

Abstract
Photosynthetic O2 production from water is catalyzed by a cluster of four manganese ions and a tyrosine residue that comprise the redox-active components of the water-oxidizing complex (WOC) of photosystem II (PSII) in all known oxygenic phototrophs. Knowledge of the oxidation states is indispensable for understanding the fundamental principles of catalysis by PSII and the catalytic mechanism of the WOC. Previous spectroscopic studies and redox titrations predicted the net oxidation state of the S0 state to be (MnIII)3MnIV. We have refined a previously developed photoassembly procedure that directly determines the number of oxidizing equivalents needed to assemble the Mn4Ca core of WOC during photoassembly, starting from free MnII and the Mn-depleted apo-WOC complex. This experiment entails counting the number of light flashes required to produce the first O2 molecules during photoassembly. Unlike spectroscopic methods, this process does not require reference to synthetic model complexes. We find the number of photoassembly intermediates required to reach the lowest oxidation state of the WOC, S0, to be three, indicating a net oxidation state three equivalents above four MnII, formally (MnIII)3MnII, whereas the O2 releasing state, S4, corresponds formally to (MnIV)3MnIII. The results from this study have major implications for proposed mechanisms of photosynthetic water oxidation.
Introduction
Oxygenic photosynthesis extracts electrons and protons from water via the water-oxidizing complex (WOC) found within photosystem II (PSII). The WOC is comprised of an inorganic core (Mn4CaO5) that cycles through a series of four intermediate states, called S states, before releasing a molecule of O2. These states are denoted as S0, S1 (dark stable), S2, and S3, with the subscript referring to the number of oxidizing equivalents that are stored through successive electron removal. Despite extensive studies, neither the individual oxidation states of the Mn ions nor their mean oxidation levels during the catalytic process have been conclusively determined in any of the S states. Identifying the oxidation states of Mn operating in the cluster is essential to understanding the catalytic potential and the molecular mechanism of this pervasive biological process, as well as the synthesis of functional water-splitting catalysts modeled on its principles.
The four Mn ions of the WOC are spin coupled, which creates a ground electronic state with a single value for the electronic spin. In the S0 and S2 intermediates, distinctive multiline electron paramagnetic resonance (EPR) spectra are seen at low temperature, and have been assigned to a spin state with one unpaired electron (total spin S = 1/2 (1,2)). This multiline structure in S2 fit simulations based on 55Mn hyperfine interactions with all four Mn ions (3), which constrained the allowed individual Mn spin states to values consistent with this total spin. On this basis, the S2 intermediate is widely accepted to contain only MnIII (S = 2) and MnIV (S = 3/2) ions, but the number of each (one or three Mn being formally MnIV) has not been determined and is not agreed upon (4–7).
Three general types of experiments have been applied to identify the Mn oxidation states: 1), spectroscopic techniques applied to the intact WOC (4,8–11); 2), biogenesis and reassembly of disassembled apo-WOC-PSII complexes from the free inorganic cofactors (12); and 3), chemical titrations using reductants to selectively reduce the PSII-WOC to MnII, which is subsequently quantified after it is released from the PSII protein (13–17). Debate over the Mn oxidation states originates from uncertainties in interpretation and calibration of these methods. For example, oxidation-state assignments derived from spectroscopic techniques are semi-empirical, relying upon comparisons with data from synthetic complexes. These databases are invariably incomplete and generally different for each technique. They are further subject to ambiguity owing to differences in the coordination number and local environment (4,18).
Three different sets of oxidation patterns for the Mn ions through the water-splitting cycle have been proposed, with each set starting from an assumed oxidation distribution for the least oxidized intermediate, S0. Subsequent S-state intermediates are derived from these sets by progressive electron withdrawals. In S0, these proposed individual oxidation states are (MnIII)3MnII, (MnIII)3MnIV, and (MnIV)2MnIIIMnII. These assignments are based on electronic spectroscopy methods, including EPR (4), 55Mn-ENDOR (8), Mn-K-edge absorption (XANES) (9), NMR relaxation (10) and Mn-Kβ emission (11). The latter two possibilities, (MnIII)3MnIV and (MnIV)2MnIIIMnII, can be considered equivalent for our purposes because they have lost the same number of electrons relative to the initial MnII ions from which they formed during the light-dependent assembly process (also called photoactivation in earlier literature). Thus, five one-electron photo-oxidation steps are required to reach S0 for this assignment of oxidation states. By contrast, if S0 has the (MnIII)3MnII oxidation states, S0 is reached after three one-electron photo-oxidation steps starting from four MnII. Consequently, with the inclusion of the four one-electron steps of the S-state water oxidation cycle, the minimum number of photons required to produce the S4 state is expected to be either 7 or 9 for the above two oxidation-state assignments. Here, we call these proposed paradigms the low oxidation (LO)-state and high oxidation (HO)-state paradigms, respectively.
Measurements of the photoassembly process involve detecting O2 after short saturating flashes of light and, to date, favor the LO-state paradigm (19), whereas XAS and ENDOR spectroscopic data have generally been interpreted within the HO-state paradigm (8,9). Photoassembly measures the yield of O2 as a function of flash delivery rate and flash duration starting from the cofactor-extracted apo-WOC-PSII. This method has identified two sequential light-induced intermediates (IM1 and IM2) below the native S0 state (19,20), which have also been characterized by EPR spectroscopy (21–23). These experiments clearly show that only seven flashes are required to advance to the O2 evolving state, using millisecond-duration pulses (10–30 ms). These pulses are long compared with the flash requirements for single oxidation state turnover in the intact WOC, but are the optimum duration for the lowest quantum yield photoassembly step in apo-WOC-PSII (QY ∼ 1%). Single turnover pulses used to advance native S states in the holoenzyme are shorter (<500 μs) (24). As a result, skepticism has prevailed about how to interpret the oxidation state assignments based on photoassembly results, because they contradict the prevailing spectroscopic interpretation.
The chemical-reduction approach for counting the number of oxidation equivalents above the MnII level that are needed to achieve functional turnover relies on titrations using reductants such as NH2OH, NH2NH2, and hydroquinone to form new reduced S states (S-1, S-2, etc.). These states have been invoked to rationalize the delayed patterns of flash O2 yield (25–27) seen on reduction, and proposed to be transiently populated during photoassembly (28). Alternatively, the number of released MnII can be measured (14). Such methods all require a large excess of reductant to observe a response, which then must be fully washed away before assaying for O2 by flashing. Photoassembly studies have revealed that excess chemical reductants invariably result in carryover that cannot be removed by washing (29), resulting in an increase in the number of flashes required to reoxidize the WOC-PSII and produce O2. By contrast, photoassembly in vivo is done without an exogenous reductant except for the native substrate, water.
Here we present an improved photoassembly method that uses a series of much shorter, single-turnover laser flashes of variable duration to eliminate the above uncertainties, coupled with higher-sensitivity detection of O2. This approach reaffirms the previously determined LO-state paradigm as deduced on the basis of photoassembly with long pulses.
Materials and Methods
Preparation of PSII membranes and apo-WOC-PSII
Apo-WOC-PSII samples depleted of MnII, Ca2+, and three extrinsic proteins (PsbO, PsbP, and PsbQ) were prepared as previously described (30), starting from BBY PSII membrane particles (31) with minor modifications. Briefly, the samples were incubated for 40–90 s in an aminosulfonic buffer (20 mM CHES/NaOH, pH 9.4) and divalent cations (200 mM MgCl2) at room temperature, followed by dilution with 50 mM MES, 35 mM NaOH, and 400 mM sucrose buffer (pH 6.0) to stop the reaction. This method reversibly inactivates O2 evolution (32–34) and results in >98% yield of apo-WOC-PSII membranes that are devoid of the three extrinsic WOC proteins as determined by SDS-PAGE (30), exhibit no residual O2 evolution activity (below ∼5 × 10−15 mol O2), and efficiently photoassemble the water-splitting system of PSII with high yield equal to 60–70% as measured by the O2 production rate observed with extrinsic protein-depleted PSII (30).
Photoassembly conditions
Freshly prepared apo-WOC-PSII membranes were diluted to a final concentration of 1 μM apo-WOC-PSII in buffer A (pH 6.0) and 2 mM K3Fe(CN)6. Optimal concentrations of inorganic cofactors were added (100 μM MnCl2, 100 mM CaCl2, and 10 mM NaHCO3) (30,35,36), mixed for ∼30 s, and loaded into the photoassembly cell. Samples were incubated in the dark for 10 min before the start of the illumination. Double hits were suppressed by lowering the MnII/PSII ratio and by using a 1000-fold higher concentration of CaCl2, which competes directly with MnII for binding to the photo-oxidizable site on apo-WOC-PSII (KD = 40–50 μM, at low Ca2+ (10 mM)) (22,31), whereas all other sites are considerably weaker (36,37). At the MnII and Ca2+ concentrations used here, there is <1 MnII bound per PSII. This produces the fastest photoassembly kinetics, requiring the fewest total flashes to reach the endpoint (n = 400). By contrast, using 10× higher MnCl2 (1 mM) and lower CaCl2 (10–20 mM) concentrations results in a considerably slower rate of photoassembly, requiring 1000–3000 flashes and proceeding via a different pathway involving a 150-ms dark process (20,38,39).
O2 electrode/photoassembly cell
O2 was measured amperometrically using an in-house-built Clark-type electrode described previously (12,29), employing a thin microcell configuration (sample cavity 0.25 mm thick, 5 μL volume) covered with a thin, low-density polyethylene membrane that allowed a rapid response time (∼100 ms). Integration of the O2 pulse (for 1.6 s) was done for improved sensitivity (∼1 nM O2). For stable illumination, a GaAs laser diode was used (665 nm, optical power 500 mW in continuous regime that generates rectangular pulses from 1 μs to 100 ms via an in-house-made current driver).
Standard Kok models
The data were modeled using a standard Kok model (40,41) implemented with a Markov chain (42). The PSII-WOC cycles through five flash-induced intermediates (S0, S1 (dark stable), S2, and S3, with O2 formation occurring during the S3-to-S0 transition via a transient S4 state). A standard Kok model includes four possible outcomes of photon excitation (transitions):
γ (hit), single-turnover advancement to the next stable S state.
α (miss), no S-state transition occurs.
β (double hit), double-turnover advancement.
ε (inactivation), enzyme inactivates, no further S-state transitions occur.
The model assigns identical transition probabilities for all Si states (average transition probabilities), allowing a unique determination of the system (27,42). In the real PSII, the flash response is S-state dependent (43–47).
Modeling photoassembly
We used both two-step and four-step models (denoted LO and HO, respectively) described by the Markov chain in Fig. 1 (IMi intermediates) with either the same model with two photoassembly intermediates (IM1∗ and IM2) as in Baranov et al. (30), or four photoassembly intermediates (IM1∗, IM2, IM3, and IM4).
Figure 1.
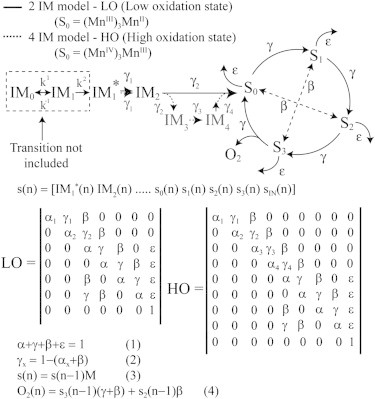
Markov models describing photoassembly processes. Photoassembly (IMi) and Kok (Si) intermediates. Parameters of the LO (solid line), and HO (dotted line) models are indicated. Previously identified kinetic steps leading to formation of IM1∗ are included (k1 and k2) (30) but precede the Markov modeling chain used herein. See Materials and Methods for definitions.
Results and Discussion
Optimizing the Kok parameters
Fig. 2, A and B, display the O2 oscillation pattern observed in intact PSII membranes that were dark-adapted in media with FeCN for 5 min and 20 min, respectively. The simulated trace offset from the data represents a least-squares fitting using a standard Kok model as described above. Factors contributing to the intensity of O2 yield oscillations were systematically investigated, and conditions were optimized for each of the following parameters: flash duration, dark preincubation period, and [FeCN]. A full description follows.
Figure 2.
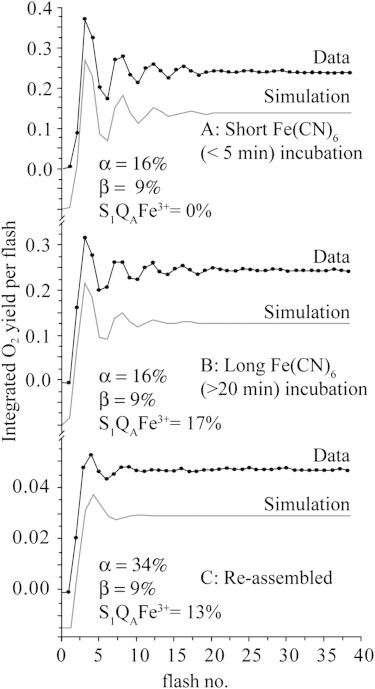
Flash O2 yields observed for intact PSII membranes after dark incubation in 2 mM FeCN/buffer for (A) 4 min and (B) 20 min. (C) Reassembled PSII (reconstituted apo-WOC). Laser flash length: 60 μs; repetition rate: 0.5 Hz. Simulated traces offset from data use a standard Kok model, with parameters indicated.
Flash duration
Titration of the flash length was performed to optimize the O2 yield of intact PSII samples. A 60 μs flash was sufficient to saturate the O2 yield while limiting the double turnovers to 9% (see Figs. 3 A and 3 A; also see optimization in Fig. S1, Fig. S2, Fig. S3, and Table S1 in the Supporting Material). This relatively high value of double turnovers arises in part from the use of FeCN as an electron acceptor during a long dark adaptation, which oxidizes the nonheme in PSII (48). In turn, it serves as an endogenous electron acceptor, causing a higher double-turnover probability (49).
Figure 3.
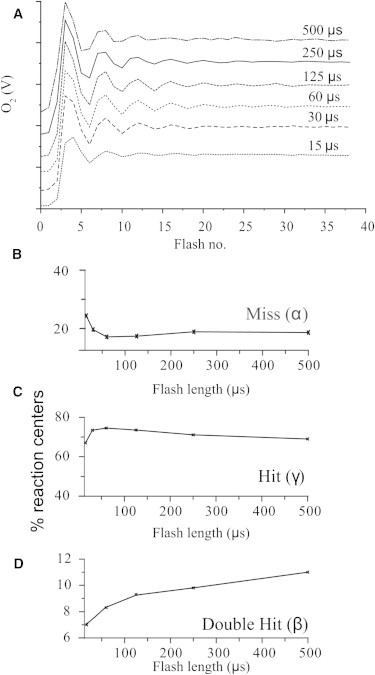
(A) Laser flash length dependence of O2 yield from intact spinach PS II membranes in the presence of 2 mM FeCN and flash lengths of 30–500 μs. (B–D) Flash length dependence of the Kok (B) miss parameter (α), (C) single-hit parameter (γ), and (D) double-hit parameter (β). Samples were dark-adapted for 4 min. Flash rate = 0.5 Hz. Parameters for the simple Kok model were described previously (40). All data traces in Figs. 1–7 are an average of three samples.
Preincubation dark time
The intact PSII enzyme requires a dark-adaptation time to allow the S states to relax to their dark-adapted populations and the plastoquinone pool to reoxidize. The S2 and S3 states decay to S1, whereas S0 remains unaltered on our timescales (41). The decay time required to maximally populate S1+S0 corresponds to that required to maximize the O2 yield on flashes 3 and 4. Here we observed the decay to be first order with a t1/2 of ∼120 s in the presence of FeCN, in agreement with the literature (50). Consequently, the initial preincubation dark time was typically set to 5 min.
Electron acceptor contribution
FeCN is the preferred electron acceptor for photoassembly because it does not interfere with formation of the photoassembly intermediates and produces the fastest rate and highest yield of photoassembly of all tested electron acceptors (29,35,36), including exogenous quinones (30). The optimum concentration of FeCN for photoassembly experiments was previously determined to be 1–2 mM based on O2 yield (29,35,36). Our studies confirm that outcome and reveal that this concentration directly increases the initial dark S2 population, as evidenced by an increase in the double-hit parameter on the first 60-μs flash only. This outcome was identified as being due to oxidation of the nonheme iron (normally ferrous) (48), and was quantified as described next and verified by EPR spectroscopy (Fig. S4).
The effects of two different dark preincubation times on the flash pattern are compared in Fig. 2, A and B. Simulations are shown assuming 0% and 17% population of the initial dark state containing the oxidized nonheme iron (S1QAFe3+). In the simulations presented in Fig. 2, the dark state is assumed to be an admixture of centers with lower and higher probability of first-flash double hits, corresponding to S1QAFe2+ and S1QAFe3+, respectively. From the perspective of the O2 simulations, the S1QAFe3+ state is equivalent to the S2QA state when using saturating flashes. Both states require only one 60-μs flash to progress to the S3 state. Simulations of the Kok S-state cycle, in which all parameters were varied, showed that the initial dark S2 population was specifically increased by dark incubation in FeCN on the timescale of oxidation of the nonheme iron (Fig. 2, A and B, and Fig. S1). This effect was included in the simulations.
O2 oscillation pattern in photoassembled PSII
The required use of excess MnII for fast photoassembly kinetics leads to competition for electron donation at other sites without reconstitution of O2 evolution, and limits the reconstitution yield to ∼70% of active centers (30). This, together with the absence of the extrinsic PSII subunits, results in less efficient turnover (i.e., the miss parameter doubles) (51), which dampens the oscillation pattern of flash-induced O2 yield compared with intact PSII (Fig. 2 C). A significant O2 yield is observed on the second flash, and peaks on both the third and fourth flashes. No oscillations are observed beyond the sixth flash even though the steady-state yield is quite high (60–70%). No unique fitting of these data could be achieved using the Kok model employed in this study. It is reasonable to assume that the double-hit probability (β) in these samples should be no higher than in native samples, because β is controlled by the intrinsic optical cross section of PSII and the pulse duration. A fit assuming the same β-value as in native samples (9%) is shown in Fig. 2 C (fitting results are given in Fig. 1).
Choosing an optimal flash regime for photoassembly
Fig. 1 describes the sequence of photoassembly steps at low MnII loading of the high-affinity site (12,30). IM0 represents all the dark precursor states in which only the MnII oxidation state is present. IM1 and IM1∗, the first light-induced intermediate, has one MnIII bound to the high-affinity site and forms reversibly. IM1∗ forms from IM1 as a result of a dark rearrangement process (k2) that increases the quantum efficiency for advancement upon the next turnover. IM2, the second light-induced intermediate, can only form after this slow, dark rearrangement step (36,37). IM1 and IM1∗ have been characterized by EPR (22,52,53), whereas IM2 and other intermediates (IM3, IM4, etc.) have not yet been detected by EPR. The dark step (k2) is rate-limiting and determines the low overall quantum yield (typically <1%) as the back reaction out competes forward assembly (19,35). The lifetime of the k2 dark step exceeds 10 s in spinach PSII and is instrument limited (19,30). Accurate measurement of k2 is described next.
k2 is intrinsically slow
Light flashes of variable duration (60–500 μs), such as those used to progress the intact PSII through the S-state cycle, do not efficiently assemble the apo-WOC. This is due to the low quantum yield of the initial MnII photooxidation step that forms IM1, and competition between the back reaction and the forward k2 step (19,20). The consequence of this competition on O2 yield is shown in Fig. 4. Using single-turnover flashes of 60 μs duration (spaced 2–3 s apart), we did not observe O2 until approximately the 20th flash. The flash number for observation of O2 rapidly decreased to two flashes as the flash length increased. A significant O2 signal was not observed on the first flash for any flash duration, consistent with the k2 step being intrinsically slow.
Figure 4.
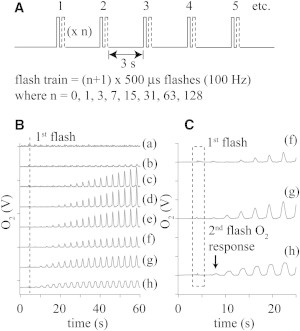
Photoassembly of apo-WOC-PSII using long laser flashes of variable length. (A) Flash sequences used in this experiment. Each flash train is comprised of 20 flashes. Each flash is comprised of a series of n shorter flashlettes (500 μs each). (B) The resulting O2 signal for the 0–50 s range. (C) Enlarged O2 signal for the 0–20 s range. Flash length in units of 500 μs (and O2 activation flash number minus the number of flashes before O2 is observed) for each trace: (a) 1 (14), (b) 2 (8), (c) 4 (6), (d) 8 (4), (e) 16 (2), (f) 32 (2), (g) 64 (2), and (h) 128 (2). Traces are scaled for clarity.
Determination of k2
We performed a titration of the dark time between the first and second flashes to determine k2 (Fig. 5) (19,30). The flash sequence used for this titration is shown in Fig. 5 A. Here, a long preflash of 1 s duration (100 × 60 μs flashlettes) was used to form IM1. Subsequent flashes were clusters of five (single turnover) 60-μs flashlettes separated by 100 ms (10 Hz). We chose to use five flashlettes per cluster to obtain a stronger signal (we obtained the same dark time regardless of the number of flashlettes). These clusters were repeated at a fixed interval of 3 s. Each cluster of five flashlettes is denoted as a flash in Fig. 5, and produced an O2 signal that was used to monitor the course of photoassembly (Fig. 5 B). Fig. 5 C plots the integrated O2 yields for flashes 3–8 as a function of the dark time between the long preflash and the second flash. This figure demonstrates that the O2 yield increased with increasing dark time and approached saturation within the 500 s measurement interval. A simple monoexponential expression gave a good fit with lifetime ∼70 s (parameter b in Y = Y0 + a(1 − e−bt)). Parameter b is equivalent to k2 for the dark rearrangement process. This lifetime provides a more accurate determination than our earlier, instrumentally limited estimate (>10 s) (19).
Figure 5.
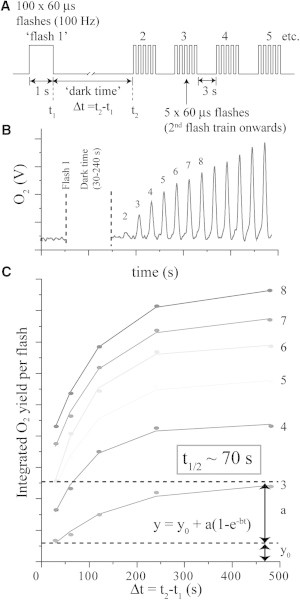
Determination of k2 by titration of the dark time between the first and second flashes used to photoassemble the apo-WOC. (A) Flash sequence used in this experiment. (B) Baseline corrected O2 data. (C) Integrated O2 signal (from B) for flashes 3–8 as a function of the dark time length. (Refer to Fig. 4 for sample.)
Optimal flash sequence
From the results in Figs. 4 and 5, we can design an optimal flash sequence for photoassembly of the apo-WOC-PSII. This sequence achieves the fastest rate of assembly given the intrinsic kinetic limitations of the process. The sequence, depicted in Fig. 6 A, includes two key changes from previous photoassembly studies: 1) a long initial preflash of 1 s duration consisting of 100 60-μs flashlettes with interflashlette separation of 10 ms; and 2), a fixed dark time of 240 s before the next (second) flash, of 60-μs duration (single-turnover duration). This long dark interval allows conversion of IM1 to form IM1∗. The 1-s duration preflash does not allow a significant proportion of the sample to advance past IM1∗, because the required dark process is much slower (k2 ∼ 70 s; Fig. 5). We estimate that no more than 1% of the total number of centers can progress past IM1∗ during the 1-s-long preflash. This experimental approach is designed so that the system must end up in either of the following two measurable states: 1), centers progress to IM1∗; or 2), centers progress past IM1∗ to form completely assembled cluster (in S0 or S1) due to the higher quantum efficiency of the later photoassembly steps (see next paragraph). Centers that do not progress beyond IM0 or decay back to IM0 from IM1 or IM1∗ are not measurable until about the 40th flash (Fig. 6 B) because only inefficient photoassembly takes place during the single 60-μs flashes due to the much slower dark process (k2 ∼ 70 s).
Figure 6.
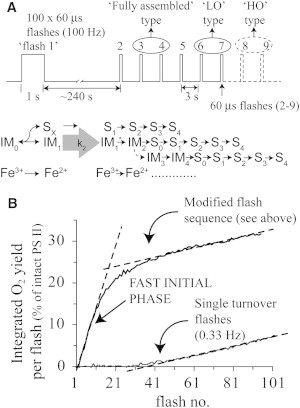
Photoassembly of the apo-WOC using the optimal laser flash sequence. (A) Flash sequence showing expected positions for first O2 release in fully assembled (intact) PSII (photo-assembly LO and HO models from Fig. 1). (B) Comparison of the photoassembly kinetics using the standard flash sequence (60 μs laser flashes spaced by 3 s) or the optimal flash sequence with long preflash (see panel A). The data shown correspond to the integrated O2 signal/flash.
Few centers progress past IM1∗
Data from Tamura et al. (20,54) and others (30,35) established that subsequent photoassembly intermediates (IM2, and potentially IM3 and IM4) form with much higher quantum efficiencies than IM1∗. As a consequence, <1% of the centers that do progress past IM1∗ during the first preflash are expected to be fully assembled by flash 2. Because there is a long dark time between preflash 1 and flash 2 (240 s), the few centers that assembled during the preflash are expected to decay back to the dark stable S states, S1 or S0. This population is accounted for in the simulations.
Photoassembly using the optimal flash sequence
The integrated O2 signal during photoassembly is shown in Fig. 6 B, using the optimal flash sequence and 1-s-long preflash to populate IM1 (Fig. 6 A). The data in Fig. 6 B show that the population of fast photoassembling centers (those that are in IM1∗ or later) is ∼24% of all PSII centers (compared with the steady-state O2 yield of intact PS II). After 20 flashes, a sharp decrease in the slope of O2 recovery occurs, which is an order of magnitude slower than the initial slope (for flashes 20–100). The O2 recovery of apo-WOC-PSII without the long preflash, but otherwise using the same flash sequence (60-μs standard), is also given in Fig. 6 B. It shows only the slow recovery phase due to centers starting from IM0. Thus the rapid phase (∼24% of centers) corresponds to centers that efficiently assembled IM1 (during the preflash) and advanced to IM1∗ (in the dark following the 240-s adaption period) and beyond to fully assemble.
Lag in O2 production during photoassembly
The first 20 flashes (i.e., the first phase) of the photoassembly curve further resolve into two temporally distinct phases when expanded (Fig. 7 A). An initial lag phase producing no O2 occurs on the first two flashes, followed by a shallow increase on flashes 3–5, followed by a sharp increase on flashes 6–9. This small signal observed on flashes 3–5 cannot be readily interpreted within any plausible photoassembly model. An O2 response on the third flash would require both double and triple hits to occur to fully assemble the WOC and evolve O2 from centers starting from IM1∗. Because a double hit for the 60-μs flash duration already has a low probability (9%), the probability of multiple double hits is vanishingly small. By contrast, an initial low amplitude phase is expected due to the low leakage of centers past IM1∗ during the 1-s-long preflash, as described above. Due to the highly cooperative nature of photoassembly, the majority of such centers are expected to be fully assembled and thus generate an O2 signal beginning on the third flash and continuing thereafter.
Figure 7.
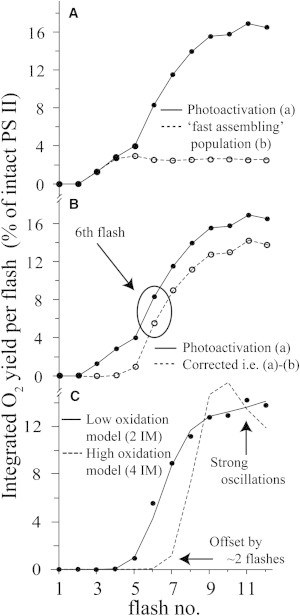
Initial kinetic phases of integrated O2 flash yields recovered during photoassembly using the optimal laser flash sequence (see Fig. 6). (A) a: During photoassembly of apo-WOC-PSII; b: after photo-assembly is complete. (B) a: The same as panel A (a), corrected photoassembly curve equal to a – b. (C) Model simulations of the corrected photoassembly curve using the LO-state model (2 IM) and the HO-state model (4 IM). Parameters used for the LO model simulation: α1 ∼ 20%, α2 ∼ 70%, IM1∗ = 100% (fixed), IM2 = 0% (fixed). Parameters used for the HO model (unconstrained) simulation: α1 ∼ 87%, α2 ∼ 0%, α3 ∼ 0%, α4 ∼ 66%, IM1∗ = 31%, IM2 = 21%, IM3 = 25%, IM4 = 24%. Parameters used for the corresponding constrained HO model simulation: α1 ∼ 0%, α2 ∼ 0%, α3 ∼ 0%, α4 ∼ 66%, IM1∗ = 100% (fixed), IM2 = 0% (fixed), IM3 = 0% (fixed), IM4 = 0% (fixed). Shared parameters for the LO and both HO simulations: α = 34%, γ = 56%, β = 9% (fixed), ε = 0.02% (fixed), S0 (QAFe2+) = 49%, S1 (QAFe2+) = 38%, S1 (QAFe3+) = 13%.
O2 from photoassembled centers is produced on the sixth flash
As expected, the magnitude of these leaked centers is small and is ∼2% of the total number of centers. This compares well with our above estimate of ∼1% leaked centers based on the value of k2 ∼70 s. The calculated O2 signal arising from this fraction of preassembled centers is shown in Fig. 7 A (denoted the fast-assembled population) using a scaled subtraction of the O2 oscillation pattern seen for reassembled PSII (Fig. 2 C). This signal was subtracted from the photoassembly curve. The amount subtracted was such that the corrected photoassembly curve had a zero third flash O2 response (∼2% of the total number of PSII centers, or 8% of the fraction of PSII centers that assembled rapidly). This scaling is not arbitrary because no plausible photoassembly model, based on current understanding, can rationalize a third flash O2 response of significant magnitude. The corrected photoassembly O2 curve is given in Fig. 7 B and reveals a sharp O2 increase beginning on the sixth flash. This number of flashes should therefore correspond to the number of electrons transferred to PSII that are required to make O2, starting from the state IM1∗. A smaller O2 response is observed on the fifth flash (∼10% of the sixth flash signal). This residual peak is expected due to the finite probability of a double-turnover event for a single flash (i.e., ∼9%) and represents the typical offset in O2 response seen for O2 oscillation patterns of the fully assembled oxygen-evolving complex (OEC; see Fig. 2 and below).
Simulation of photoassembly data
Photoassembly steps preceding normal S-state transitions were fitted with the use of transmission parameters, γI = 1− (αi + β) (defined in Fig. 1), whereas S-state transitions used the identical Kok parameters (α and β) obtained from fitting the O2 oscillation pattern of the reassembled PSII (see Fig. 2 C). The values of all parameters used to simulate the photoassembly curves, using the Markov model shown in Fig. 2, are given in the legend to Fig. 7. Details of the fitting are given in Fig. S6, Fig. S7, and Table S2.
Correction for nonheme iron oxidation
Because the dark time between the first preflash and the second flash is long (∼240 s), the nonheme iron partially reoxidizes during this time. This effect is included in the simulations, effectively as a larger double hit (∼10%) on the second flash (first 60-μs flash). For simulation of the initial phase of the corrected O2 photoassembly curve (Fig. 7 B), only centers that had progressed to IM1∗ after the preflash were considered, because only a small fraction of centers had leaked beyond this point, and these were removed as described above. The states IM2, IM3, and IM4 are expected to have vanishingly small initial populations.
LO versus HO model
Using these values for the initial populations and fit parameters, we simulated the O2 photoassembly curve using the LO and HO models (Fig. 7 C). Only the two-intermediate model is consistent with the observed data. It fits the threshold, the two limiting slopes of the corrected O2 photoassembly curve (initial rise and plateau), and the small fifth flash contribution arising from a double hit. It correctly models the first significant O2 burst on the sixth flash and the decrease in slope without oscillations beyond flash 9. By contrast, the same is not true for the four-intermediate model. In this case, IM2, IM3, and IM4 all need to be significantly populated initially to remotely approximate the photoassembly curve. If fitting to the four-intermediate model is constrained such that the initial IM1∗ population dominates, the simulated curve does not agree well with the data (see Fig. 7 C). The simulated curve is offset by two flashes, and large oscillations in O2 are predicted because the calculated miss parameters must be very small (α ∼ 0). As a consequence, our analysis indicates that the two-intermediate model is the best model for interpreting the photoassembly data. The four-intermediate model would require a significant revision of the current photoassembly literature.
Conclusions
The two-intermediate photoassembly model suggested here requires the Mn oxidation level in S0 to be (MnIII)3MnII, with all subsequent S states determined by successive one-electron removals. This result supports the validity of our earlier photoassembly data obtained using long, 30-ms flashes and a constant flash separation for photoassembly (19). In those experiments, we had to obtain seven flashes to observe the first O2 signal starting from IM0. This outcome is the expected result because the dominant factor that determines the flash number for initial O2 detection is the low (∼1%) quantum efficiency of the first photo-oxidation step from IM0. The use of long, 30-ms pulses overcame this limitation by advancing a sufficient population of centers to IM1. Additionally, the use of a relatively short interpulse delay in our earlier studies (much shorter than the intrinsic dark rearrangement time, t1/2 ∼ 70 s) did not allow a significant number of these centers to advance past IM1∗ during the long pulses. The ability of the two-intermediate photoassembly model to explain both the present and earlier results measured under very different flash conditions further supports the validity of the underlying model.
The number of holes captured during photoassembly represents the formal Mn oxidation states and is completely independent of reference standards. As noted in the Introduction, this is not the case for some spectroscopic techniques (such as XAS and ENDOR), the results of which have been interpreted as favoring the HO-state paradigm. To illustrate this ambiguity, a recent XANES study assigned the lower oxidation state model to the OEC (55). When properly structured to accommodate the differential kinetics and inefficiencies of the various steps, which may be independently determined, the photoassembly method relies only on the ability to produce one-electron photo-oxidation steps and to count flashes. However, the method does require the ability to remove all Mn ions from the WOC in PSII and to detect O2 with very high sensitivity. Both of these conditions have been fully met and demonstrated in multiple previous publications and reviews (12,29). Our conclusion that only three photoassembly intermediates can be populated below S0 in apo-WOC-PSII (IM2, IM1, and IM0 differing by one electron each) is in agreement with the conclusion reached by Messinger et al. (25), i.e., that intact PSII membranes treated with hydroxylamine also produce three sequentially reduced states (S-3, S-2, and S-1). However, the latter treatment produces PSII with all extrinsic subunits present and normal probabilities of misses and double hits, unlike the case with apo-WOC-PSII.
The two-intermediate model for photoassembly determined here specifies that the S4 state, which is a precursor to O2 release, should be assigned a formal (MnIV)3MnIII oxidation level. It implies that the mean oxidation state of the four Mn progresses from 2.75 in S0 to 3.75 in S4. No Mn or ligand oxidation to potentially dangerous levels beyond this is required in the natural system.
This clearly has important implications for the catalytic mechanism and suggests, among other things, that a MnV oxidation state (invoked in some mechanistic proposals for water oxidation chemistry) is not needed. These results should have important consequences for mechanistic proposals that rely on the possible chemistry of MnIV and MnIII ions bound within the topological heterocubane geometry suggested by EXAFS and crystallographic data. It also reinforces efforts to synthesize bioinspired catalysts based on the principles of a photosynthetic WOC to target the lower Mn oxidation state found herein.
Acknowledgments
We thank J. Dasgupta and D. Vinyard for suggestions and proofreading.
This work was supported by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences of the U.S. Department of Energy through grant DE-FG02-10ER16195. D.R.J.K. was supported by an American Chemical Society Alternative Energy postdoctoral fellowship and the Dreyfus postdoctoral fellowship in environmental chemistry.
Footnotes
Derrick R. J. Kolling's present address is Department of Chemistry, Marshall University, Huntington, WV.
Nicholas Cox's present address is Max Planck Institut für Bioanorganische Chemie, Mülheiman der Ruhr, Germany.
Supporting Material
References
- 1.Zheng M., Dismukes G.C. Orbital configuration of the valence electrons, ligand field symmetry, and manganese oxidation states of the photosynthetic water oxidizing complex: analysis of the S(2) state multiline EPR signals. Inorg. Chem. 1996;35:3307–3319. doi: 10.1021/ic9512340. [DOI] [PubMed] [Google Scholar]
- 2.Messinger J., Nugent J.H.A., Evans M.C.W. Detection of an EPR multiline signal for the S0∗ state in photosystem II. Biochemistry. 1997;36:11055–11060. doi: 10.1021/bi9711285. [DOI] [PubMed] [Google Scholar]
- 3.Dismukes G.C., Ferris K., Watnick P. EPR spectroscopic evidence for a tetranuclear manganese cluster as the site for photosynthetic oxygen evolution. Photobiochem. Photobiophys. 1982;3:243–256. [Google Scholar]
- 4.Carrell G., Tyryshkin M., Dismukes C. An evaluation of structural models for the photosynthetic water-oxidizing complex derived from spectroscopic and X-ray diffraction signatures. J. Biol. Inorg. Chem. 2002;7:2–22. doi: 10.1007/s00775-001-0305-3. [DOI] [PubMed] [Google Scholar]
- 5.Lubitz W., Reijerse E.J., Messinger J. Solar water-splitting into H2 and O2: design principles of photosystem II and hydrogenases. Energy Environ. Sci. 2008;1:15–31. [Google Scholar]
- 6.Brynda M., Britt R. The manganese-calcium cluster of the oxygen-evolving system: synthetic models, EPR studies, and electronic structure calculations. In: Hanson G., Berliner L., editors. Metals in Biology. Springer; New York: 2010. [Google Scholar]
- 7.Kuzek D., Pace R.J. Probing the oxidation states in the OEC. Insights from spectroscopic, computational and kinetic data. Biochim. Biophys. Acta. 2001;1503:123–137. doi: 10.1016/s0005-2728(00)00218-8. [DOI] [PubMed] [Google Scholar]
- 8.Kulik L.V., Epel B., Messinger J. Electronic structure of the Mn4OxCa cluster in the S0 and S2 states of the oxygen-evolving complex of photosystem II based on pulse 55Mn-ENDOR and EPR spectroscopy. J. Am. Chem. Soc. 2007;129:13421–13435. doi: 10.1021/ja071487f. [DOI] [PubMed] [Google Scholar]
- 9.Roelofs T.A., Liang W., Klein M.P. Oxidation states of the manganese cluster during the flash-induced S-state cycle of the photosynthetic oxygen-evolving complex. Proc. Natl. Acad. Sci. USA. 1996;93:3335–3340. doi: 10.1073/pnas.93.8.3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Srinivasan A.N., Sharp R.R. Flash-induced enhancements in the proton NMR relaxation rate of Photosystem II particles: response to flash trains of 1–5 flashes. Biochim. Biophys. Acta. 1986;851:369–376. [Google Scholar]
- 11.Bergmann U., Grush M.M., Cramer S.P. Characterization of the Mn oxidation states in Photosystem II by KÎ2 x-ray fluorescence spectroscopy. J. Phys. Chem. B. 1998;102:8350–8352. [Google Scholar]
- 12.Dasgupta J., Ananyev G.M., Dismukes G.C. Photoassembly of the water-oxidizing complex in photosystem II. Coord. Chem. Rev. 2008;252:347–360. doi: 10.1016/j.ccr.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Messinger J., Pauly S., Witt H.T. The flash pattern of photosynthetic oxygen evolution after treatment with low concentrations of hydroxylamine as a function of the previous S1/S0-ratio—further evidence that NH2OH reduces the water oxidizing complex in the dark. Z. Naturforsch. 1991;46:1033–1038. [Google Scholar]
- 14.Kuntzleman T., Yocum C.F. Reduction-induced inhibition and Mn(II) release from the photosystem II oxygen-evolving complex by hydroquinone or NH2OH are consistent with a Mn(III)/Mn(III)/Mn(IV)/Mn(IV) oxidation state for the dark-adapted enzyme. Biochemistry. 2005;44:2129–2142. doi: 10.1021/bi048460i. [DOI] [PubMed] [Google Scholar]
- 15.Bouges B. [Action of low concentrations of hydroxylamine on oxygen evolved by Chlorella and spinach chloroplasts] Biochim. Biophys. Acta. 1971;234:103–112. doi: 10.1016/0005-2728(71)90135-6. [DOI] [PubMed] [Google Scholar]
- 16.Shevela D.N., Khorobrykh A.A., Klimov V.V. Effect of bicarbonate on the water-oxidizing complex of photosystem II in the super-reduced S-states. Biochim. Biophys. Acta. 2006;1757:253–261. doi: 10.1016/j.bbabio.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 17.Messinger J., Wacker U., Renger G. Unusual low reactivity of the water oxidase in redox state S3 toward exogenous reductants. Analysis of the NH2OH- and NH2NH2-induced modifications of flash-induced oxygen evolution in isolated spinach thylakoids. Biochemistry. 1991;30:7852–7862. doi: 10.1021/bi00245a027. [DOI] [PubMed] [Google Scholar]
- 18.Zaleski C.M., Weng T.-C., Kessissoglou D.P. Structural and physical characterization of tetranuclear [Mn(II)3Mn(IV)] and [Mn(II)2Mn(III)2] valence-isomer manganese complexes. Inorg. Chem. 2008;47:6127–6136. doi: 10.1021/ic702109c. [DOI] [PubMed] [Google Scholar]
- 19.Zaltsman L., Ananyev G.M., Dismukes G.C. Quantitative kinetic model for photoassembly of the photosynthetic water oxidase from its inorganic constituents: requirements for manganese and calcium in the kinetically resolved steps. Biochemistry. 1997;36:8914–8922. doi: 10.1021/bi970187f. [DOI] [PubMed] [Google Scholar]
- 20.Tamura N., Cheniae G.M. Photoactivation of the water-oxidizing complex in Photosystem II membranes depleted of Mn and extrinsic proteins. I. Biochemical and kinetic characterization. Biochim. Biophys. Acta. 1987;890:179–194. [Google Scholar]
- 21.Miller A.-F., Brudvig G.W. Manganese and calcium requirements for reconstitution of oxygen-evolution activity in manganese-depleted photosystem II membranes. Biochemistry. 1989;28:8181–8190. doi: 10.1021/bi00446a033. [DOI] [PubMed] [Google Scholar]
- 22.Tyryshkin A.M., Watt R.K., Dismukes G.C. Spectroscopic evidence for Ca2+ involvement in the assembly of the Mn4Ca cluster in the photosynthetic water-oxidizing complex. Biochemistry. 2006;45:12876–12889. doi: 10.1021/bi061495t. [DOI] [PubMed] [Google Scholar]
- 23.Ananyev G.M., Dismukes G.C. Calcium induces binding and formation of a spin-coupled dimanganese(II,II) center in the apo-water oxidation complex of photosystem II as precursor to the functional tetra-Mn/Ca cluster. Biochemistry. 1997;36:11342–11350. doi: 10.1021/bi970626a. [DOI] [PubMed] [Google Scholar]
- 24.Joliot P., Joliot A., Barbieri G. Studies of system II photocenters by comparative measurements of luminescence, fluorescence and oxygen emission. Photochem. Photobiol. 1971;14:287–305. [Google Scholar]
- 25.Messinger J., Seaton G., Renger G. S(-3) state of the water oxidase in photosystem II. Biochemistry. 1997;36:6862–6873. doi: 10.1021/bi962653r. [DOI] [PubMed] [Google Scholar]
- 26.Messinger J., Schröder W.P., Renger G. Structure-function relations in photosystem II. Effects of temperature and chaotropic agents on the period four oscillation of flash-induced oxygen evolution. Biochemistry. 1993;32:7658–7668. doi: 10.1021/bi00081a009. [DOI] [PubMed] [Google Scholar]
- 27.Meunier P.C., Burnap R.L., Sherman L.A. Improved five-step modeling of the Photosystem II S-state mechanism in cyanobacteria. Photosynth. Res. 1996;47:61–76. doi: 10.1007/BF00017754. [DOI] [PubMed] [Google Scholar]
- 28.Renger G. Coupling of electron and proton transfer in oxidative water cleavage in photosynthesis. Biochim. Biophys. Acta. 2004;1655:195–204. doi: 10.1016/j.bbabio.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 29.Ananyev G.M., Zaltsman L., Dismukes G.C. The inorganic biochemistry of photosynthetic oxygen evolution/water oxidation. Biochim. Biophys. Acta. 2001;1503:52–68. doi: 10.1016/s0005-2728(00)00215-2. [DOI] [PubMed] [Google Scholar]
- 30.Baranov S.V., Tyryshkin A.M., Klimov V.V. Bicarbonate is a native cofactor for assembly of the manganese cluster of the photosynthetic water oxidizing complex. Kinetics of reconstitution of O2 evolution by photoactivation. Biochemistry. 2004;43:2070–2079. doi: 10.1021/bi034858n. [DOI] [PubMed] [Google Scholar]
- 31.Berthold D.A., Babcock G.T., Yocum C.F. A highly resolved, oxygen-evolving photosystem II preparation from spinach thylakoid membranes: EPR and electron-transport properties. FEBS Lett. 1981;134:231–234. [Google Scholar]
- 32.Ahlbrink R., Haumann M., Junge W. Function of tyrosine Z in water oxidation by photosystem II: electrostatical promotor instead of hydrogen abstractor. Biochemistry. 1998;37:1131–1142. doi: 10.1021/bi9719152. [DOI] [PubMed] [Google Scholar]
- 33.Cheniae G.M., Martin I.F. Photoactivation of manganese catalyst of O2 evolution. 1. Biochemical and kinetic aspects. Biochim. Biophys. Acta. 1971;253:167–181. doi: 10.1016/0005-2728(71)90242-8. [DOI] [PubMed] [Google Scholar]
- 34.Ono T.-A., Inoue Y. Requirement of divalent cations for photoactivation of the latent water-oxidation system in intact chloroplasts from flashed leaves. Biochim. Biophys. Acta. 1983;723:191–201. [Google Scholar]
- 35.Ananyev G.M., Dismukes G.C. Assembly of the tetra-Mn site of photosynthetic water oxidation by photoactivation: Mn stoichiometry and detection of a new intermediate. Biochemistry. 1996;35:4102–4109. doi: 10.1021/bi952667h. [DOI] [PubMed] [Google Scholar]
- 36.Ananyev G.M., Dismukes G.C. High-resolution kinetic studies of the reassembly of the tetra-manganese cluster of photosynthetic water oxidation: proton equilibrium, cations, and electrostatics. Biochemistry. 1996;35:14608–14617. doi: 10.1021/bi960894t. [DOI] [PubMed] [Google Scholar]
- 37.Chen C., Kazimir J., Cheniae G.M. Calcium modulates the photoassembly of photosystem II (Mn)4-clusters by preventing ligation of nonfunctional high-valency states of manganese. Biochemistry. 1995;34:13511–13526. doi: 10.1021/bi00041a031. [DOI] [PubMed] [Google Scholar]
- 38.Hwang H.J., McLain A., Burnap R.L. Photoassembly of the manganese cluster in mutants perturbed in the high affinity Mn-binding site of the H2O-oxidation complex of photosystem II. Biochemistry. 2007;46:13648–13657. doi: 10.1021/bi700761v. [DOI] [PubMed] [Google Scholar]
- 39.Miyao-Tokutomi M., Inoue Y. Improvement by benzoquinones of the quantum yield of photoactivation of photosynthetic oxygen evolution: direct evidence for the two-quantum mechanism. Biochemistry. 1992;31:526–532. doi: 10.1021/bi00117a032. [DOI] [PubMed] [Google Scholar]
- 40.Shinkarev V.P. Oxygen evolution in photosynthesis: simple analytical solution for the Kok model. Biophys. J. 2003;85:435–441. doi: 10.1016/S0006-3495(03)74488-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kok B., Forbush B., McGloin M. Cooperation of charges in photosynthetic O2 evolution—I. A linear four step mechanism. Photochem. Photobiol. 1970;11:457–475. doi: 10.1111/j.1751-1097.1970.tb06017.x. [DOI] [PubMed] [Google Scholar]
- 42.Lavorel J. Matrix analysis of the oxygen evolving system of photosynthesis. J. Theor. Biol. 1976;57:171–185. doi: 10.1016/s0022-5193(76)80011-2. [DOI] [PubMed] [Google Scholar]
- 43.Renger G., Hanssum B. Studies on the deconvolution of flash induced absorption changes into the difference spectra of individual redox steps within the water oxidizing enzyme system. Photosynth. Res. 1988;16:243–259. doi: 10.1007/BF00028843. [DOI] [PubMed] [Google Scholar]
- 44.Lavorel J., Lemasson C. Anomalies in the kinetics of photosynthetic oxygen emission in sequences of flashes revealed by matrix analysis. Effects of carbonyl cyanide m-chlorophenylhydrazone and variation in time parameters. Biochim. Biophys. Acta. 1976;430:501–516. doi: 10.1016/0005-2728(76)90026-8. [DOI] [PubMed] [Google Scholar]
- 45.Delrieu M.J. Evidence for unequal misses in oxygen flash yield sequence in photosynthesis. Z. Naturforsch. C. 1983;38c:247–258. [Google Scholar]
- 46.de Wijn R., van Gorkom H.J. S-state dependence of the miss probability in Photosystem II. Photosynth. Res. 2002;72:217–222. doi: 10.1023/A:1016128632704. [DOI] [PubMed] [Google Scholar]
- 47.Bouges-Bocquet B. Kinetic models for the electron donors of photosystem II of photosynthesis. Biochim. Biophys. Acta. 1980;594:85–103. doi: 10.1016/0304-4173(80)90006-3. [DOI] [PubMed] [Google Scholar]
- 48.Diner B.A., Petrouleas V. Q400, the non-heme iron of the photosystem II iron-quinone complex. A spectroscopic probe of quinone and inhibitor binding to the reaction center. Biochim. Biophys. Acta. 1987;895:107–125. [Google Scholar]
- 49.McEvoy J.P., Brudvig G.W. Redox reactions of the non-heme iron in photosystem II: an EPR spectroscopic study. Biochemistry. 2008;47:13394–13403. doi: 10.1021/bi8013888. [DOI] [PubMed] [Google Scholar]
- 50.Styring S., Miyao M., Rutherford A.W. Formation and flash-dependent oscillation of the S2-state multiline EPR signal in an oxygen-evolving photosystem-II preparation lacking the three extrinsic proteins in the oxygen-evolving system. Biochim. Biophys. Acta. 1987;890:32–38. [Google Scholar]
- 51.Ono T.-a., Inoue Y. S-state turnover in the O2 evolving system of the CaCl2-washed Photosystem II particles depleted of three peripheral proteins as measured by thermoluninescence, Removal of the 33 kDa protein inhibits S3 to S4 transition. Biochim. Biophys. Acta. 1984;806:331–340. [Google Scholar]
- 52.Dasgupta J., Tyryshkin A.M., Dismukes G.C. ESEEM spectroscopy reveals carbonate and an N-donor protein-ligand binding to Mn2+ in the photoassembly reaction of the Mn4Ca cluster in photosystem II. Angew. Chem. Int. Ed. Engl. 2007;46:8028–8031. doi: 10.1002/anie.200702347. [DOI] [PubMed] [Google Scholar]
- 53.Dasgupta J., Tyryshkin A.M., Dismukes G.C. Bicarbonate coordinates to Mn3+ during photo-assembly of the catalytic Mn4Ca core of photosynthetic water oxidation: EPR Characterization. Appl. Magn. Reson. 2010;46:8028–8031. [Google Scholar]
- 54.Tamura N., Inoue Y., Cheniae G.M. Photoactivation of the water-oxidizing complex in photosystem-II membranes depleted of Mn, Ca and extrinsic proteins. 2. Studies on the functions of Ca-2+ Biochim. Biophys. Acta. 1989;37:137–150. [Google Scholar]
- 55.Jaszewski A.R., Petrie S., Stranger R. Toward the assignment of the manganese oxidation pattern in the water-oxidizing complex of photosystem II: a time-dependent DFT study of XANES energies. Chemistry. 2011;17:5699–5713. doi: 10.1002/chem.201001996. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.