in PVC formation, regional APD lengthening, and in maintaining multifocal arrhythmias (5,6) is important for understanding EAD-mediated arrhythmogenesis in cardiac tissue.
In this study, we use computer simulation to address the question of how EADs are regionally synchronized and cause arrhythmias in heterogeneous tissue. We focus on the heterogeneities caused by cell-to-cell fluctuations in ionic conductance. Simulations were carried out in one-dimensional cable and two-dimensional tissue models to study regional synchronization of EADs and EAD-mediated triggered and reentrant arrhythmias. To avoid the confluent effects of dynamical chaos on EAD synchronization, we only gave one pacing stimulus to all cells in the tissue models at the beginning of the simulation and studied the behaviors of the system after the stimulus. This is also similar to the condition of a single long pause in the heartbeat, which has been shown to be responsible for arrhythmogenesis in long-QT syndrome (4,24–26). We studied the effects of the spatial distribution of the cell properties and gap junction coupling on EAD synchronization, PVC formation, and multiple focal arrhythmia induction.
Methods
We used the rabbit ventricular action potential model by Mahajan et al. (27) to generate phase-2 EADs following the modifications in Sato et al. (17,18). We modified this model as in Xie et al. (28) to generate phase-3 EADs that can propagate in tissue.
The one-dimensional cable and two-dimensional tissue are monodomain models. The one-dimensional cable was simulated by solving the following partial differential equation of voltage,
| (1) |
where V is the membrane potential (in mV), Cm = 1 μF/cm2 is the membrane capacitance, and D is the diffusion constant (in cm2/ms), which was computed as D = 250ggaplc/πrc/Cm, with ggap the gap junction conductance in Siemens (250 nS, unless specified otherwise), lc = 0.015 cm the cell length, and rc = 0.001 cm the cell radius. The two-dimensional tissue was simulated by solving the partial differential equation,
| (2) |
where we chose D = 0.0005 cm2/ms, which results in a conduction velocity of ∼50 cm/s. No-flux boundary conditions and a spatial discretization of Δx = Δy = 0.015 cm were used. Heterogeneities were modeled in one-dimensional cable and two-dimensional tissue by randomly varying the maximum conductance of the L-type Ca current (ICa,L) from cell to cell following a Gaussian distribution.
In all simulations, the entire cable or tissue was stimulated once at the beginning of the simulation with a current pulse of strength of 40 μA/cm2 and duration of 1 ms. Equations 1 and 2 were solved using the Euler method with an adaptive time step varying from 0.001 to 0.1 ms, using operator splitting as described previously (29). APD was defined as the time duration in which V > −70 mV in an action potential. Simulations were carried out on graphic processing units as described in Sato et al. (30). The graphic-processing-unit cards used were NVIDIA Tesla C2050 cards (NVIDIA, Santa Clara, CA).
Results
EAD synchronization in a one-dimensional cable model with phase-2 EADs
The original rabbit ventricular myocyte model was modified to exhibit phase-2 EADs following the study by Sato et al. (17). We altered the strength of the ICa,L flux (gCa) to simulate different cellular action-potential properties. As shown in Fig. 1 A, when gCa is increased from the default value, APD increases. When gCa is increased to 520 mmol/(cm C), a single EAD appears in the action potential. As gCa is increased further, a second EAD appears at another critical gCa, and so on, resulting in a staircase in APD.
Figure 1.
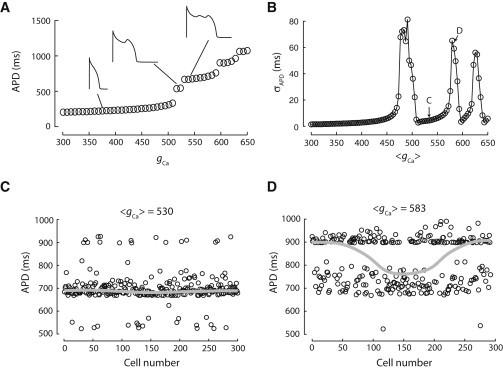
Synchronization of phase-2 EADs in a one-dimensional cable with random cell-to-cell variations. (A) APD of a single rabbit ventricular cell as a function of gCa. (B) σAPD in a cable of 300 cells versus 〈gCa〉 with random gCa drawn from a Gaussian distribution and standard deviation σgCa = 35 mmol/(cm C). (C) APD distribution with 〈gCa〉 = 530 mmol/(cm C) when uncoupled (open circles) and coupled (shaded line). (D) Same as panel C but for 〈gCa〉 = 583 mmol/(cm C).
We placed the model cells in a one-dimensional cable in which gCa was varied randomly along the cable following a Gaussian distribution. When the cells are uncoupled, the random variations in APD are very large (large standard deviation σAPD). When no (or a few) cells exhibit EADs, electrotonic coupling smoothes out these fluctuations and APD varies slightly across the cable. However, as 〈gCa〉 increases to include more cells exhibiting EADs, both small and large heterogeneities in APD distribution can develop in the cable, showing an oscillating pattern as gCa increases (Fig. 1 B). For example, Fig. 1 C shows a simulation with gCa = 530 mmol/(cm C). When the cells are uncoupled, APD varies largely. But when the cells are coupled, the APDs are almost the same in all cells and all cells have a single EAD, exhibiting a small variation. Fig. 1 D shows another simulation with gCa = 585 mmol/(cm C). The cell-to-cell APD variation in the uncoupled cells is similar to that in Fig. 1 C, but when the cells are coupled, APD distribution in the cable shows large gradients with cells exhibiting different numbers of EADs.
The observations above can be understood as follows. As shown in Fig. 1 A, as gCa increases, the action potential suddenly gains one EAD at certain gCa values, exhibiting an “all-or-none” behavior. When 〈gCa〉 is close to these critical gCa values, the cells have to choose which number of EADs to have due to the all-or-none competition between these cells. As a result of the competition, one type of action potential dominates in one region and the other type dominates in another region. When 〈gCa〉 is further away from these critical gCa values, one type of action potential wins across the whole cable, resulting in a synchronous distribution. This gives rise to the oscillation pattern shown in Fig. 1 B. It is well known that gap junction coupling affects EAD formation and propagation (28,31–33) in tissue, i.e., EADs are suppressed by stronger gap junction coupling. The APD dispersion shown in Fig. 1 can be suppressed by stronger coupling or enhanced by weaker coupling.
EAD synchronization in a one-dimensional cable model with phase-3 EADs
Because phase-2 EADs cannot propagate in our tissue models (which agrees with the experimental observations by Damiano and Rosen (34)), we used another action-potential model in which phase-3 EADs are generated (Fig. 2 A) to study EAD propagation (28). We simulated a one-dimensional cable with random cell-to-cell variations in gCa following a Gaussian distribution. When the cells are uncoupled, they may exhibit no EADs or a single phase-3 EAD (Fig. 2 A). Depending on the exact realization of the random gCa distribution, for the same mean value of gCa (〈gCa〉), EADs may either be suppressed altogether in a cable, or may be able to exist in one or more regions of the cable and form PVCs. Fig. 2 B shows the action potentials from 45 out of the 300 cells when they are uncoupled. The values of gCa for each cell were drawn from a Gaussian distribution with 〈gCa〉 = 870 mmol/(cm C) and standard deviation σgCa = 35 mmol/(cm C). Fig. 2 C shows time-space plots for two randomizations of the same 300 cells when they are coupled. A PVC forms in one of the two randomizations.
Figure 2.
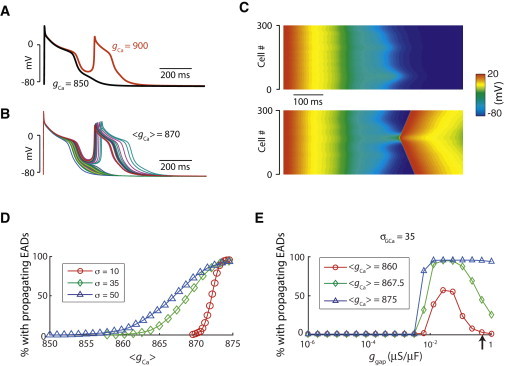
PVC formation in a one-dimensional cable of coupled phase-3 EAD cells. (A) Examples of action potentials of the phase-3 EAD model for two different values of gCa. (B) Action potentials of 45 out of 300 uncoupled cells with 〈gCa〉 = 870 mmol/(cm C) and σgCa= 35 mmol/(cm C). (C) Space-time plots of two simulations of a cable of 300 cells. Both simulations were done with the same set of gCa as in panel B, but reshuffled randomly across the cable. A single stimulus was given to all cells at t = 0. (D) Percentage of simulations exhibiting PVCs versus 〈gCa〉 for different σgCa. For each 〈gCa〉, 1000 trials (random reshuffles) were done from the same set of gCa. A propagating EAD was defined as the second activation after the initial stimulus reaching the end of the cable. (E) Same as panel D, but for varying gap junction conductance and different 〈gCa〉 with σgCa = 35 mmol/(cm C). (Arrow) Normal gap junction coupling.
We then studied the statistics of PVC occurrence as a function of 〈gCa〉. Fig. 2 D shows the percentage of simulated random trails that have one or more PVCs as a function of 〈gCa〉 for different values of the standard deviation, σgCa. As expected, the probability of having a PVC in a cable increases monotonically with 〈gCa〉, because for higher values of 〈gCa〉, more cells “want” to have an EAD and thus the chance for a PVC to occur is higher. When σGCa is increased, not only does the range of 〈gCa〉 for which PVCs can occur become larger, but the entire curve is shifted to lower values of 〈gCa〉. Therefore, when the cell-to-cell fluctuations in gCa are higher, the chance to have a PVC is higher.
When the gap junction coupling strength is reduced, the probability for PVCs increases (Fig. 2 E). This agrees with the observations that when gap junction coupling is reduced, the critical number of EAD cells required for a PVC to form is reduced (28), or with the increased occurrence of focal arrhythmias under fibrosis (35). When the gap junction conductance is reduced below a certain critical value (∼10 nS), propagation failure occurs, and the probability for PVCs drops to zero.
EAD-triggered arrhythmias in a two-dimensional tissue model with random cell-to-cell variations
To study how phase-3 EADs cause arrhythmias in tissue, we carried out simulations in a square tissue with the phase-3 EAD model cells. The values of gCa for different cells were drawn from a Gaussian distribution. The entire tissue was stimulated once at the beginning. Depending on 〈gCa〉 and the distribution of the cells, different behaviors occurred in the tissue simulations. The different outcomes are shown in Fig. 3, and detailed as follows:
Figure 3.
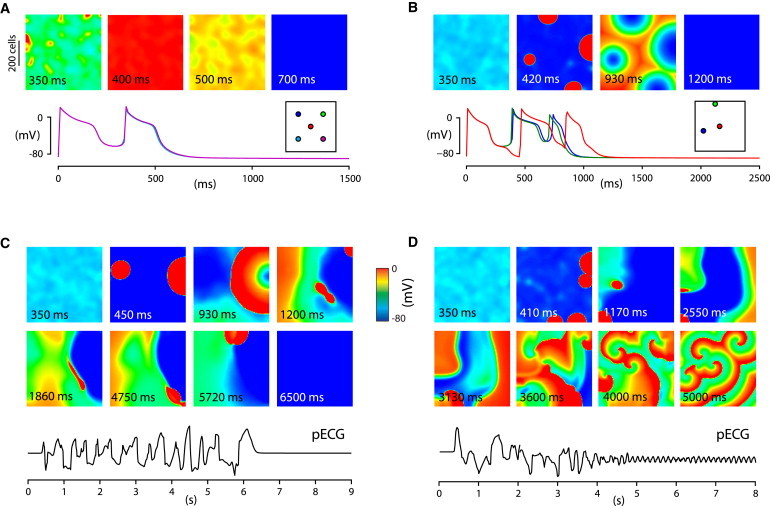
PVCs and arrhythmias triggered phase-3 EADs in two-dimensional tissue. Simulations of 600 × 600 cell tissue of phase-3 EAD model cells with σgCa = 35 mmol/(cm C) and 〈gCa〉 = 867 mmol/(cm C) (B–D) and 〈gCa〉 = 870 mmol/(cm C) (A). The entire tissue was stimulated once at t = 0. Membrane potential time series in panels A and B correspond to locations in the tissue (highlighted by circles of the same color in the inset). Pseudo- electrocardiograms are shown for panels C and D.
When 〈gCa〉 is small, the majority of the cells exhibits no EADs when they are uncoupled. When the cells are coupled via gap junction conductance, the sink effect of non-EAD cells overcomes the source effect of the cells prone to EADs, suppressing the EADs in the coupled tissue. Therefore, as expected, no PVCs or arrhythmias occur.
When 〈gCa〉 is large, the majority of the cells exhibits EADs when they are uncoupled. When they are coupled in tissue, a synchronous whole-tissue PVC occurs (Fig. 3 A, and see Movie S1 in the Supporting Material). The source effect of the cells prone to EADs overcomes the sink effect from the non-EAD cells, synchronizing the whole tissue to exhibit an EAD. The mechanism of EAD synchronization is the same as that in the case of phase-2 EADs shown in Fig. 1 C.
More interesting scenarios occur when 〈gCa〉 is in the intermediate range. In this case, different arrhythmias can occur for identical 〈gCa〉, depending on the exact distributions. Fig. 3, B–D, shows three examples with the same 〈gCa〉 and the same standard deviation σGCa but different random distributions of the cells. Fig. 3 B (see Movie S2) shows that the competition between the cells prone to EADs and non-EAD cells results in regionally synchronized EADs in multiple sites of the tissue (four sites as seen in the second panel of Fig. 3 B), which occur in roughly the same time. These regionally synchronized EADs then propagate in tissue as target waves to give rise to a PVC. Interestingly, the uncoupled cells can only exhibit a single EAD, but two PVCs (two EADs occur after the stimulated beat, see bottom panel in Fig. 3 B) occur in the coupled tissue. The second PVC originates at the same four sites. This indicates that the heterogeneous cellular properties promote EADs, a tissue-scale phenomenon that cannot be seen in single uncoupled cells or homogeneous tissue. The exact mechanism is not clear to us, but it agrees with the observations in our previous study in which we showed that phase-3 EADs occur as a result of electrical heterogeneities (36).
Fig. 3 C (see Movie S3) shows another case in which multifocal arrhythmias occur, which then terminate spontaneously after many revolutions. In this case, the competition between different types of cells causes regionally synchronized EADs. After the first PVC (second panel), regional synchronized EADs occur at locations different from the first PVC (fourth panel), differing from the case in Fig. 3 B. As this process repeats in the tissue, it results in multiple shifting focal arrhythmias. These arrhythmias last for several seconds and then terminate spontaneously. This behavior is a type of spatiotemporal chaos, similar to that shown in our previous study (17). Note that if the regionally synchronized EADs are simply due to heterogeneities, they should occur at the same locations each time; however, they occur dynamically and irregularly in space and time. As shown in our previous study (17), the beat-to-beat chaotic behavior of EADs can facilitate the formation of local PVCs and multiple shifting focal arrhythmias in a homogeneous tissue, so it is not surprising that this can also occur in a heterogeneous tissue. The mechanism of spontaneous termination of arrhythmias is the same as demonstrated in our previous studies (37,38) and those of others (39), which shows that spatiotemporal chaos in an excitable medium of finite size can terminate spontaneously to result in a quiescent tissue.
Fig. 3 D (see Movie S4) shows a case in which multifocal arrhythmias degenerate into reentrant arrhythmias. The multiple shifting foci are due to the same mechanism as in Fig. 3 C. In this case, instead of self-terminating, the multifocal arrhythmias degenerate into reentrant arrhythmias due to local wave breaks caused by heterogeneities and their interactions with the chaotic wave dynamics. Note that during the first few seconds, the action potential repolarizes fully to the resting potential (∼−80 mV). After that the system transitions to a new state in which the action potential fails to full repolarization (the color scale changes from red to green during reentry in the last two panels in Fig. 3 D). Because the sodium current only activates at voltages <−60 mV, the ICa,L is the only inward current for depolarization. Therefore, the reentry is mediated by ICa,L. One can also see from Movie S4 that the ICa,L-mediated spiral waves are stable. Because after the simulation begins, no parameters are changed, this same tissue supports both INa-mediated (e.g., wave conduction in the period of multifocal arrhythmias) and ICa,L-mediated (e.g., wave conduction in the later period of reentry) propagation. This is a “biexcitable” behavior in tissue, as shown in our previous study (40). Also note that the ICa,L-mediated reentry is much faster than was shown in our previous study due to the larger EAD amplitude (or higher ICa,L-mediated excitability) in this model (Fig. 2 A).
The probabilities of these behaviors depend on 〈gCa〉 as well as other parameters as expected. Fig. 4 shows the probabilities of different behaviors versus 〈gCa〉 for different conditions. To facilitate automatic computation of the probabilities, we classified the behaviors into three cases as follows: 1), no PVCs or arrhythmias (activity in tissue stops after the first action potential); 2), a single or a double PVCs (activity occurs after the first action potential but lasts for <2000 ms; Fig. 3, A and B); and 3), arrhythmias (activity persists for at least 2000 ms; Fig. 3, C and D). Interestingly, although each individual cell can only show a single EAD, there are two peaks in which multifocal arrhythmias occur (Fig. 4 A). These multifocal arrhythmias either spontaneously terminate or degenerate into reentry. In the two peaks, in ∼30% of the cases, arrhythmias self-terminate after ∼5000 ms, and ∼70% of the cases develop into one or more stable rotors. The two regions are widened when σgCa is increased (Fig. 4 B) or the gap junction conductance is reduced (Fig. 4 C).
Figure 4.
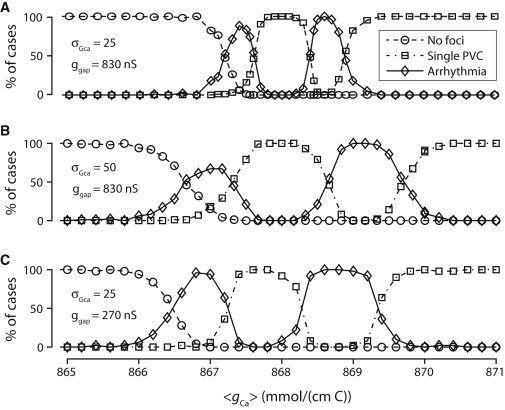
Statistics of PVCs and arrhythmias in two-dimensional tissue. Statistics for simulations of 600 × 600 cell2 tissue of phase-3 EAD model cells with random gCa drawn from a Gaussian distribution with standard deviation σgCa and with gap junction coupling ggap. Each data point corresponds to 50 simulations of 2000 ms with different realizations of the random distribution. (Dashed lines with open circles) Probability of having no EADs in the tissue. (Dashed-dotted lines with open squares) One or two single propagating PVCs with no further activity. (Solid lines with open diamonds) PVCs that lead to sustained focal activity. (A) Statistics with normal gap junctional coupling and σgCa = 25 mmol/(cm C). (B) Statistics with larger fluctuations in gCa (σgCa = 50 mmol/(cm C)). (C) Statistics with gap junction conductance reduced by 67%.
Discussion
As shown in many experimental studies, EADs tend to occur irregularly from beat to beat in a given cell (8,14–18). Our recent studies (17,18,23) showed that the irregular dynamics is likely dynamical chaos. In these studies, we show how chaos synchronization can result in regional synchronized EADs and thus PVCs and reentrant and focal arrhythmias in homogeneous tissue models. In real cardiac tissue, however, in addition to regional differences in APD (41) and calcium cycling (42), short spatial scale heterogeneities, such as random cell-to-cell variability in ionic current density (21,22) or cell size, also exist. These cell-to-cell heterogeneities cause EADs to vary irregularly from cell to cell. In this study, we used computer simulation to investigate the effects of random cell-to-cell variations in action-potential dynamics on EAD synchronization and EAD-mediated arrhythmogenesis in cardiac tissue.
Our major findings are as follows. In the absence of EADs, electrotonic coupling is effective in smoothing out the APD difference caused by random cell-to-cell variations, agreeing with our previous simulation study that random heterogeneities in small spatial scales have few effects on APD distribution and spiral wave dynamics (43). However, in the presence of EADs, random heterogeneities in small spatial scales can result in large spatial scale changes; i.e., due to the all-or-none nature of EADs, regional synchronization of EADs can occur to result in: 1), large APD gradients or dispersion of refractoriness; 2), PVCs; and 3), multifocal arrhythmias. These properties are modulated by gap junction coupling and cell distributions.
The implications of this study to arrhythmogenesis in cardiac diseases can be drawn as follows:
-
1.
Agreeing with other modeling studies (12,33) and experimental observations (34), our study shows that phase-2 EADs cannot propagate in tissue with normal gap junction coupling. Regional synchronization of EADs causes large dispersion of refractoriness, which may serve as a substrate for unidirectional conduction block to result in reentry. On the other hand, due to its lower take-off potential and higher amplitude, phase-3 EADs can propagate in tissue with normal gap junction coupling. Under this condition, arrhythmia-trigger (PVC) and substrate can occur simultaneously to result in focal and reentrant arrhythmias.
-
2.
EAD-related arrhythmias have historically been called “triggered arrhythmias” (10,11), however, how triggered arrhythmias are subsequently maintained in tissue for longer periods of time is not clear. In other words, a single cell often exhibits one or several EADs in a stimulated action potential, but in tissue, once an arrhythmia is triggered by EADs, it lasts for seconds to minutes or even becomes sustained. In many cases, the arrhythmias are maintained by multiple shifting foci (5,6). In this study, we show that even though the uncoupled cells can only exhibit a single EAD, in heterogeneous tissue, long-lasting arrhythmias can occur due to the complex spatiotemporal dynamics, a tissue phenomenon that cannot be understood at the single-cell level. More specifically, when multiple PVC sites fire at roughly the same time (as in Fig. 3 A), they run into each other, after which the tissue becomes quiescent. However, when fewer sites fire, or when the PVC sites do not fire at the same time, a PVC in one location propagates to another location and triggers a new beat at that location. This new beat will then propagate as a PVC and trigger a PVC in another location (as in Fig. 3 C), and so on. Although the heterogeneities in cellular parameters are fixed, the foci occur dynamically and irregularly in space and time. This behavior is similar to what we showed in a previous study (17) in which spatiotemporal chaotic EAD dynamics maintains the multiple shifting foci to manifest polymorphic ventricular tachycardia (PVT) and Torsade de Pointes (TdP) in homogeneous tissue. Therefore, this study and the previous one can provide mechanistic insight into the shifting foci that maintain PVT or TdP in long-QT syndrome observed in experimental studies (5,6) and in clinical settings. In addition, spontaneous termination of the shifting foci in our simulation study can also provide mechanistic insights into spontaneous termination of TdP that are widely observed clinically (44–46).
-
3.
PVT and TdP in long-QT syndrome are mainly triggered by a single pause in the heartbeat (4,24–26). Although we argue that chaos synchronization can explain the maintenance and spontaneous termination of the multiple shifting foci, it cannot well explain the pause-induced PVT and TdP because it takes several-to-many beats for the instability to develop to cause localized PVCs or foci in homogeneous tissue. In this study, we show that in tissue with random cell-to-cell variations, local PVCs self-organize immediately after a single stimulus. These PVCs may cause reentry or undergo the processes as shown in Fig. 3 to cause focal arrhythmias. The combined effects of preexisting heterogeneities and nonlinear dynamics can better explain the pause-induced PVT and TdP observed clinically and experimentally.
-
4.
Our previous study (40) and this study show that when EADs can propagate in tissue, because they occur at a voltage range where INa is not available, their propagation is mediated by ICa,L. We show that under such conditions, both INa-mediated conduction and ICa,L-mediated conduction can occur in the same tissue; we call this “biexcitability” (40). Although a direct demonstration of biexcitability in the heart is so far not available, microelectrode recordings during pause-induced TdP show that the voltage only partially repolarizes (4), providing indirect evidence for the ICa,L-mediated conduction during PVT and TdP.
Limitations
In this study, we investigated the effects of random cell-to-cell variations on EAD synchronization and arrhythmogenesis in cardiac tissue models. We only varied the maximum conductance of ICa,L to obtain different EAD properties and to model random cell-to-cell heterogeneities. In real tissue, EADs and their heterogeneous distributions can result from alterations and heterogeneities of many other ion currents, e.g., the reduction of potassium currents or increase in sodium or calcium currents and heterogeneities in long QT syndrome (1–6). However, altering one or more of these ionic currents randomly from cell to cell might result in similar action-potential properties and EAD behaviors, and thus possibly the same tissue-scale EAD dynamics.
The random cell-to-cell distribution may be a too-idealized setting for real tissue because macroscopic regional heterogeneities and gradients in the ventricles (41,47,48) exist, which may have a critical number of cells to exhibit EADs or triggered activity simultaneously to form PVCs as shown in previous modeling studies (28,49,50). However, preexisting fixed heterogeneities alone cannot explain the dynamical behaviors of PVCs seen clinically (e.g., they vary from beat to beat) (51,52) and shifting foci in multifocal arrhythmias (5,6). In addition to the mechanisms of multiple shifting focal arrhythmias shown in our previous study (17) and this study, it is also possible that multiple shifting foci can arise from synchronization of random-calcium-wave-induced delayed afterdepolarizations in tissue (53,54).
In addition, phase-2 EADs were generated by altering the ICa,L kinetics and reducing of IKs (17), whereas phase-2 EADs may arise from different mechanisms (55–59), which may affect our conclusions obtained in this study. The phase-3 EADs were generated by adding a Ca-activated nonselective cation current (Ins(Ca)) to the action potential model (17,28). Although this current has been identified in different species (60–62), its existence is controversial and its physiological or pathophysiological roles remain unclear. However, EADs have been shown to be able to propagate in cardiac tissue (5), and large-magnitude EADs have been observed (34). As long as the EADs propagate, our observations of PVC formation and multifocal arrhythmias may still occur even when the phase-3 EADs are due to other mechanisms. Our changes in parameters to generate phase-2 and phase-3 EADs, especially the changes in ICa,L kinetics and conductance, were not validated with experimental data, which raises an issue of how realistic these parameters are.
On the other hand, recent studies from Marder and Goaillard (63) have found that in neural circuits, similar bursting behaviors could result from combinations of very different ion channel conductances. In ventricular myocytes, Banyasz et al. (22) found that the ion channel conductances could differ in many folds while the variation in action potential was much smaller. This is supported by simulations showing that drastically different parameter sets in an action potential model can give rise to almost identical action potentials (64). These observations indicate that multiple choices in a wide range of parameters exist for a biological function, but also raise serious issues on how to select proper parameters and model formulations in mathematical modeling of biological systems.
Nevertheless, the purpose of our study is to understand in general how random cell-to-cell variations affect the manifestation of EADs in the tissue scale and thus the genesis of PVCs and arrhythmias. To study these general behaviors at the tissue scale, it is important for the cell model to exhibit certain characteristic properties though they may result from different combinations of parameters or even different cell models. In fact, the same results as shown in Fig. 1 were obtained (see Fig. S1 in the Supporting Material) using the phase I of the Luo and Rudy model (65) with modifications to generate phase-2 EADs (23). However, whether the theoretical predictions are applicable to real heart needs to be validated in experimental studies.
Acknowledgments
This work is supported by National Institutes of Health/National Heart, Lung, and Blood Institute P01 HL078931 and R01 HL093205, a fellowship award for advanced researchers from the Swiss Foundation for Grants in Biology and Medicine (to E.d.L.), and a postdoctoral fellowship award from the American Heart Association, Western States affiliate (to Y.X.).
Footnotes
Enno de Lange's present address is Department of Knowledge Engineering, Maastricht University, Maastricht, The Netherlands.
Yuanfang Xie's present address is Department of Pharmacology, University of California, Davis, California.
Contributor Information
Enno de Lange, Email: enno.delange@maastrichtuniversity.nl.
Zhilin Qu, Email: zqu@mednet.ucla.edu.
Supporting Material
References
- 1.Roden D.M., Lazzara R., Vincent G.M., The SADS Foundation Task Force on LQTS Multiple mechanisms in the long-QT syndrome. Current knowledge, gaps, and future directions. Circulation. 1996;94:1996–2012. doi: 10.1161/01.cir.94.8.1996. [DOI] [PubMed] [Google Scholar]
- 2.El-Sherif N., Caref E.B., Restivo M. The electrophysiological mechanism of ventricular arrhythmias in the long QT syndrome. Tridimensional mapping of activation and recovery patterns. Circ. Res. 1996;79:474–492. doi: 10.1161/01.res.79.3.474. [DOI] [PubMed] [Google Scholar]
- 3.Antzelevitch C., Sicouri S. Clinical relevance of cardiac arrhythmias generated by afterdepolarizations. Role of M cells in the generation of U waves, triggered activity and torsade de pointes. J. Am. Coll. Cardiol. 1994;23:259–277. doi: 10.1016/0735-1097(94)90529-0. [DOI] [PubMed] [Google Scholar]
- 4.Yan G.-X., Wu Y., Kowey P.R. Phase 2 early afterdepolarization as a trigger of polymorphic ventricular tachycardia in acquired long-QT syndrome : direct evidence from intracellular recordings in the intact left ventricular wall. Circulation. 2001;103:2851–2856. doi: 10.1161/01.cir.103.23.2851. [DOI] [PubMed] [Google Scholar]
- 5.Choi B.R., Burton F., Salama G. Cytosolic Ca2+ triggers early afterdepolarizations and torsade de pointes in rabbit hearts with type 2 long QT syndrome. J. Physiol. 2002;543:615–631. doi: 10.1113/jphysiol.2002.024570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asano Y., Davidenko J.M., Jalife J. Optical mapping of drug-induced polymorphic arrhythmias and torsade de pointes in the isolated rabbit heart. J. Am. Coll. Cardiol. 1997;29:831–842. doi: 10.1016/s0735-1097(96)00588-8. [DOI] [PubMed] [Google Scholar]
- 7.Clancy C.E., Rudy Y. Linking a genetic defect to its cellular phenotype in a cardiac arrhythmia. Nature. 1999;400:566–569. doi: 10.1038/23034. [DOI] [PubMed] [Google Scholar]
- 8.Li G.R., Lau C.P., Nattel S. Transmural action potential and ionic current remodeling in ventricles of failing canine hearts. Am. J. Physiol. Heart Circ. Physiol. 2002;283:H1031–H1041. doi: 10.1152/ajpheart.00105.2002. [DOI] [PubMed] [Google Scholar]
- 9.Pogwizd S.M., Bers D.M. Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc. Med. 2004;14:61–66. doi: 10.1016/j.tcm.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 10.Wit A.L., Rosen M.R. Pathophysiologic mechanisms of cardiac arrhythmias. Am. Heart J. 1983;106:798–811. doi: 10.1016/0002-8703(83)90003-0. [DOI] [PubMed] [Google Scholar]
- 11.Rosen M.R. Mechanisms for arrhythmias. Am. J. Cardiol. 1988;61:2A–8A. doi: 10.1016/0002-9149(88)90735-7. [DOI] [PubMed] [Google Scholar]
- 12.Yang P.C., Kurokawa J., Clancy C.E. Acute effects of sex steroid hormones on susceptibility to cardiac arrhythmias: a simulation study. PLOS Comput. Biol. 2010;6:e1000658. doi: 10.1371/journal.pcbi.1000658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scarle S., Clayton R.H. Early afterdepolarizations and ventricular arrhythmias in cardiac tissue: a computational study. Med. Biol. Eng. Comput. 2009;47:291–300. doi: 10.1007/s11517-008-0405-0. [DOI] [PubMed] [Google Scholar]
- 14.Gilmour R.F., Jr., Moïse N.S. Triggered activity as a mechanism for inherited ventricular arrhythmias in German shepherd dogs. J. Am. Coll. Cardiol. 1996;27:1526–1533. doi: 10.1016/0735-1097(95)00618-4. [DOI] [PubMed] [Google Scholar]
- 15.Song Y., Thedford S., Belardinelli L. Adenosine-sensitive afterdepolarizations and triggered activity in guinea pig ventricular myocytes. Circ. Res. 1992;70:743–753. doi: 10.1161/01.res.70.4.743. [DOI] [PubMed] [Google Scholar]
- 16.Sridhar A., Nishijima Y., Carnes C.A. Repolarization abnormalities and afterdepolarizations in a canine model of sudden cardiac death. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008;295:R1463–R1472. doi: 10.1152/ajpregu.90583.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato D., Xie L.H., Qu Z. Synchronization of chaotic early afterdepolarizations in the genesis of cardiac arrhythmias. Proc. Natl. Acad. Sci. USA. 2009;106:2983–2988. doi: 10.1073/pnas.0809148106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato D., Xie L.H., Qu Z. Irregularly appearing early afterdepolarizations in cardiac myocytes: random fluctuations or dynamical chaos? Biophys. J. 2010;99:765–773. doi: 10.1016/j.bpj.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lemay M., de Lange E., Kucera J.P. Effects of stochastic channel gating and distribution on the cardiac action potential. J. Theor. Biol. 2011;281:84–96. doi: 10.1016/j.jtbi.2011.04.019. [DOI] [PubMed] [Google Scholar]
- 20.Tanskanen A.J., Greenstein J.L., Winslow R.L. The role of stochastic and modal gating of cardiac L-type Ca2+ channels on early after-depolarizations. Biophys. J. 2005;88:85–95. doi: 10.1529/biophysj.104.051508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pueyo E., Corrias A., Rodríguez B. A multiscale investigation of repolarization variability and its role in cardiac arrhythmogenesis. Biophys. J. 2011;101:2892–2902. doi: 10.1016/j.bpj.2011.09.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banyasz T., Horvath B., Chen-Izu Y. Sequential dissection of multiple ionic currents in single cardiac myocytes under action potential-clamp. J. Mol. Cell. Cardiol. 2011;50:578–581. doi: 10.1016/j.yjmcc.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tran D.X., Sato D., Qu Z. Bifurcation and chaos in a model of cardiac early afterdepolarizations. Phys. Rev. Lett. 2009;102:258103. doi: 10.1103/PhysRevLett.102.258103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Viskin S., Fish R., Barron H.V. Arrhythmias in the congenital long QT syndrome: how often is torsade de pointes pause-dependent? Heart. 2000;83:661–666. doi: 10.1136/heart.83.6.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu J., Laurita K.R. The mechanism of pause-induced torsade de pointes in long QT syndrome. J. Cardiovasc. Electrophysiol. 2005;16:981–987. doi: 10.1111/j.1540-8167.2005.40677.x. [DOI] [PubMed] [Google Scholar]
- 26.Halkin A., Roth A., Viskin S. Pause-dependent torsade de pointes following acute myocardial infarction: a variant of the acquired long QT syndrome. J. Am. Coll. Cardiol. 2001;38:1168–1174. doi: 10.1016/s0735-1097(01)01468-1. [DOI] [PubMed] [Google Scholar]
- 27.Mahajan A., Shiferaw Y., Weiss J.N. A rabbit ventricular action potential model replicating cardiac dynamics at rapid heart rates. Biophys. J. 2008;94:392–410. doi: 10.1529/biophysj.106.98160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie Y., Sato D., Weiss J.N. So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys. J. 2010;99:1408–1415. doi: 10.1016/j.bpj.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qu Z., Garfinkel A. An advanced numerical algorithm for solving partial differential equation in cardiac conduction. IEEE Trans. Biomed. Eng. 1999;49:1166–1168. doi: 10.1109/10.784149. [DOI] [PubMed] [Google Scholar]
- 30.Sato D., Xie Y., Sanderson A.R. Acceleration of cardiac tissue simulation with graphic processing units. Med. Biol. Eng. Comput. 2009;47:1011–1015. doi: 10.1007/s11517-009-0514-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huelsing D.J., Spitzer K.W., Pollard A.E. Electrotonic suppression of early afterdepolarizations in isolated rabbit Purkinje myocytes. Am. J. Physiol. Heart Circ. Physiol. 2000;279:H250–H259. doi: 10.1152/ajpheart.2000.279.1.H250. [DOI] [PubMed] [Google Scholar]
- 32.Saiz J., Ferrero J.M., Jr., Thakor N.V. Influence of electrical coupling on early afterdepolarizations in ventricular myocytes. IEEE Trans. Biomed. Eng. 1999;46:138–147. doi: 10.1109/10.740876. [DOI] [PubMed] [Google Scholar]
- 33.Viswanathan P.C., Rudy Y. Cellular arrhythmogenic effects of congenital and acquired long-QT syndrome in the heterogeneous myocardium. Circulation. 2000;101:1192–1198. doi: 10.1161/01.cir.101.10.1192. [DOI] [PubMed] [Google Scholar]
- 34.Damiano B.P., Rosen M.R. Effects of pacing on triggered activity induced by early afterdepolarizations. Circulation. 1984;69:1013–1025. doi: 10.1161/01.cir.69.5.1013. [DOI] [PubMed] [Google Scholar]
- 35.Morita N., Sovari A.A., Karagueuzian H.S. Increased susceptibility of aged hearts to ventricular fibrillation during oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2009;297:H1594–H1605. doi: 10.1152/ajpheart.00579.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maruyama M., Lin S.F., Chen P.S. Genesis of phase 3 early afterdepolarizations and triggered activity in acquired long-QT syndrome. Circ. Arrhythm. Electrophysiol. 2011;4:103–111. doi: 10.1161/CIRCEP.110.959064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qu Z. Critical mass hypothesis revisited: role of dynamical wave stability in spontaneous termination of cardiac fibrillation. Am. J. Physiol. Heart Circ. Physiol. 2006;290:H255–H263. doi: 10.1152/ajpheart.00668.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qu Z. Chaos in the genesis and maintenance of cardiac arrhythmias. Prog. Biophys. Mol. Biol. 2011;105:247–257. doi: 10.1016/j.pbiomolbio.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Strain M.C., Greenside H.S. Size-dependent transition to high-dimensional chaotic dynamics in a two-dimensional excitable medium. Phys. Rev. Lett. 1998;80:2306–2309. [Google Scholar]
- 40.Chang M.G., Sato D., Qu Z. Bi-stable wave propagation and early afterdepolarization-mediated cardiac arrhythmias. Heart Rhythm. 2012;9:115–122. doi: 10.1016/j.hrthm.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Antzelevitch C., Sicouri S., Liu D.W. Heterogeneity within the ventricular wall. Electrophysiology and pharmacology of epicardial, endocardial, and M cells. Circ. Res. 1991;69:1427–1449. doi: 10.1161/01.res.69.6.1427. [DOI] [PubMed] [Google Scholar]
- 42.Laurita K.R., Katra R., Koo M.H. Transmural heterogeneity of calcium handling in canine. Circ. Res. 2003;92:668–675. doi: 10.1161/01.RES.0000062468.25308.27. [DOI] [PubMed] [Google Scholar]
- 43.Xie F., Qu Z., Weiss J.N. Electrophysiological heterogeneity and stability of reentry in simulated cardiac tissue. Am. J. Physiol. Heart Circ. Physiol. 2001;280:H535–H545. doi: 10.1152/ajpheart.2001.280.2.H535. [DOI] [PubMed] [Google Scholar]
- 44.Krikler D.M., Curry P.V. Torsade de pointes, an atypical ventricular tachycardia. Br. Heart J. 1976;38:117–120. doi: 10.1136/hrt.38.2.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.El-Sherif N., Turitto G. Torsade de pointes. Curr. Opin. Cardiol. 2003;18:6–13. doi: 10.1097/00001573-200301000-00002. [DOI] [PubMed] [Google Scholar]
- 46.Drew B.J., Ackerman M.J., Zareba W., American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology, the Council on Cardiovascular Nursing, and the American College of Cardiology Foundation Prevention of torsade de pointes in hospital settings: a scientific statement from the American Heart Association and the American College of Cardiology Foundation. Circulation. 2010;121:1047–1060. doi: 10.1161/CIRCULATIONAHA.109.192704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng J., Kamiya K., Kodama I. Heterogeneous distribution of the two components of delayed rectifier K+ current: a potential mechanism of the proarrhythmic effects of methanesulfonanilide class III agents. Cardiovasc. Res. 1999;43:135–147. doi: 10.1016/s0008-6363(99)00061-9. [DOI] [PubMed] [Google Scholar]
- 48.Akar F.G., Yan G.X., Rosenbaum D.S. Unique topographical distribution of M cells underlies reentrant mechanism of torsade de pointes in the long-QT syndrome. Circulation. 2002;105:1247–1253. doi: 10.1161/hc1002.105231. [DOI] [PubMed] [Google Scholar]
- 49.Wilders R., Wagner M.B., Jongsma H.J. Effects of anisotropy on the development of cardiac arrhythmias associated with focal activity. Pflugers Arch. 2000;441:301–312. doi: 10.1007/s004240000413. [DOI] [PubMed] [Google Scholar]
- 50.Tveito A., Lines G.T. A condition for setting off ectopic waves in computational models of excitable cells. Math. Biosci. 2008;213:141–150. doi: 10.1016/j.mbs.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 51.Lerma C., Lee C.F., Goldberger A.L. The rule of bigeminy revisited: analysis in sudden cardiac death syndrome. J. Electrocardiol. 2007;40:78–88. doi: 10.1016/j.jelectrocard.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 52.Glass L. Multistable spatiotemporal patterns of cardiac activity. Proc. Natl. Acad. Sci. USA. 2005;102:10409–10410. doi: 10.1073/pnas.0504511102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen W., Aistrup G., Shiferaw Y. A mathematical model of spontaneous calcium release in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2011;300:H1794–H1805. doi: 10.1152/ajpheart.01121.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen W., Asfaw M., Shiferaw Y. The statistics of calcium-mediated focal excitations on a one-dimensional cable. Biophys. J. 2012;102:461–471. doi: 10.1016/j.bpj.2011.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clancy C.E., Tateyama M., Kass R.S. Insights into the molecular mechanisms of bradycardia-triggered arrhythmias in long QT-3 syndrome. J. Clin. Invest. 2002;110:1251–1262. doi: 10.1172/JCI15928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saucerman J.J., Healy S.N., McCulloch A.D. Proarrhythmic consequences of a KCNQ1 AKAP-binding domain mutation: computational models of whole cells and heterogeneous tissue. Circ. Res. 2004;95:1216–1224. doi: 10.1161/01.RES.0000150055.06226.4e. [DOI] [PubMed] [Google Scholar]
- 57.Luo C.H., Rudy Y. A dynamic model of the cardiac ventricular action potential. II. Afterdepolarizations, triggered activity, and potentiation. Circ. Res. 1994;74:1097–1113. doi: 10.1161/01.res.74.6.1097. [DOI] [PubMed] [Google Scholar]
- 58.Corrias A., Giles W., Rodriguez B. Ionic mechanisms of electrophysiological properties and repolarization abnormalities in rabbit Purkinje fibers. Am. J. Physiol. Heart Circ. Physiol. 2011;300:H1806–H1813. doi: 10.1152/ajpheart.01170.2010. [DOI] [PubMed] [Google Scholar]
- 59.Zhao Z., Wen H., Xie L.H. Revisiting the ionic mechanisms of early afterdepolarizations in cardiomyocytes: predominant by Ca waves or Ca currents? Am. J. Physiol. Heart Circ. Physiol. 2012;302:H1636–H1644. doi: 10.1152/ajpheart.00742.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ehara T., Noma A., Ono K. Calcium-activated non-selective cation channel in ventricular cells isolated from adult guinea-pig hearts. J. Physiol. 1988;403:117–133. doi: 10.1113/jphysiol.1988.sp017242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Giles W., Shimoni Y. Comparison of sodium-calcium exchanger and transient inward currents in single cells from rabbit ventricle. J. Physiol. 1989;417:465–481. doi: 10.1113/jphysiol.1989.sp017813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Doerr T., Denger R., Trautwein W. Ionic currents contributing to the action potential in single ventricular myocytes of the guinea pig studied with action potential clamp. Pflugers Arch. 1990;416:230–237. doi: 10.1007/BF00392058. [DOI] [PubMed] [Google Scholar]
- 63.Marder E., Goaillard J.M. Variability, compensation and homeostasis in neuron and network function. Nat. Rev. Neurosci. 2006;7:563–574. doi: 10.1038/nrn1949. [DOI] [PubMed] [Google Scholar]
- 64.Sarkar A.X., Christini D.J., Sobie E.A. Exploiting mathematical models to illuminate electrophysiological variability between individuals. J. Physiol. 2012;590:2555–2567. doi: 10.1113/jphysiol.2011.223313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luo C.H., Rudy Y. A model of the ventricular cardiac action potential. Depolarization, repolarization, and their interaction. Circ. Res. 1991;68:1501–1526. doi: 10.1161/01.res.68.6.1501. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.