

Abstract
Hsp90, the most abundant cellular protein, has been implicated in numerous physiological and pathological processes. It controls protein folding and prevents aggregation, but it also plays a role in cancer and neurological disorders, making it an attractive drug target. Experimental efforts have demonstrated its remarkable structural flexibility and conformational complexity, which enable it to accommodate a variety of clients, but have not been able to provide a detailed molecular description of the conformational transitions. In our molecular dynamics simulations, Hsp90 underwent dramatic structural rearrangements into energetically favorable stretched and compact states. The transitions were guided by key electrostatic interactions between specific residues of opposite subunits. Nucleotide-bound structures showed the same conformational flexibility, although ADP and ATP seemed to potentiate these interactions by stabilizing two different closed conformations. Our observations may explain the difference in dynamic behavior observed among Hsp90 homologs, and the atomic resolution of the conformational transitions helps elucidate the complex chaperone machinery.
Introduction
The biological activity of macromolecules depends on their structure and dynamics, which is why most proteins become fully functional only after they attain their well-defined tertiary structure. Although it is believed that the path to reaching the native state is encoded in a protein's primary sequence, in the cellular environment crowding effects may impede the folding process and promote aggregation. Moreover, during translation, the hydrophobic residues of the nascent polypeptide chains of larger proteins are exposed for periods of seconds to minutes, which is more than enough time for unfavorable interactions to form with other emerging chains or aggregates (1). Molecular chaperones play a key role in helping proteins to mature and keeping them continually functional by ensuring proper de novo folding and correct multimeric assembly, and by helping to restore aberrantly formed structures (2). A group of these proteins, termed heat shock proteins (Hsp), were initially observed to be overexpressed under conditions of cellular stress (3); however, the full extent of their role in the cell has only recently begun to emerge.
Assisted folding is a concerted action of many chaperones, usually accompanied by ATP binding and hydrolysis, with the mechanism and substrate selectivity differing among different chaperones. The 70-kDa Hsp (Hsp70) and a complex multimeric ring known as GroEL, formed mainly from 57-kDa units (Hsp60), operate by making hydrophobic interactions with many nonnative substrates and consequently assist in the formation of the native state. In contrast, the 90-kDa Hsp (Hsp90) is more selective and interacts with well-defined clients in later stages of protein folding (4,5). It is a highly conserved molecular chaperone that is found in all kingdoms of life but archaea (6) and plays diverse biochemical roles under normal physiological conditions. This is not surprising considering that it is the most abundant cellular protein, accounting for 1–2% of all proteins in an unstressed cell (7). A large number of client proteins spanning a wide range of sizes and functions, such as protein kinases, nuclear steroid receptors, telomerase, α-synuclein, and actin (6,8) (http://www.picard.ch), interact with Hsp90. Many of these are oncoproteins, and in fact, cancer cells use the antistress and chaperoning activity of Hsp90 to protect their proteins from misfolding and degradation (9). Additionally, Hsp90 is overexpressed on the surface of cancer cells, correlating with cell motility and metastasis. For these reasons, Hsp90 is a very attractive target in cancer therapy and currently dozens of potential Hsp90 inhibitors are undergoing various oncology and clinical trials (10). The activity of Hsp90 has further been linked to several presently incurable progressive neurodegenerative disorders, such as Alzheimer's disease, Parkinson's disease, and Huntington's disease, and other conditions caused by incorrectly folded or aggregated proteins (11). As a result, interest in investigating the complex chaperone machinery and elucidating its place in the cell metabolism has increased significantly in recent years.
Although investigators have devoted a considerable amount of effort to elucidate how Hsp90 and other chaperones function, some basic aspects of the mechanisms by which they operate remain elusive. Specifically, it is unclear how Hsp90 interacts with its clients at the molecular level, although recent studies have shed light on the manner in which client loading affects its structural arrangement, and identified new substrate contact areas (12,13). Moreover, although it has become clear that Hsps rely on their high conformational flexibility for client recognition and the chaperone cycle (14), little is known about the progression of large-scale protein movements in atomic detail. Much of this lack of detail may be attributed to the delayed discovery of the protein's full-length structure (15,16). Over the last several years, the perception of the chaperone's conformational cycle has grown from a simple open-and-closed state model dictated by nucleotide binding (17,18) into a complex multistate cycle, the direction of which is influenced by client loading, interactions with the substrates and nucleotides, and environmental factors such as acidity and the presence of osmolytes (19–23). As part of the growing effort to elucidate the molecular details of the conformational mechanism, recent experimental work on human Hsp90 implicated the importance of electrostatic interactions in the flexibility and functionality of the protein (24).
The exact role of nucleotides in the conformational cycle of Hsp90 also remains undetermined. However, it is becoming increasingly clear that, rather than irreversibly determining the conformational state of the chaperone, nucleotide binding stabilizes an otherwise accessible state, consistent with the conformational selection mechanism (25). Interestingly, although the conformational equilibrium is conserved between species, the degree to which different conformational ensembles are populated varies strongly (20). At the same time, the remarkably slow ATPase rate (less than one reaction per minute) suggests that ATP hydrolysis is only weakly coupled to the conformational transition, which is observed to occur orders of magnitude faster in the yeast homolog (26,27). Moreover, recent evidence indicates that ATP binding to yeast Hsp90 has little or no effect (28). This leaves open questions regarding the driving force of the conformational change and where the protein gets the energy to power its chaperone activity.
Previous computational work on Hsp90 by Colombo and co-workers (29) yielded important insights into the dynamics of the protein. In their initial simulations of the isolated N-terminal domain (NTD), the authors described a local structural rearrangement of a flexible lid covering the active site upon binding of various substrates. In a subsequent study, they identified interdomain communication pathways from a coarse-grained analysis based on all-atom simulations of the full-length dimer (30). Most recently, they employed a novel comparative scheme to analyze dynamic differences among Hsp90 homologs, which demonstrated the common aspects of the functional dynamics (31).
In this work, we aimed to quantify the thermodynamics of the conformational flexibility of Hsp90, identify specific molecular interactions involved in structural transitions, and unify the currently disparate experimental observations. We captured large-scale structural rearrangements in all of our simulation systems and identified key interactions governing the conformational change. We observed a remarkable flexibility in the protein that allowed it to undergo considerable and complex rigid-body movements within 100 ns of simulation time. By varying the presence of bound nucleotide (ADP and ATP) in open and closed conformations, we were able to discern how nucleotide binding shifts the direction of the conformational change and the effect it has on the structural stability of the protein. Finally, we defined a path of structural change and performed umbrella sampling to calculate the free energy associated with the conformational cycle.
Methods
Structure modeling
The full-length structures of Escherichia coli Hsp90 were previously determined by Shiau and co-workers (15) using x-ray crystallography (PDB IDs: 2IOQ (apo structure) and 2IOP (ADP-bound structure)). Both models contained several gaps in the structure, none of which were >20 residues long (apo structure: residues 1–14, 100–114, 494–500, and 544–564 in both chains; ADP-bound structure: residues 494–499 in both chains and 118–120 in chain B only). These gaps were patched using Modeler with loop refinement (32). The missing residues from the NTD of the apo structure were modeled using NTD in the ADP-bound structure as reference, and the remaining missing residues were treated as unstructured.
Molecular dynamics
We performed molecular dynamics simulations using Gromacs 4.5 (33) with the Gromos 45a3 force field (34). We derived the parameters for ADP from the existing ATP parameters in the same force field by removing the β-phosphate. Constant temperature was maintained by velocity rescaling with a coupling time of 0.1 ps (35), and constant pressure was maintained by a barostat at 1 atm, with a coupling time of 0.5 ps (36). Electrostatic interactions were treated by particle-mesh Ewald summation (37) with a cutoff of 0.9 nm, and the pair list was updated every five steps. Newton's equations of motion were integrated by means of the leapfrog algorithm with a step size of 2 fs. Initial velocities were taken from the Maxwell-Boltzmann distribution at the reference temperature of the simulation. Center-of-mass motion and rotation around the center of mass were removed every 10 steps. All bonds were constrained using LINCS with a tolerance of 0.0001 (38).
Structure optimization
The structures were solvated with SPC water molecules (39) and neutralized with Mg2+ ions in a rectangular box with edges 1.2 nm away from the farthest atom in each direction. An extensive minimization protocol was then conducted to alleviate the strains from the crystal structure and introduced by modeling. Initially, the minimization was carried out by keeping the protein's backbone and the ligand (if present) restrained with the potential of 10 000 kJ mol−1 nm−2, after which unrestrained minimization was performed. This was followed by 120-ps-long simulated annealing, with cooling from 450 K to 200 K in 50-K intervals, each 15 ps long. Finally, the structures were minimized one more time.
Preequilibration
Structures were prepared for long molecular dynamics simulations using a 100-ps-long preequilibration period. During this time, the system was heated in the NVT ensemble from 100 K to 300 K in 50-K intervals, each 20 ps long, while the restraining potential on the protein's backbone atoms was simultaneously relaxed from 25 000 kJ mol−1 nm−2 to 5000 kJ mol−1 nm−2 in 5000 kJ mol−1 nm−2 intervals. Finally, the system was simulated under constant NPT conditions, unrestrained, for 20 ps at 300 K.
Simulation systems
In addition to the two principal, initial configurations (apo structure (open conformation) and ADP-bound structure (closed extended conformation)), we generated three more systems: ADP was removed from the binding site in the extended conformation, and ADP and ATP were inserted into the binding site of the open conformation based on alignment against the NTD of the ADP-bound structure. A magnesium ion was placed in the coordination sphere of each nucleotide to coincide with the observed coordination in the isolated NTD (40) (PBD ID: 1y4s). Each of the five different systems was simulated for 100–150 ns in multiple copies that differed in initial velocities (for a list of trajectories, see Table S1 of the Supporting Material).
Analysis
Protein sequences of the E. coli (HtpG), yeast (Hsp82), and human (Hsp90α, two isoforms) homologs were taken from GenBank (www.ncbi.nlm.nih.gov/genbank/) and aligned on the ClustalW2 web server using the slow method with default settings (41). Structures were visualized with VMD (42).
Free-energy simulations
Using snapshots from the unbiased apo Hsp90 simulations, we generated 35 umbrella sampling windows with conformational angles ranging from 0.01 rad to 2.46 rad, each separated by 0.07 rad. The configurations were minimized and preequilibration was done as described above, with the time of the final NPT step extended to 300 ps. Umbrella sampling simulations of each window (43) were run for 4 ns, with the minimum of the potential set at the corresponding conformational angle, using a force constant of 2000 kJ mol−1 nm−2. We calculated the potential of mean force using the weighted histogram analysis method (44) by splitting the sampled space in 500 bins. We calculated errors using the Monte Carlo bootstrap analysis, accounting for a correlation time of 100 steps (45). The histogram of the simulation is shown in Fig. S1. It is worth noting that we tried other collective variables (i.e., root mean-square deviation (RMSD), absolute position of atoms, radius of gyration, and an alternative conformational angle (with planes passing through the other domains)), and we also attempted to use replica-exchange molecular dynamics. Using the conformational angle as the collective variable gave the clearest and most instructive results.
Results and Discussion
The open state of Hsp90 favors the stretched and compact states
In its functional form, Hsp90 is a homodimer, with each subunit composed of three independent domains: the C-terminal domain (CTD), which acts as the permanent dimerization site; the middle domain (MD), which is largely implicated in client binding; and the NTD, which contains the nucleotide-binding pocket (Fig. 1). The domains are linked with long strands, suggesting high rotational mobility, especially in the case of the MD and CTD, which are connected with an unstructured link 10 residues long. This confers a great flexibility to the NTD/MD with respect to the dimerization region. In fact, solution small-angle x-ray scattering (SAXS) experiments and electron microscopy imaging showed the remarkable flexibility of these domains, especially in apo (ligand-free) conditions (19,20). Most likely, this conformational flexibility precluded earlier determination of the full-length x-ray crystal structure at atomic resolution.
Figure 1.
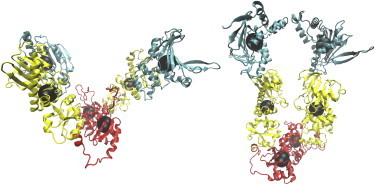
Initial configurations of Hsp90. Left: Apo (open) state. Right: ADP-bound (extended) state. Black spheres represent centers of mass of the corresponding domains. Color-coding (online) or shading (print) highlights the individual domains: cyan (top, NTD), yellow (middle, MD), and red (bottom, CTD).
The initial configurations used in the molecular dynamics simulations were based on the structure of E. coli Hsp90 (also known as HtpG) determined by x-ray crystallography (15) in both apo (open) and ADP-bound (closed extended) states (Fig. 1). We believe this model is more appropriate than the alternative yeast full-length structure for studying the protein's behavior in solution (16), because the E. coli homolog was not crystalized with cochaperones or other substrates. In addition, having two configurations in different structural states (free and bound to its natural nucleotide, ADP) allows a more comprehensive analysis of the conformational properties of the system. Five systems (as detailed in Materials and Methods) were simulated in five copies of the two principal systems (from the crystal structure) and two copies of the three variations in nucleotide binding. Each copy was run for 100–150 ns, yielding 1.75 μs of total simulated time (Table S1).
Even on the 100-ns timescale, the apo Hsp90 simulations showed dramatic conformational rearrangements characterized by a propelling motion of the subunits around the dimerization region in two directions, resulting in two different structures: a stretched state and a closed compact state (Fig. 2). We traced the conformational change by measuring the degree of openness of the two subunits, defined by the angle between two planes that intersect at the line passing through the centers of masses of the CTDs, and each comprising the center of mass of the corresponding NTD. Essentially, this is a dihedral angle defined by the consecutive sequence of terminal domains, and we refer it as the conformational angle, ϑ (Fig. 2).
Figure 2.
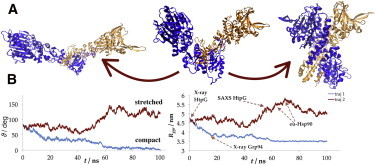
Large-scale conformational rearrangement of apo Hsp90. (A) Middle: Initial configuration showing the conformational angle (ϑ) defined as the angle between planes intersecting at the dimerization region and each passing through the center of mass of its NTD. Left and right: Final configurations of two independent trajectories. Color-coding (online) or shading (print) reflects different subunits. (B) Tracing the conformational angle (left) and the radius of gyration (right) in two independent trajectories. Trajectory 1 corresponds to A-right, trajectory 2 to A-left. Grp-94 is the endoplasmic reticulum and eu-Hsp90 eukaryotic homolog of Hsp90. X-ray stands for the value calculated from the crystallographic structure.
The measurement of ϑ clearly demonstrates the progression of the conformational change (Fig. 2 B): the conformational angle increased from 80° to 140° during the course of a stretching motion (in the alternative setup even to 180°; see “How is nucleotide binding coupled with conformational change?” section below), and decreased to essentially 0° in the closure movement. Such rotational flexibility of the NTD and MD about the hinge region connecting CTD with MD agrees well with solution scattering experiments (19) in which the radius of gyration (Rgyr) of the free Hsp90 in solution (5.2 nm) was found to be considerably higher than the one calculated from the full-length crystal structure (4.4 nm). This suggests that the solution structure is highly dynamic and different from the one predicted by x-ray crystallography, clearly in support of the results of our simulations (Fig. 2 B). Moreover, the eukaryotic homolog (in Fig. 2 denoted as eu-Hsp90) shows an even higher Rgyr in solution (5.5 nm or 5.8 nm depending on the calculation method) that corresponds with the highest values seen in our simulations (46). Our simulations demonstrate that subunits of Hsp90 in solution may undergo stretching motions well beyond the 120° angle predicted by SAXS experiments, without compromising structural integrity.
The structure appears to be equally flexible with respect to the closure motion. The closed conformation of the protein from our simulations (Fig. 2 A, right) bears a resemblance to a more compact structure observed in Grp-94, the endoplasmic reticulum homolog of Hsp90 (47). In this conformation, the NTDs show significant contact between the binding pockets, unlike the closed extended state from the ADP-bound crystal structure, where the interactions are established only via a short helical segment from a region termed the lid (see below). Furthermore, our simulations predict that the closure proceeds far beyond what is seen in the Grp-94 structure into a completely closed and compact state. The final configurations from other independent apo Hsp90 simulations that demonstrated various intermediate degrees of openness are shown in Fig. S2. In two trajectories, only a modest change in the conformational angles was observed, although other indicators show that a rearrangement indeed occurred. In particular, the distance between the MDs increased by 1 nm in trajectory 3, and by 2 nm in trajectory 4, corresponding to opening and closing, respectively. Additionally, an RMSD analysis showed the same behavior as in the trajectories, where a dramatic rearrangement was observed: individual domains did not deviate too much, whereas taking the calculation over the entire structure, considerable deviation was observed, indicating large-scale motions (Fig. S3). These observations demonstrate the complexity of motion of such a large protein system and the difficulty of analyzing it, which warranted our thorough equilibration scheme and a long total sampling time.
The twofold fate of the conformations of apo Hsp90 suggests that the closed compact and stretched states are more favorable than the open crystal state. We carried out umbrella sampling simulations along the path of the conformational angle, ranging from 0.5° to 140°, by taking the initial configurations from the unbiased simulations. The results show (Fig. 3) that the barrier between the stretched and the compact states lies very close to the crystal open conformation (at 77°, compared with 81° in the crystal state). This raises doubts as to the stability of the conformation seen in the crystal structure and its physiological relevance. Binding of competent states most likely corresponds to regions on the shoulders of the free-energy profile, observed on both sides of the maximum, corresponding to angles of 20–46° and 110–132°. At this range, the protein is fairly free to change the domain arrangement and accommodate its structure to receive clients.
Figure 3.
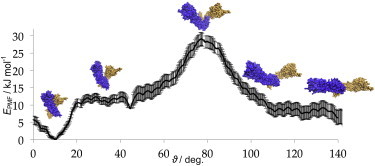
Free-energy simulation of apo Hsp90. The potential of mean force with respect to the conformational angle (the angle between planes going through the center of masses of the NTD and CTD of their respective monomers) is shown. The maximum and minimum are observed at 77° and 9°, respectively.
According to our calculations, the stretched state needs to overcome a slightly higher barrier (18.5 kJ/mol (4.4 kcal/mol) high) to reach the compact state compared with the opposite transition (16.0 kJ/mol (3.8 kcal/mol)). The existence of a barrier in the middle of the conformational space may explain the slow transition from the fully open (probably similar to the stretched structure seen in this work) to a compact state, measured in the yeast homolog. The yeast homolog can be assumed to show a similar free-energy profile, based on the conservation of all but one of the residues involved in the conformational change.
Strong electrostatic attractions (see next section), possibly enhanced by overall increased hydrophobic interactions, appear to push the protein into a free-energy well centered at 9°, ∼27.3 kJ/mol (6.5 kcal/mol) deep with respect to the maximum, and roughly 11.5 kJ/mol (2.8 kcal/mol) lower in energy than the flat segment of the closed state. It appears that the transition into a fully compact conformation may trap the protein in this state for an extended period of time, as suggested by single-molecule fluorescence experiments (26). Importantly, the way collective variables were defined makes the free-energy profile very coarse, and thus it should be taken as a qualitative demonstration of the process. We attempted to use other, more complex collective variables as well as different free-energy simulation methods (such as replica-exchange molecular dynamics), but the complexity of the protein's structure and dynamics prevented convergence. Nonetheless, our free-energy profile agrees with observations from unbiased simulations, and it seems to explain the most likely range of conformational angles the protein adopts in the solution.
Key electrostatic interactions guide Hsp90 into a closed compact conformation
In apo Hsp90 simulations, the structure showed a very high RMSD, implying that large-scale structural rearrangements were taking place (Fig. S3). When the calculations were performed separately on each subunit, the RMSD values decreased significantly, indicating that the chains behave as independent rigid bodies and that in the conformational change, the secondary structure remains undisturbed. Even when individual domains were being aligned, the CTD showed significant deviation in one of the chains. We traced the discrepancy to a long unstructured region spanning residues 545–565, known as H21. After this region was omitted from the RMSD calculations, the deviation of the CTDs decreased to a level comparable to that observed for other domains.
Although the crystal structure of the isolated CTD shows this region to be α-helical (and hence H21 or the 21st helix), in the full-length structure (and in our work) H21 is disordered (15). In our simulations, it formed a long, unstructured loop that projected into the cleft separating the two subunits and made significant contacts with the NTD of the opposite chain, specifically with the lid region (Fig. 4). It appears that the predominantly polar interactions between H21 loop and the lid guide the progression of the conformational change (Fig. 4, A and B). Interactions were formed between Gln-559 (H21) and Asp-112 (lid), Lys-552 (H21) and Glu-107 (lid), and Lys-560 (H21) and Asp-116 (lid). Moreover, the two regions were held together by backbone interactions between residues 106–112 in the lid and 553–562 from the loop. Occasionally, the loop would project into the solution, leaving one site of the cleft exposed. This may make the protein accessible to clients and possibly guide them toward their binding site (Fig. 4 C, left). The helical portion of the two lids was kept in contact through interactions between Lys-103 from the lid and Asp-292 from the opposite MD, and intermittently by interactions with Asp-287 and Glu-384, also from the opposite chain. These interactions appear to be crucial in helping to maintain the protein's closed conformation. Interestingly, although Lys-103 is conserved in human and yeast Hsp90, Asp-292 is replaced by lysine, which would cause unfavorable electrostatic repulsions and ultimately prevent the tight interaction between the two lids (Fig. S4). This may explain why the closed state is minimally populated in human and yeast homologs (20). Here we focus on the global aspects of the structural transitions, potentially applicable to other homologs, and give their molecular justifications. Other, finer motions in the protein accompany these changes, but due to the complex nature of the structure, a full description of these motions is beyond the scope of this work.
Figure 4.
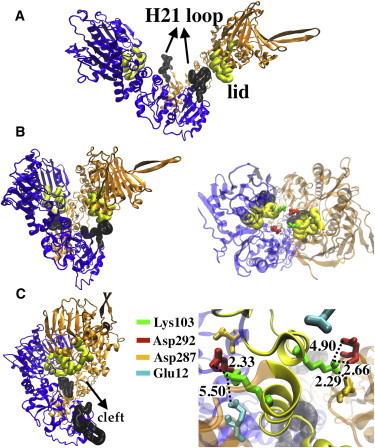
Interactions leading to the closed conformation of apo Hsp90. Black bold tubes represent the H21 loop, and yellow (in print light shaded) tubes and ribbons represent the lid. (A) Initial configuration of apo Hsp90, showing the projection of the H21 into the cleft. (B) The conformation immediately before the fully compact state, showing interactions between H21 and the lid from the opposite chain. Right: Top view of the same configuration with the interlocking residues Lys-103 (colored green, in print light shaded) and Asp-292 (colored red, in print dark shaded). (C) The fully compact state. Left: One H21 loop occasionally projects into the solution, leaving the cleft exposed. Right: A close-up of the lid-lid interaction site shows the electrostatic interactions between Lys-103 and Asp-292 that are seen in all trajectories. Also, in some simulations, contacts are established between Lys-103 and Glu-12 and Asp-287. The distances are denoted in angstroms.
We propose that the mechanism of the conformational change toward the compact state proceeds as follows: One or both H21 loops establish contact with the lid of the opposite NTD through mostly polar interactions. The conformational angle decreases and both NTDs bend toward the opposite CTDs (distinguishing this state from the closed extended state). Finally, the helical segments of lids bind together by means of electrostatic attractions between a lysine in the lid and an aspartate from the MD of the opposite subunit. As a result, a large interface between the NTDs is created in which the binding pockets face each other, a characteristic of the compact state.
How is nucleotide binding coupled with conformational change?
The NTD carries the nucleotide-binding site, which is characterized by an uncommon structural motif known as the Bergerat fold (48). This motif is found among members of the GHKL superfamily, which consists of only four protein classes: gyrases, Hsp90, histidine kinases, and MutL (DNA mismatch repair proteins). The nucleoside moiety of the ligand (base and sugar) fits in a hydrophobic pocket, whereas the phosphates are exposed to the cleft separating the subunits. In all simulations that started from the closed extended conformation, ADP was consistently coordinated by the magnesium ion and a set of residues from both subunits, namely, Lys-45, Ser-100, His-93, Asp-41, and occasionally Lys-99 and Lys-103 (Fig. 5, left). It is likely that these residues participate in ATP hydrolysis, given that similar residues are known to be catalytically important in other biochemical reactions (e.g., the catalytic triad in proteases). In contrast to the closed compact state seen in the apo simulations, in the closed extended state Asp-287 and Asp-292 are far from the binding site and both lids. Interestingly, the two residues are the primary points of contact between the MDs, where the aspartates are bridged by a magnesium ion and water molecules (Fig. 5, right).
Figure 5.
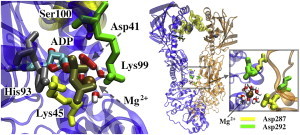
Simulations of the extended state (ADP-bound). Left: Nucleotide coordination sphere. The phosphate moiety is coordinated by Lys-45, Asp-41, Lys-99, His-93, Ser-100, and a magnesium ion. Right: MDs come in contact via Asp-287 and Asp-292 bridged by a magnesium ion and water molecules (inset).
In the ADP-bound closed structure, the NTDs are considerably farther away from the CTDs of the opposite chains than in the compact state (∼8 nm compared with 5.7 nm, hence termed the extended state). Simulations of the extended state showed no large-scale rearrangements, and instead the protein motions were confined to local motions within the domains. Both the primary dimerization site (via the CTDs) and the secondary dimerization region between the lids remained intact during the course of our simulations. We observed that the lids shifted toward the nucleotide early in simulations, with the NTD bending toward the cleft and moving 0.5–1.5 nm closer to the opposite NTD (Fig. 6). However, the MDs showed the most significant movements, either by establishing contacts with each other or by twisting around the NTD/MD interface (Fig. 6; for all trajectories, see Fig. S5, Fig. S6, and Fig. S7). We quantified the latter case by measuring the alternative conformational angle, defined by the planes encompassing the MDs rather than the NTDs, which increased by 15–20°, whereas the dihedral angle between the NTDs remained constant. Therefore, the initial rotating motion of the NTDs and the more extensive bending and twisting movements of the MDs may be events that precede the detachment of the NTDs, marking the initiation of the conformational change into the open state.
Figure 6.
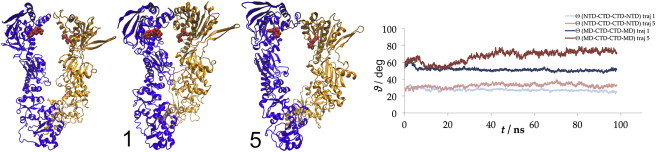
Simulation of the extended (ADP-bound) state. Left: The initial configuration (1 and 5 mark the final configurations of the individual trajectories; the numbers correspond to Table S1 and Fig. S4). ADP molecules are represented as van der Waals spheres; color-coding (in print shading) highlights the subunits. Right: Conformational angle measurements for the shown trajectories done in two ways, with planes passing through the NTDs (light lines) and alternatively through the MDs (dark lines).
To study the global and local effects of nucleotide binding on both crystal states, we generated additional systems by removing the ligand from the ADP-bound state and by inserting ADP and ATP into the binding site of the open configuration. In one of the two simulated cases, where the ADP was removed from the closed extended state, the NTD bent toward the MD of the opposite chain, moving into the cleft. This motion appears to be prompted by the interactions of Lys-45 (which normally coordinates the phosphate moiety of ADP) and Arg-49 from the NTD, with Asp-292 and Asp-287 from the other MD (Fig. 7). This NTD collapse was not observed in any of the ADP-bound simulations, and we believe that it could mark the initiation of the conformational transition into the closed compact state, observed in apo Hsp90. Nucleotide binding in the closed extended state appears to impede key electrostatic interactions required for the conformational transition. Interestingly, Asp-287 and Asp-292, which were implicated in driving the conformational change into the closed compact state (see above), participated in the intersubunit interactions in the closed extended state as well.
Figure 7.
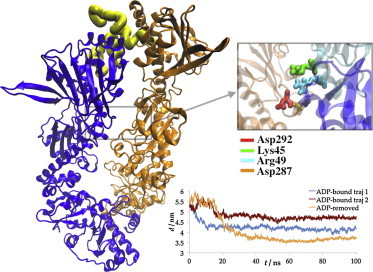
Simulations of the extended state with ADP removed. Left: A collapse is observed with NTD of one chain coming close to the NTD and MD of the opposite chain. The inset shows a magnified view of the interactions formed between the opposite chains mainly via Lys-45 of the collapsing chain and Asp-292 and Asp-287 of the opposite chain. Interestingly, Asp-287 and Asp-292 again play a role in intersubunit interactions. Bottom right: NTD–NTD distance measurements for the two trajectories of the ADP-bound crystal structure compared with the one with ADP removed.
When ADP and ATP were inserted into the binding site of the open crystal state, the protein showed behavior similar to that observed in the apo Hsp90 simulations. Both stretching and closure motions were observed with either nucleotide (Fig. 8 and Fig. S7). Of note, in the stretched conformation of the ATP-inserted system, the NTDs were closer to the opposite CTDs than in the apo or ADP-inserted systems (4.4 nm compared with 6.4 nm average distance; Fig. 8, bottom right). During closing, the distance stayed essentially the same. We interpret this to mean that the stretching of the structure is a propelling motion that may be accompanied by sliding of the subunits. In simulations with ATP inserted into the apo state, in addition to transitioning into the closed conformation, the subunits extended with an increase of the NTD–CTD distance from 6.4 nm to 7 nm, a distance similar to that seen in the ADP-bound crystal structure, and did not create an interface between the binding sites, as was observed in simulations starting from the open state (both apo and ADP-bound). Thus, ATP binding may stabilize the extended closed state, rather than a compact state.
Figure 8.
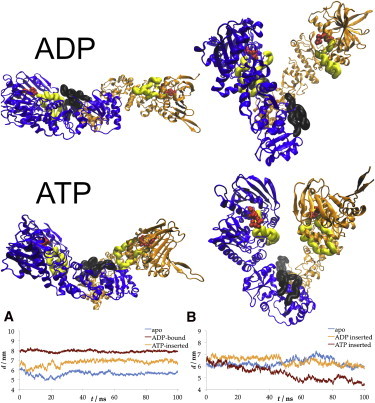
Final configurations of the simulations with ADP and ATP inserted into the apo state. Both setups show stretching and closing motions. In the ATP-inserted simulations, the protein proceeds into a more extended structure, in similarity to the ADP-bound crystal state (middle right). Bottom: Average NTD–CTD distance measurements (between opposite chains). (A) Closing motion in apo, extended (with ADP bound), and ATP-inserted simulations. The ATP-inserted state approaches the extended state rather than the compact state. (B) Stretching motion in simulations of apo ADP-inserted and ATP-inserted states. In ATP-bound simulations, in addition to stretching, subunits slide a distance of 2 nm against each other.
Conclusions
The intricate role played by molecular chaperones in controlling protein maturation and trafficking, repairing aberrant structures, preventing the formation of aggregates, and ultimately affecting cellular viability has attracted much attention in recent years, and it is becoming clear that the effect of Hsp90 on proteostasis is far more pervasive than originally thought. The remarkable structural features of Hsp90, characterized by relatively rigid segments separated by long unstructured strands, allow the subunits and domains to undergo rigid-body movements and consequently form a variety of conformational states. We carried out molecular dynamics simulations, starting from two different states of the full-length crystal structure of HtpG, to gain more comprehensive structural and thermodynamic insights into Hsp90 and further our understanding of the conformational properties of this chaperone at atomic resolution. The results of our simulations show that Hsp90 possesses an extraordinary dynamic flexibility that is independent of the initial conformational state or the presence of nucleotides. The most dramatic rearrangements occurred starting from the open state, where in independent simulations that were identical except for the initial velocity distribution of atoms, the protein experienced two different fates. A thermodynamic analysis showed that a structure very similar to the crystal state lies on top of the free-energy barrier separating the more-favorable stretched and compact conformations. It also suggested that the protein might easily sample various conformational angles, allowing it to bind an impressive array of clients that differ largely in size and function. Experimental efforts have shown how the ensemble average structural parameters of the protein are easily influenced by various environmental parameters, so we do not expect the average Rgyr in our simulations to match any single reported value. We show, however, that the model is able to capture the full scope of the observed conformational freedom and therefore link configurations of the protein to the underlying molecular interactions.
On the basis of our simulations, we propose the following conformational cycle: The apo state is free to shift between the compact and fully stretched states, with a free-energy barrier at a conformational angle of 77°. ATP binding stabilizes the more extended closed state, which may be maintained by the nucleotide even after hydrolysis. In the ADP-bound state, the protein may undergo a transition into an open conformation, induced by outward twisting of the MDs. Release of the nucleotide from the extended state leads to the formation of the compact structure, guided by interactions between Asp-287 and Asp-292 from the MD and Lys-103 from the NTD, which is normally involved in nucleotide coordination. It is worth noting that in vivo, Hsp90 interacts with cochaperones and its clients, which may quantitatively or qualitatively change the free-energy profile and consequently the conformational cycle.
Because several key local interactions significantly affect the global motions of the protein, Hsp90 appears to be amenable to evolutionary fine-tuning by point mutations that would regulate the populations of different conformational states between homologs. The results of our work suggest that the electrostatic interactions between residues in different subunits and the magnesium cation are key for guiding the conformational change. Binding of nucleotide potentiates the interactions between the subunits because the charged phosphate moiety projects into the cleft. Therefore, even if the ATP hydrolysis itself is not directly involved in the dynamic flexibility of Hsp90, small, hydrophobic molecule inhibitors that attack the nucleotide-binding site could affect the chaperone cycle by preventing the favorable electrostatic interactions. Alternatively, directly targeting the key polar residues described here may be an effective means of inhibiting Hsp90.
The molecular and thermodynamic description of the progression of the conformational transitions presented in this work enhances our understanding of the conformational diversity of Hsp90. These results may help broaden our understanding of the complex roles the chaperone plays in proteostasis and cell proliferation, and ultimately could lead to methods capable of modulating Hsp90 activity, a development that has the potential to affect a wide range of diseases.
Acknowledgments
We thank Marissa Saunders and John Grime for critically reading the manuscript. We also thank Lanyuan Lu and other members of our group for insightful discussions.
This work was supported by a Collaborative Research in Chemistry grant from the National Science Foundation (grant CHE-1047323).
Supporting Material
References
- 1.Hartl F.U., Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- 2.Hartl F.U., Bracher A., Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- 3.De Maio A. Heat shock proteins: facts, thoughts, and dreams. Shock. 1999;11:1–12. doi: 10.1097/00024382-199901000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Young J.C., Agashe V.R., Hartl F.U. Pathways of chaperone-mediated protein folding in the cytosol. Nat. Rev. Mol. Cell Biol. 2004;5:781–791. doi: 10.1038/nrm1492. [DOI] [PubMed] [Google Scholar]
- 5.Mayer M.P. Gymnastics of molecular chaperones. Mol. Cell. 2010;39:321–331. doi: 10.1016/j.molcel.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 6.Taipale M., Jarosz D.F., Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010;11:515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 7.Csermely P., Schnaider T., Nardai G. The 90-kDa molecular chaperone family: structure, function, and clinical applications. A comprehensive review. Pharmacol. Ther. 1998;79:129–168. doi: 10.1016/s0163-7258(98)00013-8. [DOI] [PubMed] [Google Scholar]
- 8.Taiyab A., Rao ChM. HSP90 modulates actin dynamics: inhibition of HSP90 leads to decreased cell motility and impairs invasion. Biochim. Biophys. Acta. 2011;1813:213–221. doi: 10.1016/j.bbamcr.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 9.Whitesell L., Lindquist S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 10.Trepel J., Mollapour M., Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer. 2010;10:537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sajjad M.U., Samson B., Wyttenbach A. Heat shock proteins: therapeutic drug targets for chronic neurodegeneration? Curr. Pharm. Biotechnol. 2010;11:198–215. doi: 10.2174/138920110790909641. [DOI] [PubMed] [Google Scholar]
- 12.Southworth D.R., Agard D.A. Client-loading conformation of the Hsp90 molecular chaperone revealed in the cryo-EM structure of the human Hsp90:Hop complex. Mol. Cell. 2011;42:771–781. doi: 10.1016/j.molcel.2011.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Street T.O., Lavery L.A., Agard D.A. Cross-monomer substrate contacts reposition the Hsp90 N-terminal domain and prime the chaperone activity. J. Mol. Biol. 2012;415:3–15. doi: 10.1016/j.jmb.2011.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krukenberg K.A., Street T.O., Agard D.A. Conformational dynamics of the molecular chaperone Hsp90. Q. Rev. Biophys. 2011;44:229–255. doi: 10.1017/S0033583510000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shiau A.K., Harris S.F., Agard D.A. Structural analysis of E. coli hsp90 reveals dramatic nucleotide-dependent conformational rearrangements. Cell. 2006;127:329–340. doi: 10.1016/j.cell.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 16.Ali M.M.U., Roe S.M., Pearl L.H. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature. 2006;440:1013–1017. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prodromou C., Panaretou B., Pearl L.H. The ATPase cycle of Hsp90 drives a molecular ‘clamp’ via transient dimerization of the N-terminal domains. EMBO J. 2000;19:4383–4392. doi: 10.1093/emboj/19.16.4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Young J.C., Moarefi I., Hartl F.U. Hsp90: a specialized but essential protein-folding tool. J. Cell Biol. 2001;154:267–273. doi: 10.1083/jcb.200104079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krukenberg K.A., Förster F., Agard D.A. Multiple conformations of E. coli Hsp90 in solution: insights into the conformational dynamics of Hsp90. Structure. 2008;16:755–765. doi: 10.1016/j.str.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Southworth D.R., Agard D.A. Species-dependent ensembles of conserved conformational states define the Hsp90 chaperone ATPase cycle. Mol. Cell. 2008;32:631–640. doi: 10.1016/j.molcel.2008.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krukenberg K.A., Southworth D.R., Agard D.A. pH-dependent conformational changes in bacterial Hsp90 reveal a Grp94-like conformation at pH 6 that is highly active in suppression of citrate synthase aggregation. J. Mol. Biol. 2009;390:278–291. doi: 10.1016/j.jmb.2009.04.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Street T.O., Krukenberg K.A., Agard D.A. Osmolyte-induced conformational changes in the Hsp90 molecular chaperone. Protein Sci. 2010;19:57–65. doi: 10.1002/pro.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Street T.O., Lavery L.A., Agard D.A. Substrate binding drives large-scale conformational changes in the Hsp90 molecular chaperone. Mol. Cell. 2011;42:96–105. doi: 10.1016/j.molcel.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsutsumi S., Mollapour M., Neckers L.M. Charged linker sequence modulates eukaryotic heat shock protein 90 (Hsp90) chaperone activity. Proc. Natl. Acad. Sci. USA. 2012;109:2937–2942. doi: 10.1073/pnas.1114414109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsai C.J., Kumar S., Nussinov R. Folding funnels, binding funnels, and protein function. Protein Sci. 1999;8:1181–1190. doi: 10.1110/ps.8.6.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mickler M., Hessling M., Hugel T. The large conformational changes of Hsp90 are only weakly coupled to ATP hydrolysis. Nat. Struct. Mol. Biol. 2009;16:281–286. doi: 10.1038/nsmb.1557. [DOI] [PubMed] [Google Scholar]
- 27.Hessling M., Richter K., Buchner J. Dissection of the ATP-induced conformational cycle of the molecular chaperone Hsp90. Nat. Struct. Mol. Biol. 2009;16:287–293. doi: 10.1038/nsmb.1565. [DOI] [PubMed] [Google Scholar]
- 28.Ratzke C., Berkemeier F., Hugel T. Heat shock protein 90's mechanochemical cycle is dominated by thermal fluctuations. Proc. Natl. Acad. Sci. USA. 2012;109:161–166. doi: 10.1073/pnas.1107930108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colombo G., Morra G., Verkhivker G. Understanding ligand-based modulation of the Hsp90 molecular chaperone dynamics at atomic resolution. Proc. Natl. Acad. Sci. USA. 2008;105:7976–7981. doi: 10.1073/pnas.0802879105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morra G., Verkhivker G., Colombo G. Modeling signal propagation mechanisms and ligand-based conformational dynamics of the Hsp90 molecular chaperone full-length dimer. PLOS Comput. Biol. 2009;5:e1000323. doi: 10.1371/journal.pcbi.1000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morra G., Potestio R., Colombo G. Corresponding functional dynamics across the Hsp90 Chaperone family: insights from a multiscale analysis of MD simulations. PLOS Comput. Biol. 2012;8:e1002433. doi: 10.1371/journal.pcbi.1002433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 33.Hess B., Kutzner C., Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 34.Schuler L.D., Daura X., Van Gunsteren W.F. An improved GROMOS96 force field for aliphatic hydrocarbons in the condensed phase. J. Comput. Chem. 2001;22:1205–1218. [Google Scholar]
- 35.Bussi G., Donadio D., Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 36.Berendsen H.J.C., Postma J.P.M., Haak J.R. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 37.Darden T., York D., Pedersen L. Particle mesh Ewald—an N.Log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 38.Hess B., Bekker H., Fraaije J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 39.Berendsen H.J.C., Postma J.P., Herman J. Interaction models for water in relation to protein hydration. In: Pullman B., editor. Intermolecular Forces. D. Reidel Publishing; Dordrecht: 1981. pp. 331–342. [Google Scholar]
- 40.Huai Q., Wang H.C., Ke H. Structures of the N-terminal and middle domains of E. coli Hsp90 and conformation changes upon ADP binding. Structure. 2005;13:579–590. doi: 10.1016/j.str.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 41.Larkin M.A., Blackshields G., Higgins D.G. Clustal W and clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 42.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 43.Kumar S., Bouzida D., Rosenberg J.M. The weighted histogram analysis method for free-energy calculations on biomolecules. 1. The Method. J. Comput. Chem. 1992;13:1011–1021. [Google Scholar]
- 44.Grossfield, A. 2011. WHAM: the weighted histogram analysis method. Version 2.05, http://membrane.urmc.rochester.edu/content/wham.
- 45.Efron B., Tibshirani R. Chapman and Hall/CRC; New York: 1994. An Introduction to the Bootstrap. [Google Scholar]
- 46.Bron P., Giudice E., Garnier C. Apo-Hsp90 coexists in two open conformational states in solution. Biol. Cell. 2008;100:413–425. doi: 10.1042/BC20070149. [DOI] [PubMed] [Google Scholar]
- 47.Dollins D.E., Warren J.J., Gewirth D.T. Structures of GRP94-nucleotide complexes reveal mechanistic differences between the hsp90 chaperones. Mol. Cell. 2007;28:41–56. doi: 10.1016/j.molcel.2007.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dutta R., Inouye M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem. Sci. 2000;25:24–28. doi: 10.1016/s0968-0004(99)01503-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.