

Abstract
Modulation of aryl hydrocarbon receptor (AHR) activity by a class of ligands termed selective AHR modulators (SAhRMs) has been demonstrated to attenuate proinflammatory gene expression and signaling, including repression of cytokine-mediated induction of acute-phase genes (e.g., Saa1). These effects are observed to occur through an AHR-dependent mechanism that does not require canonical signaling through dioxin response elements. Previously, we have demonstrated that the SAhRM 3′,4′-dimethoxy-α-naphthoflavone (DiMNF) can repress the cytokine-mediated induction of complement factor genes. Here, we report that the activation of the AHR with DiMNF can suppress cytokine-mediated induction of the membrane complement regulatory protein CD55. When CD55 is expressed on host cells, it facilitates the decay of the complement component 3 (C3) convertase, thereby protecting the cell from complement-mediated lysis. Tumor cells often exhibit elevated CD55 expression on the cell surface in the inflammatory microenvironment of the tumor, and such enhanced expression could represent a means of escaping immune surveillance. DiMNF can repress the cytokine-mediated induction of CD55 mRNA and protein. Luciferase reporter analysis has identified possible response elements on the CD55 promoter, which may be targets for this repression. A C3 deposition assay with [125I]C3 revealed that repression of cytokine-mediated CD55 expression by DiMNF led to an increase of C3 deposition on the surface of Huh7 cells, which would likely stimulate the formation of the membrane attack complex. These results suggest that SAhRMs such as DiMNF have therapeutic potential in regulating the immune response to tumor formation.
Introduction
The aryl hydrocarbon receptor (AHR) is a cytosolic ligand-activated transcription factor belonging to the basic helix-loop-helix/Per-Arnt-Sim family of transcription factors, which mediate a wide range of tissue-dependent responses upon exposure to AHR agonists (Kewley et al., 2004). In the absence of ligand, the AHR resides in the cytosol, bound to a dimer of 90-kDa heat shock protein and X-associated protein 2 (Perdew, 1988; Meyer et al., 1998). Upon binding of the AHR to its prototypical ligand, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), the AHR translocates to the nucleus where it dissociates from its chaperone proteins and heterodimerizes with the AHR nuclear translocator (ARNT) (Reyes et al., 1992). The AHR/ARNT complex can then bind specific recognition sites, called dioxin response elements (DREs), which stimulate the transcription of the AHR target gene battery, including but not limited to those involved in phase I metabolism (Beischlag et al., 2008). In addition to the DRE-dependent mode of action, ligand activation of the AHR can influence gene expression. Such mechanisms probably depend on protein-protein interactions and have been demonstrated most notably with the estrogen receptor and others, including NF-κB, signal transducers and activators of transcription, and cyclin-dependent kinase 4 (Kimura et al., 2008; Ahmed et al., 2009; Vogel and Matsumura, 2009; Barhoover et al., 2010).
Persistent activation of the AHR by the environmental contaminant TCDD is known to result in a myriad of toxic effects, including potent immune-suppressive effects. However, such AHR-mediated immune suppression suggests the potential for the AHR as a viable therapeutic target for the pharmacological intervention of uncontrolled inflammation and autoimmune disease (Patel et al., 2009). Although AHR agonists can elicit immune suppression, their therapeutic usefulness is somewhat compromised by long-established side effects associated with DRE-dependent gene expression, including biotransformation of procarcinogens. Thus, despite efficacy, agonists fail to fulfill the appropriate therapeutic criteria required for pharmacological agents. However, a new class of AHR ligands has been described and termed selective AHR modulators (SAhRMs), which fail to mediate potentially toxic DRE-mediated gene expression while still stimulating and maintaining AHR-dependent, noncanonical activity associated with the suppression of inflammatory gene expression. SAhRMs have been demonstrated to have the capacity to suppress the cytokine-mediated induction of genes associated with the acute phase and complement responses through a DRE-independent mechanism (Patel et al., 2009). The SAhRMs 4-[1-allyl-7-(trifluoromethyl)-1H-indazol-3-yl]benzene-1 (WAY-169916) and its methoxy derivative 1-allyl-3–4-dimethoxyphenyl)-7-(trifluromethyl)-1H-indazole (SGA360) have been demonstrated to effectively suppress interleukin 1β (IL1β)-mediated induction of the acute-phase targets including serum amyloid A1 and C-reactive protein (CRP) (Murray et al., 2010a,b). In addition, 3′,4′-dimethoxy-α-naphthoflavone (DiMNF), a derivative of the AHR partial agonist α-naphthoflavone, has been shown to possess SAhRM activity in the suppression of IL1β-mediated induction of the complement factors C3, C1S, and C4 (Murray et al., 2011). It is noteworthy that gene expression analysis using a non-DRE binding AHR mutant, which mimics SAhRM-like AHR activity, reveals numerous complement factors as being potential targets for SAhRM-activated AHR, including the inhibitory complement factors CD55 or decay-accelerating factor, CD46, and CFH (Patel et al., 2009).
The complement system is a critical component of the innate immune system and is a first line of defense against invading pathogens and altered host cells. Activation of the complement system can occur through three different pathways, the classic, alternative, and mannose-binding lectin, all of which converge on the formation of the C3 and C5 convertases. Formation of the convertases leads to the deposition of complement proteins, which ultimately stimulates the formation of the membrane attack complex (MAC), causing lysis of the target cell (Walport, 2001).
Sustained activation of complement is associated with chronic inflammation and damage to normal tissue; therefore, the complement cascade is tightly regulated at each level by inhibitory proteins (Zipfel and Skerka, 2009). Most proteins involved with the complement cascade are synthesized by hepatocytes and secreted into serum; therefore, liver dysfunction can lead to alterations in complement-mediated lysis of damaged cells and immune suppression (Whaley and Schwaeble, 1997). Membrane complement regulatory proteins are anchored in the cell membrane and include membrane cofactor protein (CD46), protectin (CD59), and decay-accelerating factor (CD55), all of which regulate complement activation on the cell surface (Kim and Song, 2006). These factors essentially exhibit global basal expression as a means of preventing attack on host cells. CD55 is a glycosylphosphatidylinositol-anchored protein, which is expressed on the surface of nearly all nucleated cells (Lublin and Atkinson, 1989). Expression of CD55 on cell surfaces prevents the formation of the C3 convertase or stimulates the degradation of pre-existing C3 convertase, thereby blocking the complement cascade and inhibiting formation of the MAC, ultimately protecting cells from autologous complement attack (Medof et al., 1984; Fujita et al., 1987).
Tumor cells have been characterized as having increased expression of membrane complement regulatory proteins, including CD55 (Li et al., 2001). In fact, enhanced expression of CD55 has been observed in colorectal, breast, and prostate cancers (Niehans et al., 1996) and has been associated with poor prognosis in patients with breast and colorectal cancers (Durrant et al., 2003; Ikeda et al., 2008). It has been suggested that the elevated expression of inhibitory complement factors such as CD55 may arise as a consequence of the proinflammatory microenvironment generated by solid tumors. Enhanced levels of the cytokines IL1β, tumor necrosis factor α, and IL6 derived from both tumor and surrounding stromal cells have been demonstrated to enhance CD55 expression. It is believed that such cytokine-mediated induction may contribute to a protective environment by attenuating complement-mediated attack, thus allowing tumor survival and expansion (Spiller et al., 2000). Supporting evidence for this notion can be derived from studies illustrating that siRNA-mediated ablation of CD55 sensitizes prostate tumor cell lines to complement-mediated lysis (Loberg et al., 2006). Here, we demonstrate that such attenuation can be achieved by using DiMNF acting as a SAhRM. More importantly, we demonstrate that DiMNF-mediated suppression of CD55 expression is associated with enhanced C3 opsonization of tumor cells. Furthermore, we reveal that DiMNF specifically attenuates cytokine but not basal CD55 expression, suggesting that tumor cells embedded in a proinflammatory microenvironment will be uniquely sensitized to complement-mediated attack. Such data suggest that the use of selective AHR modulators, such as DiMNF, may have a therapeutic benefit, particularly as an adjuvant during immunotherapy.
Materials and Methods
Materials.
The flavonoid compound DiMNF was obtained commercially (Indofine Chemicals, Hillsbourough, NJ). TCDD was a generous gift from Dr. Stephen Safe (Texas A&M University, College Station, TX). N-(2-(1H-indol-3-yl)ethyl)-9-isopropyl-2-(5-methylpyridin-3-yl)-9H-purin-6-amine (GNF351) and SGA360 were synthesized as described previously (Murray et al., 2010a; B. D. DiNatale, K. Smith, G. Krisdahalli, S. G. Amin, and G. H. Perdew, unpublished work). Human recombinant IL1β was obtained commercially (PeproTech, Rocky Hill, NJ). Forskolin (3R,4aR,5S,6S,6aS,10S,10aR,10bS)-6,10,10b-trihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-3-vinyldodecahydro-1H-benzo[f]chromen-5-yl acetate) was obtained from Sigma (St Louis, MO). Human serum was obtained commercially (Innovative Research of America, Inc., Novi, MI).
Cell Culture.
Huh7 and Hep3B human hepatoma cell lines were maintained in α-modified essential media (Sigma) supplemented with 8% HyClone fetal bovine serum (Thermo Fisher Scientific, Waltham, MA), 100 units/ml penicillin, and 100 μg/ml streptomycin (Sigma). Cells were cultured at 37°C in a humidified atmosphere containing 95% air/5% CO2.
Plasmids.
The promoter region of CD55 was amplified from MCF-7 human genomic DNA by using the primer pair CD55FL (forward and reverse) (Integrated DNA Technologies, Inc., Coralville, IA) listed in Supplemental Table 1. The 1008-base pair fragment was subcloned into the XhoI/HindIII site of pGL3basic-Luc (Promega, Madison, WI) to generate the pGL3basic/CD55WT-Luc construct. The construct was sequenced to confirm that the appropriate sequence was cloned. The −423/+71 deletion construct of CD55 was generated by digestion of the full-length CD55 promoter with the restriction enzymes PstI and HindIII (New England Biolabs, Ipswich, MA), and the fragment was subcloned into the pGL3basic-Luc vector by using PstI and HindIII. The −209/+71 deletion mutant of the CD55 promoter was generated by amplifying the desired fragment by PCR and specific primers for the sequence that contained XhoI and HindIII restriction sites at the 5′ and 3′ ends, respectively (Supplemental Table 2) and subcloning the resulting fragment into the pGL3basic-Luc vector by using XhoI and HindIII (New England Biolabs). Generation of point mutations in the CD55 promoter was performed by using a QuikChange site-direct mutagenesis kit (Agilent Technologies, Santa Clara, CA) within the pGL3basic/CD55WT-Luc construct and primers for the desired sequence (Supplemental Table 2). The constructs were sequenced to verify that the appropriate sequences had been cloned.
Transient Transfection and Luciferase-Based Reporter Assays.
Huh7 cells were cultured in six-well plates and transfected by using Geneporter 3000 transfection reagent (Genlantis, San Diego, CA) following the manufacturer's protocol. Cells were pretreated with either vehicle (DMSO) or 10 μM DiMNF for 1 h and then treated with either 10 ng/ml IL1β or 1 μM forskolin for 24 h. Cells were lysed in 200 μl of lysis buffer [25 mM Tris-phosphate, pH 7.8, 2 mM dithiothreitol, 2 mM EDTA, 10% (v/v) glycerol, and 1% (v/v) Triton X-100]. Lysate (20 μl) was combined with 80 μl of luciferase reporter substrate (Promega), and luciferase activity was measured with a TD-20e luminometer (Turner Designs, Sunnyvale, CA). Luciferase activity was normalized with respect to β-galactosidase activity.
RNA Isolation and Reverse Transcription.
Total RNA was isolated from cells cultured in six-well plates by using TRIzol (Invitrogen, Carlsbad, CA) and reverse-transcribed to cDNA by using the High-Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA).
Quantitative Real-Time PCR.
Expression levels of specific mRNA for all genes examined were measured by quantitative real-time PCR with the Quanta SYBR Green kit (Quanta Biosciences, Gaithersburg, MD) on an iCycler DNA engine equipped with the MyiQ single-color real-time PCR detection system (Bio-Rad Laboratories, Hercules, CA). Expressed quantities of mRNA were normalized to L13A mRNA levels. Oligonucleotide sequences are shown in Supplemental Table 2.
siRNA-Mediated Knockdown.
siRNA-mediated knockdown of AHR expression in Hep3B cells was performed by using the Amaxa nucleofection system (Lonza Walkersville, Inc., Walkersville, MD) with either AHR-specific oligonucleotide sequence (GCACGAGAGGCUCAGGUUA) on-target, RelA/p65-specific oligonucleotide sequence (CCCACGAGCUUGUAGGAAA) on-target, or control siRNA (Dharmacon RNA Technologies, Lafayette, CO). Cells were washed and suspended at a concentration of 2.0 × 106 per 100 μl of nucleofection solution. Control or targeted siRNA was added to the sample for a final concentration of 1.5 μM. Samples were electroporated with program T16 and plated into six-well dishes in complete media. Cells were allowed to recover for 24 h after electroporation before further treatments.
Protein Expression Analysis and Immunoblotting.
Huh7 cells were cultured to ∼80% confluence in six-well plates and treated with vehicle (DMSO) or 10 nM DiMNF for 1 h before treatment with 10 ng/ml IL1β for 24 h. Cells were lysed with 25 mM MOPS, 2 mM EDTA, 0.02% NaN3, and 10% glycerol, pH 7.5/20 mM sodium molybdate/1% Igepal CA-630/protease inhibitor cocktail (Sigma). Lysates were centrifuged (13,000g, 30 min, 4°C), and protein concentrations of supernatants were determined by use of the bicinchoninic acid kit (Thermo Fisher Scientific). Protein samples were resolved on 8% SDS-polyacrylamide gel electrophoresis gels and transferred to polyvinylidene difluoride membrane (Millipore Corporation, Billerica, MA). Membranes were probed at the recommended dilutions for 1 h with the following primary antibodies: anti-CD55 rabbit IgG (Santa Cruz Biotechnology, Inc., Santa Cruz, Ca) and anti-β-actin mouse IgG (Santa Cruz Biotechnology, Inc.). Secondary antibody detection was achieved by using species-appropriate biotin-conjugated IgG (Jackson ImmunoResearch Laboratories Inc., West Grove, PA). Tertiary detection was achieved through incubation with 0.03 μCi/ml [125I]streptavidin. Blots were exposed to BioMAX film (Eastman Kodak, Rochester, NY) and developed.
Iodination of C3 and Streptavidin.
Sodium [125I]iodide was obtained from PerkinElmer Life and Analytical Sciences (Waltham, MA), and iodobeads were obtained from Thermo Fisher Scientific. C3 (EMD Biosciences, San Diego, CA) and streptavidin (Jackson Immunoresearch Laboratories Inc.) were iodinated essentially by the method of Markwell (1982). Iodination of C3 was verified through autoradiography of protein resolved by SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membrane (Supplemental Fig. 3).
C3 Deposition Assay.
Huh7 cells were cultured in 24-well plates in 500 μl of α-minimal essential medium containing reduced serum (5% fetal bovine serum) at a density of 5 × 104 cells/well for 24 h. Cells were treated with either vehicle (DMSO) or 10 μM DiMNF for 1 h, followed by treatment with 10 ng/ml IL1β for 24 h. Cells were washed three times with phosphate-buffered saline and incubated for 30 min in 250 μl of α-minimal essential medium supplemented with either 5% normal human serum (Innovative Research of America, Inc.), 5% normal human serum that had been heat-inactivated for 20 min at 55°C, or 5% BSA. Cells were then exposed to [125I]C3 (10 μl) for 1 h. The media were collected, and cells were washed three times with phosphate-buffered saline. Cells were lysed with 150 μl of lysis buffer [25 mM Tris-phosphate, pH 7.8, 2 mM dithiothreitol, 2 mM EDTA, 10% (v/v) glycerol, and 1% (v/v) Triton X-100], and protein was quantified by using the bicinchoninic acid protein assay kit. Lysate was measured for radioactivity by using a gamma counter, and counts were normalized to protein content.
Statistical Analysis.
In all cases, studies were performed in triplicate. Statistical analyses of data were performed by using Prism 4 graphing and statistical analysis software (GraphPad Software Inc., San Diego, CA). Data were analyzed by using one-way analysis of variance and Tukey's multiple comparison tests or Student's unpaired t test for a statistically significant difference. In the figures an asterisk indicates all cases in which a statistically significant change has occurred.
Results
DiMNF Represses IL1β-Induced Expression of CD55 mRNA and Protein.
We have previously characterized DiMNF as a SAhRM with the capacity to attenuate cytokine-mediated C3 expression in Huh7 cells through binding to the AHR. In addition, preliminary global DNA microarray evidence, arising from studies with a non-DRE binding mutant of AHR that mimics SAhRM activity, indicated that the inhibitory complement factor CD55 may represent a target of non-DRE-dependent SAhRM action (data not shown). To validate SAhRM-mediated suppression of CD55, Huh7 cells were exposed to vehicle (DMSO) or 10 μM DiMNF 1 h before administration of 10 ng/ml IL1β. After incubation for 24 h, total RNA was isolated, and CD55 expression was assessed through quantitative PCR (Fig. 1A). Treatment with IL1β resulted in a 2-fold enhancement in the expression of CD55 compared with control. Combinatorial treatment of IL1β with DiMNF prompted a complete suppression of CD55 expression compared with IL1β alone. Exposure to DiMNF in isolation failed to significantly influence basal CD55 expression. In an effort to determine whether the suppressive activity of DiMNF with regard to IL1β-induced CD55 expression is cell line-dependent, Hep3B cells were exposed to the same treatment regime as Huh7 cells. Indeed, results in Hep3B cells were similar to those obtained with Huh7 cells. Exposure to IL1β resulted in a significant 75% increase in CD55 expression. Cotreatment with DiMNF prompted almost complete attenuation of CD55 mRNA levels comparable with that observed with untreated Huh7 cells (Fig. 1B). In addition, treatment of Hep3B with DiMNF alone yielded a similar lack of effect on basal CD55 expression as that observed with Huh7 cells. Such data indicate that CD55 is responsive to IL1β-mediated induction and such induction is sensitive to suppression mediated by DiMNF. Furthermore, this confirms that DiMNF-dependent attenuation is not cell line-specific.
Fig. 1.

DiMNF represses the IL1β-mediated induction of CD55 expression in Huh7 and Hep3B hepatoma cell lines. Huh7 or Hep3B cells were treated for 1 h with vehicle or 10 μM DiMNF and subsequently treated for 24 h with 10 ng/ml of IL1β. A and B, CD55 mRNA expression in Huh7 (A) and Hep3B (B) cells was measured by quantitative real-time PCR. Data represent mean mRNA expression level S.E.M. fold induction normalized to constitutively expressed ribosomal L13 mRNA. C, protein expression analysis of CD55 after treatment of Huh7 cells with 10 μM DiMNF for 1 h followed by treatment with 10 ng/ml of IL1β for 24 h. β-Actin was used as a control. D, protein was quantified by using a gamma counter and normalized to β-actin. Statistical significance is indicated: ***, p < 0.001; *, p < 0.05.
Having established the responsiveness of human hepatoma cell lines to IL1β with regard to CD55 mRNA and subsequent attenuation by DiMNF, we wanted to determine whether these effects are restricted to the level of mRNA or are reflected at the protein level. Therefore quantitative CD55 protein expression analyses were performed on Huh7 cells treated as indicated previously (Fig. 1, C and D). Analyses of CD55 protein levels identified a significant increase after exposure to IL1β, which was then attenuated by coexposure to DiMNF. The degree of CD55 protein induction after IL1β treatment proved to be less marked compared with that observed at the level of mRNA.
Attenuation of IL1β-Mediated CD55 Expression by DiMNF Is AHR-Dependent.
To evaluate the effects of additional AHR ligands on the repression of CD55 gene expression in an inflammatory microenvironment, Huh7 cells were pretreated with either 10 nM TCDD, an AHR agonist, or 10 μM SGA360, a SAhRM, for 1 h before exposure of cells to IL1β for 24 h. Quantitative PCR analysis revealed that the administration of TCDD before treatment with IL1β resulted in a significant and complete attenuation of CD55 mRNA levels induced by treatment with IL1β alone (Fig. 2A). Treatment of Huh7 cells with TCDD alone resulted in a modest increase in CD55 mRNA. Activation of the AHR with TCDD is known to enhance the expression of IL1β mRNA (Vogel et al., 2004); therefore, treatment of cells with AHR agonists such as TCDD may enhance the induction of CD55 mRNA through an indirect mode of action involving an increase in cytokine expression. Administration of the SAhRM SGA360, which has been shown to repress the expression of proinflammatory genes before treatment with IL1β, demonstrated a 50% repression of IL1β-induced CD55 mRNA expression (Fig. 2B), lending further credence to the hypothesis that the DiMNF-mediated repression of CD55 expression occurs via the AHR. To further substantiate the requirement for SAhRM activity, in lieu of generalized AHR ligand binding, as a prerequisite to facilitate attenuation of CD55 expression, we used GNF351, an AHR antagonist, which inhibits both DRE-dependent and independent AHR activity (Smith et al., 2011). Huh7 cells were treated with 500 nM GNF351 for 1 h, and cells were then exposed to IL1β for 24 h. Quantitative PCR demonstrated that the treatment of cells with GNF and IL1β yielded no repression in IL1β-mediated CD55 mRNA levels (Fig. 2C).
Fig. 2.

DiMNF-mediated repression of CD55 mRNA expression occurs through an AHR-dependent pathway. A to C, Huh7 cells were pretreated with vehicle or 10 nM TCDD (A), 10 nM SGA360 (B), or 500 nM GNF351 (C) for 1 h, followed by treatment with 10 ng/ml of IL1β, and quantitative real-time PCR was performed to measure the expression of CD55 mRNA. D, Hep3B cells targeted with either control or AHR-specific siRNA were incubated with vehicle or 10 μM DiMNF for 1 h before treatment with IL1β for 24 h, and CD55 mRNA expression was measured by quantitative real-time PCR. Data represent mean mRNA expression level ± S.E.M. normalized to constitutively expressed ribosomal L13 mRNA. Statistical significance is indicated: *, p < 0.05.
To establish the dependence for AHR expression in the context of DiMNF-mediated attenuation of cytokine-induced CD55 expression, Hep3B cells were electroporated with small interfering RNA specific for the AHR. Hep3B cells were selected because Huh7 cells yielded modest levels of siRNA knockdown. Partial knockdown of AHR was verified by protein immunoblot analysis (Supplemental Fig. 1A), and functional repression of AHR transcriptional activity was confirmed by measuring CYP1A1 mRNA induction upon treatment with 10 nM TCDD (Supplemental Fig. 1B). Forty-eight hours after electroporation, cells were treated with 10 μM DiMNF for 1 h before treatment with IL1β for 24 h. Quantitative real-time PCR of CD55 mRNA revealed that in cells electroporated with AHR siRNA IL1β induced a 2-fold increase in CD55 mRNA. Coexposure of cells electroporated with AHR to DiMNF and IL1β yielded no repression of IL1β-mediated induction of CD55 mRNA (Fig. 2D). These data suggest an absolute requirement for AHR expression in DiMNF-mediated suppression of cytokine-inducible CD55 expression.
Characterization of the CD55 Promoter.
To attempt to identify the response elements involved in the AHR-mediated repression of CD55 gene expression, truncation mutants were generated in the CD55 promoter based on the previously described transcription start site (Ewulonu et al., 1991). Transfection of the full-length CD55 promoter into Huh7 cells resulted in a 2.5-fold induction of promoter activity upon 24 h treatment with 10 ng/ml IL1β and repression of this induction to control levels upon 1-h treatment with 10 μM DiMNF before administration of IL1β, as measured by luciferase expression normalized to β-galactosidase activity. (Fig. 3) Transfection of the CD55 −423/+71 construct into these cells resulted in a 2-fold induction of CD55 promoter activity upon treatment with IL1β; however, pretreatment with DiMNF demonstrated no repression of IL1β-mediated induction (Fig. 3). Transfection of the CD55 −209/+71 construct resulted in complete ablation of CD55 promoter activity upon treatment with IL1β; therefore, the repressive effects of DiMNF on the CD55 promoter were also eliminated (Fig. 3). These results indicate that the AHR probably is mediating repression of CD55 expression through response elements located between −858 and −423 nucleotides from the transcription start site.
Fig. 3.

DiMNF mediates repression of IL1β-mediated induction of CD55 promoter activity in Huh7 cells. Huh7 cells were transfected with pGL3-basic reporter plasmids containing either the −858/+71, −423/+71, or −209/+71 CD55 promoter reporter constructs by using the GenePorter3000 transfection reagent. Twenty-four hours after transfection, cells were pretreated with either vehicle or 10 μM DiMNF for 1 h followed by treatment for 24 h with 10 ng/ml of IL1β. Luciferase reporter activity was measured and normalized to β-galactosidase activity. Data are reported as mean relative luciferase units ± S.E.M. normalized to vehicle treated. Statistical significance is indicated: ***, p < 0.001.
CD55 expression has been determined to be induced by activation of the NF-κB pathway (Andoh et al., 2001) in intestinal epithelial cells. To study whether the IL1β-mediated induction of CD55 expression occurs though NF-κB, point mutations were individually created in the CD55 promoter for three NF-κB response elements, at −605, −480, and −395 relative to the transcription start site (termed CD55Δ-395, CD55Δ-480, and CD55Δ-605) (Fig. 4A). These constructs were transfected into Huh7 cells, which were then treated with 10 μM DiMNF for 1 h before challenge with 10 ng/ml of IL1β for 24 h. Transfection of CD55Δ-395 led to a significant 4-fold induction of reporter activity upon treatment with IL1β and a significant and complete repression of IL1β-mediated induction upon coexposure with DiMNF and IL1β (Fig. 4B), indicating that this response element is not critical for IL1β-mediated expression of CD55. Treatment with IL1β after transfection of Huh7 cells with CD55Δ-480 demonstrated no induction of reporter activity, and no repression of cytokine-mediated induction was observed upon pretreatment with DiMNF (Fig. 4C). Transfection of the CD55Δ-605 construct into Huh7 cells followed by treatment with IL1β revealed no induction in reporter activity and no repression of IL1β-mediated induction upon treatment with DiMNF before treatment with IL1β (Fig. 4D).
Fig. 4.

Identification of response elements involved in IL1β-mediated induction of CD55 expression. A, schematic illustration of the positions of point mutations created in NF-κB binding sites and a CRE binding site on the CD55 promoter. B to D, Huh7 cells were transfected with either CD55Δ-395 (B), CD55Δ-480 (C), or CD55Δ-605 (D) by using the GenePorter3000 transfection reagent. Twenty-four hours after transfection, cells were treated with either vehicle or 10 μM DiMNF for 1 h followed by 24-h treatment with 10 ng/ml of IL1β. Luciferase activity was measured and normalized to β-galactosidase activity. Data represent mean relative luciferase units ± S.E.M. fold induction relative to control. E, Hep3B cells targeted with either control or RelA/p65-specific siRNA were incubated with vehicle or 10 μM DiMNF for 1 h before treatment with 10 ng/ml of IL1β for 24 h, and CD55 mRNA expression was measured by quantitative real-time PCR. Data represent mean mRNA expression level ± S.E.M. normalized to constitutively expressed ribosomal L13 mRNA. Statistical significance is indicated: ***, p < 0.001.
To confirm whether CD55 promoter activity requires the NF-κB-dependent pathway, Hep3B cells were electroporated with small interfering RNA targeted either for control or RelA/p65, and knockdown of RelA was verified by protein immunoblot analysis (Supplemental Fig. 2). Forty-eight hours after electroporation, cells were pretreated for 1 h with 10 μM DiMNF and subsequently treated with 10 ng/ml of IL1β for 24 h. Quantitative PCR revealed that IL1β failed to induce CD55 mRNA in cells treated with RelA/p65 siRNA, and no repression was observed upon pretreatment with DiMNF (Fig. 4E). Taken together, these results indicate that NF-κB is required for IL1β-induced CD55 reporter activity, which is mediated through two NF-κB response elements.
A cAMP response element (CRE) was identified in the CD55 promoter region proximal to the transcription start site (Ewulonu et al., 1991). Therefore, it was hypothesized that the induction of CD55 expression could involve the cAMP/CRE pathway. To evaluate this possibility, Huh7 cells were transfected with the CD55 promoter and treated with 10 μM DiMNF for 1 h before treatment with 1 μM forskolin, thereby raising the levels of cAMP. Luciferase reporter analysis revealed that administration of forskolin caused a 3-fold induction of reporter activity and pretreatment with DiMNF before exposure to forskolin yielded a significant repression of the forskolin-mediated induction of reporter activity (Fig. 5A). To explore this further, a point mutation was made in the CRE-binding site (termed CD55Δ-71), and this construct was transfected into Huh7 cells, followed by pretreatment with 10 μM DiMNF for 1 h and subsequent treatment with 1 μM forskolin for 24 h. Mutation of this element caused no induction of reporter activity upon treatment with forskolin, suggesting that this element is necessary for cAMP-mediated induction of CD55 activity (Fig. 5B). To assess the effect of siRNA-mediated knockdown of RelA/p65 on forskolin-mediated induction of CD55 expression, Hep3B cells, which had been electroporated with small-interfering RNA targeted to RelA, were subsequently treated with 10 μM DiMNF for 1 h followed by treatment with 1 μM forskolin. Upon knockdown of RelA/p65, treatment with forskolin still induced mRNA levels of CD55 3-fold, and coexposure with DiMNF and forskolin completely repressed forskolin-mediated induction of CD55 mRNA to basal levels (Fig. 5C), suggesting that the CRE element can induce CD55 transcription independently of the presence of p65.
Fig. 5.

Effect of DiMNF on forskolin-mediated induction of CD55 expression. A and B, Huh7 cells were transfected with either the −858/+71 CD55 promoter (A) or the CD55 promoter with a point mutation in a CRE binding site (−71) (B) in the pGL3 basic reporter vector by using the GenePorter 3000 transfection reagent. Twenty-four hours after transfection, cells were treated with either vehicle or 10 μM DiMNF followed by 24-h treatment with 1 μM forskolin. Luciferase reporter activity was measured and normalized to β-galactosidase activity. Data represent mean ± S.E.M. fold induction relative to control. C, Hep3B cells targeted with either control or RelA/p65-specific siRNA were incubated with vehicle or 10 μM DiMNF for 1 h before treatment with 1 μM forskolin for 24 h, and CD55 mRNA expression was measured by quantitative real-time PCR. Data represent mean mRNA expression level ± S.E.M. normalized to constitutively expressed ribosomal L13 mRNA. Statistical significance is indicated: ***, p < 0.001; **, p < 0.01.
Activation of the AHR by DiMNF Can Repress the Cytokine-Mediated Expression of CD46.
The CD55 gene is expressed on the long arm of chromosome 1q32, in a cluster termed regulators of complement activity, along with CD46 and CFH (Riley-Vargas et al., 2004). We have reported previously that SAhRM-mediated activation of the AHR represses cytokine-induced CFH mRNA expression (Patel et al., 2009); therefore, it was thought that activation of the AHR with DiMNF could repress IL1β-mediated induction of CD55 expression. Huh7 cells were treated for 1 h with either vehicle (DMSO) or 10 μM DiMNF and subsequently exposed to 10 ng/ml of IL1β for 24 h. After incubation, total mRNA was isolated, and quantitative real-time PCR was performed to measure CD46 mRNA expression. CD46 mRNA was induced 7-fold upon treatment with IL1β alone, and this induction was significantly repressed 100% to basal levels upon cotreatment with DiMNF and IL1β (Fig. 6). However, treatment with DiMNF alone did not significantly alter the basal levels of CD46 mRNA expression. Therefore, it can be hypothesized that DiMNF-mediated AHR activation regulated cytokine-induced CD46 mRNA expression and not basal levels of CD46 mRNA under the conditions tested.
Fig. 6.
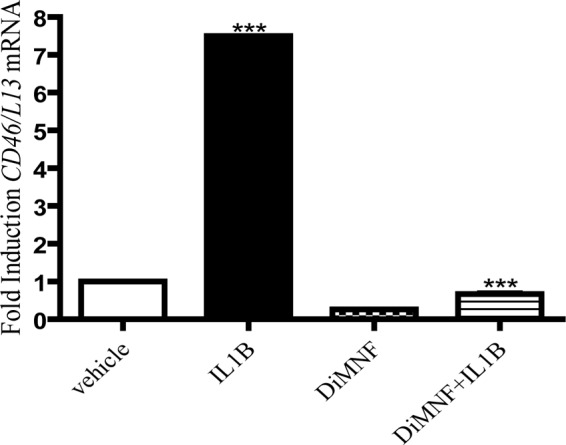
DiMNF attenuates cytokine-mediated expression of the membrane complement regulatory protein CD46. Huh7 cells were pretreated for 1 h with 10 μM DiMNF and subsequently treated for 24 h with 10 ng/ml of IL1β. Quantitative real-time PCR was performed to measure expression of CD46 mRNA. Data represent mean mRNA expression level ± S.E.M. fold induction normalized to constitutively expressed ribosomal L13 mRNA. Statistical significance is indicated: ***, p < 0.001.
AHR-Mediated Repression of CD55 Expression Leads to an Increased Deposition of the C3 Fragments on Cells.
CD55 expression on the surface of cells promotes the decay of the C3 convertases, which normally serve to cleave C3 into C3a and C3b, which deposits on the surface of cells and leads to formation of the MAC and complement-mediated cytotoxicity. Inactivation of the C3 convertase inhibits deposition of C3b; therefore, complement-mediated lysis cannot occur. To assess the functional significance of AHR-mediated repression on CD55 and CD46 expression, the deposition of C3 on the surface of Huh7 cells was assessed after treatment with 10 μM DiMNF for 1 h, followed by exposure to 10 ng/ml IL1β for 24 h. Cells were incubated in media containing either 5% normal human serum, which contains the components of the complement pathway, 5% heat-inactivated normal human serum, or 5% BSA for 30 min. Cells were then exposed to [125I]C3 for 1 h, followed by cell lysis. Deposition of [125I]C3b was determined by measuring radioactivity in a gamma counter, and cpm were normalized to milligrams of protein. Upon treatment of Huh7 cells with DiMNF before the administration of IL1β, there was a 1.5-fold increase in C3 deposition, which was statistically significant compared with cells treated with IL1β alone on the surfaces of cells incubated with normal human serum (Fig. 7). This difference was not observed in cells incubated in heat-inactivated normal human serum or BSA before the addition of [125I]C3, both of which do not have active complement components and therefore cannot cleave C3 into its fragments. Therefore, activation of the AHR with DiMNF functionally alters deposition of complement component proteins on the surfaces of cells.
Fig. 7.
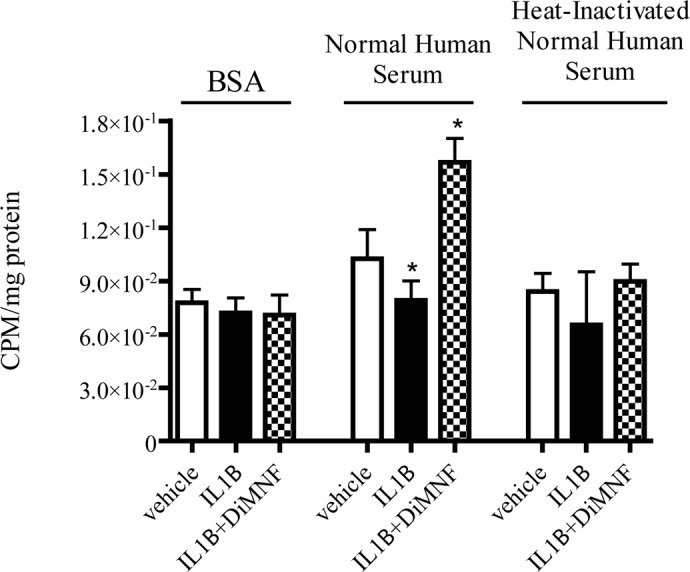
[125I]C3b deposition on the surface of Huh7 cells. Huh7 cells were cultured in 24-well places and pretreated for 1 h with either vehicle or 10 μM DiMNF before treatment with 10 ng/ml of IL1β for 24 h. Cells were then incubated in media containing either 5% BSA, 5% normal human serum, or 5% heat-inactivated normal human serum for 30 min followed by administration of [125I]C3. [125I]C3b deposition was measured on the surface of cells by a gamma counter and normalized to protein. Data represent mean ± S.E.M. Statistical significance is indicated: *, p < 0.05.
Discussion
The efficacy of current therapeutic regimes, radio, chemo, and immuno therapy, for the treatment of cancer, is largely hindered by continuous selective adaptive mechanisms inherent to neoplastic cells. Tumors have evolved to escape detection by the immune system, generating a microenvironment characterized by the presence of stromal cells, such as tumor-associated macrophages and myeloid-derived suppressor cells, which increase production of immunosuppressive factors, including transforming growth factor-β2 (Oft et al., 1998) and cyclooxygenase-2 derived prostaglandin E2 (Soslow et al., 2000). Tumor cells also secrete proinflammatory cytokines, including IL-6 and IL1β, which increase the production of amyloid precursor proteins such as amyloid A1 and CRP. This then leads to enhanced tumor growth, angiogenesis, antiapoptotic phenotype, and evasion of immune surveillance (Spiller et al., 2000). The latter effect is mediated in the inflammatory microenvironment, which promotes enhanced expression of the membrane complement regulatory proteins, CD55, CD46, and CD59. These factors inhibit the complement-mediated lysis of the tumor cells and represent another challenge for immune surveillance and treatment of tumors with immune therapy (Fishelson et al., 2003; Loberg et al., 2006).
Recently, there has been a focus on AHR activation by its ligands and the differing effects on T cell proliferation, which could affect the immune response to tumors. AHR activation by TCDD, its prototypical ligand, has been shown to induce the differentiation of CD4+CD25+ T cells, which are similar to regulatory T cells, suggesting that TCDD plays a role in suppressing the T cell response (Funatake et al., 2008). Recent evidence has shown that human glioma cells constitutively degrade tryptophan to kynurenine via tryptophan 2,3-dioxygenase (Opitz et al., 2011). Kynurenine is an AHR agonist that activates the receptor to mediate immunosuppressive effects including the differentiation of CD25+/CD4+/FoxP3+ regulatory T cells (Mezrich et al., 2010). Antagonism of the AHR by 2-methyl-2H-pyrazole-3-carboxylic acid (2-methyl-4-o-tolylazo-phenyl)-amide (CH-223191) represses T helper 17 differentiation and leads to lower levels of IL-22 and IL-17, suggesting that selectively targeting the AHR represents a mechanism for regulating the adaptive immune response and inhibiting the proinflammatory cytokine production that is a hallmark of tumor progression (Ramirez et al., 2010). Expression of CD55 has also been shown to suppress T cell immunity, because Daf(−/−) mice demonstrate a stronger T cell-mediated response to immunization through a complement-mediated mechanism (Liu et al., 2006). The increased expression of CD55 on the surfaces of tumor cells might not only serve to protect tumors against complement activation, but also protect tumors against T cell-mediated death. Therefore, repression of CD55 expression on tumor cells by DiMNF could not only have consequences for complement-mediated cell lysis of tumor cells, but also may promote T cell activation and inhibit the immunosuppression characteristics of the tumor microenvironment.
Selective activation of the AHR has been demonstrated to repress expression of genes that code for acute-phase proteins, including complement factors (Murray et al., 2011). Upon activation by these SAHRMs, AHR exerts its effects without binding its cognate response elements, thus avoiding the potentially deleterious effects associated with DRE-mediated activity, such as CYP1A1 expression, and unlike AHR activation by an agonist, such as TCDD. We have demonstrated here that activation of the AHR with DiMNF can repress the expression of the CRPs CD55 and CD46 in an inflammatory tumor cell microenvironment. However, the mechanism by which the AHR is repressing this expression remains to be determined. In contrast to the activity of a SAhRM such as DiMNF, antagonism of the AHR by GNF351 demonstrated no repression of IL1β-mediated CD55 gene expression, demonstrating that the activity of a SAHRM is distinct from that of a complete antagonist of the AHR. The response elements critical for induction of IL1β-mediated CD55 expression have been identified to be in the proximal region of the promoter. Mutagenesis analysis of three identified NF-κB response elements revealed two elements that are critical for IL1β-mediated induction; however, conclusions cannot be drawn about whether DiMNF-mediated repression is occurring at those elements. Prior studies indicated that induction of CD55 expression can also occur through a cAMP/Cre-dependent pathway (Holla et al., 2005). Indeed, DiMNF was demonstrated to repress forskolin-induced CD55 expression. Although, upon siRNA-mediated knockdown of RelA/p65, IL1β failed to induce CD55 gene expression; however, induction was observed with the administration of forskolin. This induction was repressed by pretreatment with DiMNF in the absence of RelA, suggesting that the transcriptional regulation of complement control proteins is complex and proceeds through multiple pathways. However, DiMNF is capable of repressing activity independent of the transcriptional stimulus provided. This suggests that SAhRM-mediated repression may occur at the level of coactivators or corepressors and their presence at the promoter. Clearly, further studies are needed to determine the precise mechanisms of repression.
DiMNF was demonstrated to repress expression of CD55 and another complement regulatory protein, CD46, both of which act to prevent the formation of the C3 convertase. CD46 acts as a cofactor for the plasma serine protease cofactor I, which mediates proteolysis of the complement components C3b and C4b when they are bound to host cells (Riley-Vargas et al., 2004). Prior microarray analysis has demonstrated that DiMNF treatment has the potential to repress the expression of a soluble complement factor, factor H, which also inhibits complement at the level of the C3 convertase (Patel et al., 2009). Such data suggest that selective activation of the AHR may exert a concerted or coordinated attenuation of inhibitory complement factors during inflammatory signaling. Furthermore, the observation that AHR agonists also have the potential to suppress the expression of CD55, CD46, and complement factor H may contribute to the immunotoxicity associated with such ligands.
Activation of the AHR by DiMNF was shown to increase the deposition of complement factor C3 on the surfaces of cells, which could have the potential to stimulate formation of the MAC complement-mediated lysis. It has previously been shown that DiMNF represses the expression of cytokine-mediated complement factor C3 gene expression (Murray et al., 2010a); however, the increased C3 deposition observed here upon administration of DiMNF suggests that DiMNF is acting directly on the ability of [125I]C3 to deposit on the surface of cells. These results indicate that DiMNF-mediated activation of the AHR is down-regulating cytokine-mediated expression of CD55 mRNA and protein on the surface of cells, which leads to a decreased decay of the C3 convertase. The active C3 convertase then cleaves C3 into its active components, C3a and C3b, which deposits on the surface of cells and stimulates the progression of the complement cascade, potentially leading to cytotoxicity of the cell. It is important to note that DiMNF is blocking cytokine-mediated CD55 expression and not basal expression of CD55. Thus, DiMNF should be effective at blocking CD55 expression in the inflammatory microenvironment of the tumor, but have little effect on basal expression in normal tissue. Further studies need to focus on antibody-directed, complement-mediated cytotoxicity upon repression of CD55 expression by DiMNF. In conclusion, we describe the use of the selective AHR modulator DiMNF to suppress the expression of the inhibitory complement factors CD55 and CD46 by tumor cell lines exposed to a proinflammatory environment. Such repression facilitates enhanced complement factor opsonization at the cell surface. These data indicate the involvement of the AHR in complement signaling and suggest the potential therapeutic benefit of SAhRM as an adjuvant during antitumor immunotherapy.
Supplementary Material
Acknowledgments
We thank Dr. Stephen Safe for the TCDD; Kelly Wagner for technical assistance; and Marcia H. Perdew for editorial assistance.
This work was supported by the National Institutes of Health National Institute of Environmental Health Sciences [Grants ES004869, ES019964].
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
 The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
- AHR
- aryl hydrocarbon receptor
- SAhRM
- selective AHR modulator
- ARNT
- AHR nuclear translocator
- DRE
- dioxin response element
- TCDD
- 2,3,7,8-tetrachlorodibenzo-p-dioxin
- DiMNF
- 3′,4′-dimethoxy-α-naphthoflavone
- SGA360
- 1-allyl-3–4-dimethoxyphenyl)-7-(trifluromethyl)-1H-indazole
- GNF351
- N-(2-(1H-indol-3-yl)ethyl)-9-isopropyl-2-(5-methylpyridin-3-yl)-9H-purin-6-amine
- DMSO
- dimethyl sulfoxide
- IL
- interleukin
- IL1B
- IL-1β
- C3
- complement component 3
- NF-κB
- nuclear factor κB
- CRP
- C-reactive protein
- CFH
- complement factor H
- MAC
- membrane attack complex
- siRNA
- small interfering RNA
- PCR
- polymerase chain reaction
- MOPS
- 4-morpholinepropanesulfonic acid
- BSA
- bovine serum albumin
- CRE
- cAMP response element
- CH-223191
- 2-methyl-2H-pyrazole-3-carboxylic acid (2-methyl-4-o-tolylazo-phenyl)-amide
- RLU
- relative light units
- BGal
- β-galactosidase.
Authorship Contributions
Participated in research design: Narayanan, Murray, and Perdew.
Conducted experiments: Narayanan and Murray.
Contributed new reagents or analytic tools: Krishnegowda and Amin.
Performed data analysis: Narayanan and Murray.
Wrote or contributed to the writing of the manuscript: Narayanan, Murray, and Perdew.
References
- Ahmed S, Valen E, Sandelin A, Matthews J. (2009) Dioxin increases the interaction between aryl hydrocarbon receptor and estrogen receptor α at human promoters. Toxicol Sci 111:254–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andoh A, Kinoshita K, Rosenberg I, Podolsky DK. (2001) Intestinal trefoil factor induces decay-accelerating factor expression and enhances the protective activities against complement activation in intestinal epithelial cells. J Immunol 167:3887–3893 [DOI] [PubMed] [Google Scholar]
- Barhoover MA, Hall JM, Greenlee WF, Thomas RS. (2010) Aryl hydrocarbon receptor regulates cell cycle progression in human breast cancer cells via a functional interaction with cyclin-dependent kinase 4. Mol Pharmacol 77:195–201 [DOI] [PubMed] [Google Scholar]
- Beischlag TV, Luis Morales J, Hollingshead BD, Perdew GH. (2008) The aryl hydrocarbon receptor complex and the control of gene expression. Crit Rev Eukaryot Gene Expr 18:207–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant LG, Chapman MA, Buckley DJ, Spendlove I, Robins RA, Armitage NC. (2003) Enhanced expression of the complement regulatory protein CD55 predicts a poor prognosis in colorectal cancer patients. Cancer Immunol Immunother 52:638–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewulonu UK, Ravi L, Medof ME. (1991) Characterization of the decay-accelerating factor gene promoter region. Proc Natl Acad Sci U S A 88:4675–4679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishelson Z, Donin N, Zell S, Schultz S, Kirschfink M. (2003) Obstacles to cancer immunotherapy: expression of membrane complement regulatory proteins (mCRPs) in tumors. Mol Immunol 40:109–123 [DOI] [PubMed] [Google Scholar]
- Fujita T, Inoue T, Ogawa K, Iida K, Tamura N. (1987) The mechanism of action of decay-accelerating factor (DAF). DAF inhibits the assembly of C3 convertases by dissociating C2a and Bb. J Exp Med 166:1221–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funatake CJ, Marshall NB, Kerkvliet NI. (2008) 2,3,7,8-Tetrachlorodibenzo-p-dioxin alters the differentiation of alloreactive CD8+ T cells toward a regulatory T cell phenotype by a mechanism that is dependent on aryl hydrocarbon receptor in CD4+ T cells. J Immunotoxicol 5:81–91 [DOI] [PubMed] [Google Scholar]
- Holla VR, Wang D, Brown JR, Mann JR, Katkuri S, DuBois RN. (2005) Prostaglandin E2 regulates the complement inhibitor CD55/decay-accelerating factor in colorectal cancer. J Biol Chem 280:476–483 [DOI] [PubMed] [Google Scholar]
- Ikeda J, Morii E, Liu Y, Qiu Y, Nakamichi N, Jokoji R, Miyoshi Y, Noguchi S, Aozasa K. (2008) Prognostic significance of CD55 expression in breast cancer. Clin Cancer Res 14:4780–4786 [DOI] [PubMed] [Google Scholar]
- Kewley RJ, Whitelaw ML, Chapman-Smith A. (2004) The mammalian basic helix-loop-helix/PAS family of transcriptional regulators. Int J Biochem Cell Biol 36:189–204 [DOI] [PubMed] [Google Scholar]
- Kim DD, Song WC. (2006) Membrane complement regulatory proteins. Clin Immunol 118:127–136 [DOI] [PubMed] [Google Scholar]
- Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T. (2008) Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci U S A 105:9721–9726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Spendlove I, Morgan J, Durrant LG. (2001) CD55 is over-expressed in the tumour environment. Br J Cancer 84:80–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Miwa T, Hilliard B, Chen Y, Lambris JD, Wells AD, Song WC. (2006) The complement inhibitory protein DAF (CD55) suppresses T cell immunity in vivo. J Exp Med 201:567–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loberg RD, Day LL, Dunn R, Kalikin LM, Pienta KJ.(2006) Inhibition of decay-accelerating factor (CD55) attenuates prostate cancer growth and survival in vivo. Neoplasia 8:69–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lublin DM, Atkinson JP. (1989) Decay accelerating factor: biochemistry, molecular biology, and function. Annu Rev Immunol 7:35–58 [DOI] [PubMed] [Google Scholar]
- Markwell MA. (1982) A new solid-state reagent to iodinate proteins. I. Conditions for the efficient labeling of antiserum. Anal Biochem 125:427–432 [DOI] [PubMed] [Google Scholar]
- Medof ME, Kinoshita T, Nussenzweig V. (1984) Inhibition of complement activation on the surface of cells after incorporation of decay-accelerating factor (DAF) into their membranes. J Exp Med 160:1558–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer BK, Pray-Grant MG, Vanden Heuvel JP, Perdew GH. (1998) Hepatitis B virus X-associated protein 2 is a subunit of the unliganded aryl hydrocarbon receptor core complex and exhibits transcriptional enhancer activity. Mol Cell Biol 18:978–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. (2010) An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol 185:3190–3198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray IA, Flaveny CA, Chiaro CR, Sharma AK, Tanos RS, Schroeder JC, Amin SG, Bisson WH, Kolluri SK, Perdew GH. (2011) Suppression of cytokine-mediated complement factor gene expression through selective activation of the Ah receptor with 3′,4′-dimethoxy-α-naphthoflavone. Mol Pharmacol 79:508–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray IA, Krishnegowda G, DiNatale BC, Flaveny C, Chiaro C, Lin JM, Sharma AK, Amin S, Perdew GH. (2010a) Development of a selective modulator of aryl hydrocarbon (Ah) receptor activity that exhibits anti-inflammatory properties. Chem Res Toxicol 23:955–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray IA, Morales JL, Flaveny CA, Dinatale BC, Chiaro C, Gowdahalli K, Amin S, Perdew GH. (2010b) Evidence for ligand-mediated selective modulation of aryl hydrocarbon receptor activity. Mol Pharmacol 77:247–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehans GA, Cherwitz DL, Staley NA, Knapp DJ, Dalmasso AP. (1996) Human carcinomas variably express the complement inhibitory proteins CD46 (membrane cofactor protein), CD55 (decay-accelerating factor), and CD59 (protectin). Am J Pathol 149:129–142 [PMC free article] [PubMed] [Google Scholar]
- Oft M, Heider KH, Beug H. (1998) TGFβ signaling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol 8:1243–1252 [DOI] [PubMed] [Google Scholar]
- Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller M, et al. (2011) An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 478:197–203 [DOI] [PubMed] [Google Scholar]
- Patel RD, Murray IA, Flaveny CA, Kusnadi A, Perdew GH. (2009) Ah receptor represses acute-phase response gene expression without binding to its cognate response element. Lab Invest 89:695–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdew GH. (1988) Association of the Ah receptor with the 90-kDa heat shock protein. J Biol Chem 263:13802–13805 [PubMed] [Google Scholar]
- Ramirez JM, Brembilla NC, Sorg O, Chicheportiche R, Matthes T, Dayer JM, Saurat JH, Roosnek E, Chizzolini C. (2010) Activation of the aryl-hydrocarbon receptor reveals distinct requirements for IL-22 and IL-17 production by human T helper cells. Eur J Immunol 40:2450–2459 [DOI] [PubMed] [Google Scholar]
- Reyes H, Reisz-Porszasz S, Hankinson O. (1992) Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science 256:1193–1195 [DOI] [PubMed] [Google Scholar]
- Riley-Vargas RC, Gill DB, Kemper C, Liszewski MK, Atkinson JP. (2004) CD46: expanding beyond complement regulation. Trends Immunol 25:496–503 [DOI] [PubMed] [Google Scholar]
- Smith KJ, Murray IA, Tanos R, Tellew J, Boitano AE, Bisson WH, Kolluri SK, Cooke MP, Perdew GH. (2011) Identification of a high-affinity ligand that exhibits complete aryl hydrocarbon receptor antagonism. J Pharmacol Exp Ther 338:318–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soslow RA, Dannenberg AJ, Rush D, Woerner BM, Khan KN, Masferrer J, Koki AT. (2000) COX-2 is expressed in human pulmonary, colonic, and mammary tumors. Cancer 89:2637–2645 [DOI] [PubMed] [Google Scholar]
- Spiller OB, Criado-García O, Rodríguez De Córdoba S, Morgan BP. (2000) Cytokine-mediated up-regulation of CD55 and CD59 protects human hepatoma cells from complement attack. Clin Exp Immunol 121:234–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel CF, Matsumura F. (2009) A new cross-talk between the aryl hydrocarbon receptor and RelB, a member of the NF-κB family. Biochem Pharmacol 77:734–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel CF, Sciullo E, Matsumura F. (2004) Activation of inflammatory mediators and potential role of ah-receptor ligands in foam cell formation. Cardiovasc Toxicol 4:363–373 [DOI] [PubMed] [Google Scholar]
- Walport MJ. (2001) Complement. First of two parts. N Engl J Med 344:1058–1066 [DOI] [PubMed] [Google Scholar]
- Whaley K, Schwaeble W. (1997) Complement and complement deficiencies. Semin Liver Dis 17:297–310 [DOI] [PubMed] [Google Scholar]
- Zipfel PF, Skerka C. (2009) Complement regulators and inhibitory proteins. Nat Rev Immunol 9:729–740 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.