

Abstract
The melanocortin 1 receptor (MC1R) is a highly polymorphic G protein-coupled receptor, which is known to modulate pigmentation and inflammation. In the current study, we investigated the pharmacological effects of select single-nucleotide polymorphisms (SNPs) (V60L, R163Q, and F196L). After transient expression of MC1Rs in human embryonic kidney 293 cells, basal and ligand-induced cAMP signaling and mitogen-activated protein kinase (MAPK) activation were assessed by using luciferase reporter gene assays and Western blot analysis, respectively. All receptor variants showed decreased basal cAMP activity. With the V60L and F196L variants, the decrease in constitutive activity was attributable, at least in part, to a reduction in surface expression. The F196L variant also displayed a significant reduction in potency for both the peptide agonist α-melanocyte-stimulating hormone (α-MSH) and the small-molecule agonist 1-[1-(3-methyl-l-histidyl-O-methyl-d-tyrosyl)-4-phenyl-4-piperidinyl]-1-butanone (BMS-470539). In MAPK signaling assays, the F196L variant showed decreased phospho-extracellular signal-regulated kinase levels after stimulation with either α-MSH or BMS-470539. In contrast, the R163Q variant displayed a selective loss of α-MSH-induced MAPK activation; whereas responsiveness to the small-molecule agonist BMS-470539 was preserved. Further assessment of MC1R variants in A549 cells, an in vitro model of inflammation, revealed an enhanced inflammatory response resulting from expression of the F196L variant (versus the wild-type MC1R). This alteration in function was restored by treatment with BMS-470539. Overall, these studies illustrate novel signaling profiles linked to distinct MC1R SNPs. Furthermore, our investigations highlight the potential for small-molecule drugs to rescue the function of MC1R variants that show reduced basal and/or α-MSH stimulated activity.
Introduction
The melanocortin 1 receptor (MC1R) is one of five members of the melanocortin subfamily of G protein-coupled receptors (GPCRs). The majority of research on the MC1R has focused on its role in regulating pigmentation through the activation of melanocytes (Roberts et al., 2006). The MC1R is coupled to Gαs; stimulation of this receptor triggers agonist-induced activation of adenylate cyclase with subsequent production of cAMP. High levels of constitutive activity are observed with expression of either the human or mouse MC1R orthologs (Sánchez-Más et al., 2004). In addition to adenylate cyclase, stimulation of the MC1R activates the mitogen-activated protein kinase (MAPK) pathway, leading to activation of the serine/threonine kinases ERK1 and ERK2 (Buscà et al., 2000). Once activated, ERKs translocate to the nucleus where they modulate gene transcription. MC1R variants leading to alterations in these signaling pathways have been associated with both red hair phenotypes and melanoma risk (Cohen et al., 2002; Robinson and Healy, 2002).
A further increase in second-messenger signaling (above basal levels) occurs in response to stimulation by α-melanocyte-stimulating hormone (α-MSH), the primary endogenous agonist for the MC1R. α-MSH is derived from the processing of the pro-opiomelanocortin precursor protein to adrenocorticotrophic hormone, which is in turn cleaved to yield α-MSH (Getting, 2006). Many derivatives of the mature form of endogenous α-MSH have been synthesized and studied, including NDP-MSH. This analog contains a d-phenylalanine at position 7 of α-MSH and has increased stability and a higher potency for the MC1R, facilitating use of this compound as a radioligand (Sawyer et al., 1980). In addition to peptide agonists, a small-molecule ligand for the MC1R has been synthesized based on the structure of a substituted piperidine melanocortin 4 receptor agonist. Using a piperidine linked to an O-methyltyrosine and a histidine as a template, modification of either the α-amino group or imidazole ring of the histidine resulted in varying degrees of activity and selectivity for the MC1R. The compound used in this study, 1-[1-(3-methyl-l-histidyl-O-methyl-d-tyrosyl)-4-phenyl-4-piperidinyl]-1-butanone (BMS-470539), was characterized and found to be a potent, specific MC1R ligand (Herpin et al., 2003). The affinity of this molecule, as determined by [125I]NDP-MSH competition binding experiments, was in the nanomolar range (IC50 = 120 nM).
Efforts to identify and characterize small-molecule agonists for the MC1R are in part motivated by the emerging role of this receptor in regulating inflammatory processes. In addition to its expression on melanocytes, the MC1R is found on macrophages, monocytes, dendritic cells, and mast cells where agonist stimulation attenuates inflammatory responses (Getting, 2002). Among the putative underlying mechanisms, modulation of nuclear factor κB (NFκB)-mediated transcription by the MC1R is of particular interest in light of the regulation of select target genes involved in inflammation through this pathway (Wikberg et al., 2000). To this end, both peptide and small-molecule agonists of the MC1R have been shown to attenuate NFκB activity in response to various inflammatory agents [e.g., tumor necrosis factor-α (TNFα), lipopolysaccharide] (Manna and Aggarwal, 1998; Yoon et al., 2003; Kang et al., 2006).
The MC1R is a highly polymorphic GPCR with more than 70 variants reported to date (García-Borrón et al., 2005). The majority of pharmacological studies of this receptor have focused on single-nucleotide polymorphisms (SNPs) associated with decreased pigmentation and melanoma risk (Beaumont et al., 2009). In contrast, the primary focus of this article is on an MC1R variant, F196L, which is found in a significant portion (11%) of sub-Saharan Africans (John et al., 2003). To our knowledge, this is the first report demonstrating the pharmacological characterization of this polymorphism. To enable side-by-side comparisons with other variants occurring with increased frequency in distinct ethnic populations, we have characterized in parallel a V60L variant found with a frequency of 15% in whites (Flanagan et al., 2000) and a R163Q variant, identified in 70% of those of East/Southeast Asian descent (Rana et al., 1999). With each of these three variants, we demonstrate distinct alterations in cAMP signaling, MAPK activity, and/or NFκB-mediated transcription. Furthermore, we show that selected SNPs have signaling defects that can be restored by BMS-470539, suggesting the potential utility of small-molecule drugs for rescuing clinical phenotypes resulting from aberrant MC1R signaling.
Materials and Methods
Materials
Dulbecco's modified Eagle's medium, fetal bovine serum, and Lipofectamine were obtained from Invitrogen (Carlsbad, CA). RPMI 1640 medium was purchased from Lonza (Chicago, IL). MegaTran 1.0 was purchased from OriGene (Atlanta, GA). Polyethylenimine and 2-nitrophenyl β-d-galactopyranoside were obtained from Sigma (St. Louis, MO). α-MSH was purchased from Bachem California (Torrance, CA), human TNFα was from Cell Signaling Technology (Danvers, MA), and [125I]NDP-MSH was from PerkinElmer Life and Analytical Sciences (Waltham, MA). BMS-470539 was a generous gift from Bristol Myers Squibb (New York, NY). The chemical structure of BMS-470539 is illustrated in Fig. 1. The plasmid encoding a cAMP response element (CRE6x) ligated upstream of a luciferase reporter gene has been described previously (Al-Fulaij et al., 2007). The plasmid encoding a NFκB response element upstream of a luciferase reporter gene was purchased from Agilent Technologies (Santa Clara, CA).
Fig. 1.
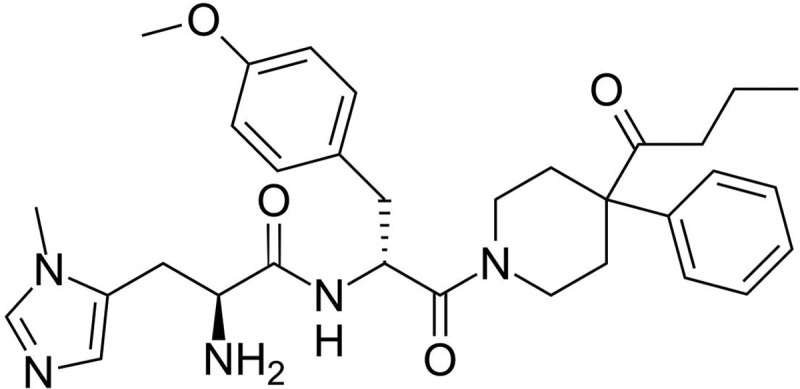
Chemical structure of BMS-470539.
Cell Culture
Human embryonic kidney (HEK) 293 cells were grown in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. A549 cells were grown in RPMI 1640 medium (Lonza) supplemented with 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were maintained at 37°C in a humidified environment containing 5% CO2.
Construction of Receptor Plasmids
Complementary DNA encoding the human MC1R was obtained from the UMR cDNA Resource Center (Rolla, MO) and subcloned into pcDNA1.1. Single amino acid substitutions were introduced by using oligonucleotide-directed, site-specific mutagenesis as described previously (Fortin et al., 2009). A hemagglutinin (HA) epitope tag (YPYDYPDYA) was introduced after the initiator methionine of each receptor construct by polymerase chain reaction. The nucleotide sequences of all receptor constructs were confirmed by automated DNA sequencing.
Luciferase Reporter Gene Assays
CRE-Luciferase.
Receptor-mediated signaling was assessed by slight modification of a previously described method (Al-Fulaij et al., 2008). In brief, HEK293 cells were plated at a density of 6000 cells/well onto clear-bottom, white 96-well plates (Corning Life Sciences, Lowell, MA) and grown to ∼80% confluence. Cells were transiently transfected in serum-free media by using polyethylenimine (0.1 μl/well of a 1 mg/ml solution in distilled H2O) (Boussif et al., 1995; Ehrhardt et al., 2006) with cDNAs encoding 1) wild-type or mutant MC1R, 2) a CRE-luciferase reporter gene (CRE6x-Luc), and 3) β-galactosidase to enable correction of interwell variability. Twenty-four hours after transfection, cells were incubated with or without ligand for 4 h in serum-free media. After agonist treatment, the medium was aspirated, and luciferase activity was measured by using SteadyLite Reagent (PerkinElmer Life and Analytical Sciences). A β-galactosidase assay was then performed by using the substrate 2-nitrophenyl β-d-galactopyranoside. After incubation at 37°C for 30 to 60 min, substrate cleavage was quantified by measurement of optical density at 420 nm with a SpectraMax microplate reader (Molecular Devices, Sunnyvale, CA). Luciferase activity was then normalized to the corresponding β-galactosidase activity levels in each well.
NFκB Luciferase.
A549 cells were plated at a density of 10,000 cells/well onto clear-bottom, white 96-well plates (Corning Life Sciences) and grown to ∼80% confluence. Cells were transiently transfected by using a previously described method with slight modification (Bonnans et al., 2006). In brief, A549 cells were transfected in serum-free media using MegaTran (0.2 μl/well) with cDNAs encoding 1) wild-type or mutant MC1R, 2) a NFκB-luciferase reporter gene, and 3) β-galactosidase to enable correction of interwell variability. Twenty-four hours after transfection, cells were incubated with BMS-470539 for 30 min followed by TNFα (0.5 ng/ml) for 4 h in serum-free media. Luciferase activity was determined and analyzed according to the protocol for CRE-luciferase.
ELISA of MC1R Expression
Receptor expression levels were determined as described previously (Al-Fulaij et al., 2008). In brief, HEK293 cells were grown in clear 96-well plates (BD Biosciences Discovery Labware, Bedford, MA) and transiently transfected with a plasmid encoding an HA-tagged wild-type or mutant MC1R. Twenty-four hours after transfection, the medium was aspirated, and cells were fixed with 4% paraformaldehyde in PBS for 10 min at room temperature. To enable measurement of total receptor expression, cells were permeabilized with 0.1% Triton X-100 in PBS for 2 min, whereas cell-surface expression was determined without the permeabilization step. After washing with 100 mM glycine in PBS, cells were incubated for 30 min in blocking solution (PBS containing 20% fetal bovine serum). A horseradish peroxidase-conjugated antibody (clone 3F10; Roche Applied Science, Indianapolis, IN) directed against the HA epitope tag was then added to the cells (1:500 dilution in blocking solution). After 2 h, cells were washed five times with PBS. The peroxidase substrate BM-blue (3.3′-5.5′-tetramethylbenzidine; Roche Applied Science) was then added at 50 μl per well. After incubation for 30 min at room temperature, conversion of this substrate by antibody-linked horseradish peroxidase was terminated by adding 2.0 M sulfuric acid (50 μl/well). Converted substrate was quantified by measuring light absorbance at 450 nm on a SpectraMax microplate reader (Molecular Devices). To correct for background, the absorbance values of cells transfected with empty pcDNA1.1 plasmid were subtracted from those observed with the HA-tagged MC1Rs.
Radioligand Binding Assays
Radioligand binding studies were performed as described previously (Schiöth et al., 1995; Beaumont et al., 2005; Fortin et al., 2010). In brief, 35,000 cells/well were seeded into 24-well plates that had been coated with poly-l-lysine. Cells were grown to 80% confluence before transient transfection with selected receptor cDNA using Lipofectamine reagent (Invitrogen). Whole-cell binding studies were initiated 48 h later by washing cells twice with cold (4°C) assay buffer (Dulbecco's modified Eagle's medium with 0.25% bovine serum albumin and 20 mM HEPES), followed by addition of the same buffer with 20,000 cpm of [125I]NDP-MSH and varying concentrations of α-MSH. After a 2-h incubation at room temperature, cells were placed on ice and washed three times with ice-cold PBS. Cells were then dissolved in 0.1 N NaOH, and the lysates were neutralized with an equal volume of 0.1 N HCl. The samples were then counted by using a Packard Cobra Quantum γ-counter (PerkinElmer Life and Analytical Sciences) to measure cell-associated radioactivity.
Phospho-ERK and Western Blotting
A total of 300,000 cells/plate were seeded in 60-mm dishes and grown to 80% confluence followed by transient transfection of receptor cDNA or pcDNA1.1 (vector control) by using Lipofectamine reagent (Invitrogen). Two days later, cells were treated with ligand for 0, 5, or 20 min in serum-free media. Cells were then placed on ice and, after aspiration of media, lysed with radioimmunoprecipitation assay buffer according to the manufacturer's protocol (Cell Signaling Technology). In brief, 200 μl of radioimmunoprecipitation assay buffer was added per dish for 5 min followed by cell scraping and disruption by repeated forceful passage through a 26-gauge needle. Lysates were centrifuged at 4°C for 10 min at 14,000g, and the supernatant was removed. Protein content in the supernatant was determined by using a commercially available bicinchoninic acid protein assay kit (Thermo Fisher Scientific, Waltham, MA). Protein samples for Western blot analysis (20 μg) were mixed with a 5× Laemmli buffer and subjected to SDS-polyacrylamide gel electrophoresis on a 10% Tris-glycine gel (running buffer: 25 mM Tris, 192 mM glycine, and 0.02% SDS). Samples were then electroblotted onto polyvinylidene difluoride membranes in 1× transfer buffer (25 mM Tris, 192 mM glycine, 20% methanol, and 0.02% SDS). After transfer, membranes were blocked with a TBST-milk solution (10 mM Tris, 150 mM NaCl, pH 8.0, 5% nonfat dry milk, and 0.05% Tween 20) for 1 h at room temperature. Membranes were then incubated overnight at 4°C in 5% milk containing primary antibodies. Corresponding antibody dilutions and sources were as follows: phospho-p44/42 MAPK (ERK1/2) 1:1000 (Cell Signaling Technology); p44/42 MAPK (ERK1/2) 1:2500 (Cell Signaling Technology). Membranes were washed four times in 1× TBST for 3 min each, then incubated with a horseradish peroxidase-conjugated bovine anti-rabbit secondary antibody (1:2500; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) in TBST-milk solution for 1 h at room temperature. Membranes were subsequently washed as described above and treated with enhanced chemiluminescent detection reagents (PerkinElmer Life and Analytical Sciences). Band intensities (phospho-p44/42 MAPK) were analyzed by using Kodak Gel Logic 100 image analysis software (Eastman Kodak, Rochester, NY) and normalized to wild-type control.
Data Analysis
In experiments in which results are presented as percentages of wild-type receptor control the value for each variant receptor was divided by the value of the corresponding control (e.g., effect on wild-type receptor) and multiplied by 100. The corresponding means were calculated across experimental days. Statistical comparisons of pharmacological parameters observed with wild-type versus variant MC1R were made by one-way analysis of variance with Tukey's post hoc test. Half-maximal effective concentrations (EC50 values) and dissociation constants (Ki) were determined by nonlinear curve fitting using Prism 5.0 software (GraphPad Software Inc., San Diego, CA). Results with a p value < 0.05 were considered statistically significant.
Results
Human Melanocortin 1 Receptor Variants.
Receptor constructs encoding the naturally occurring MC1R SNPs V60L, R163Q, and F196L were generated. A cartoon representation of the amino acid substitutions in the MC1R is illustrated in Fig. 2. All SNPs were reported in the Natural Variants database, a catalog of known human GPCR polymorphisms (Kazius et al., 2008). The reference SNP ID and corresponding nucleotide substitution are presented in Table 1.
Fig. 2.

A cartoon of the MC1R illustrating the position of missense mutations within the receptor protein. Respective residue substitutions in the wild-type MC1R are indicated by the single-letter amino acid code.
TABLE 1.
MC1R missense variants, SNP reference identification number, and corresponding nucleotide substitutions
| SNP | Reference ID Number | Nucleotide Substitution |
|---|---|---|
| V60L | rs1805005 | G/T |
| R163Q | rs885479 | G/A |
| F196L | rs3212366 | T/C |
MC1R Variants Exhibit Decreased Basal Activity.
Basal signaling of wild-type versus variant receptors was assessed by using a cAMP-responsive luciferase construct. As illustrated in Fig. 3, when expressed in HEK293 cells all receptors exhibit constitutive activity. However, relative to wild type, this function is significantly decreased for each variant. At the highest level of transfected cDNA, the V60L and R163Q variants show ∼80% and the F196L variant exhibits ∼50% of the basal signaling observed with the wild-type receptor. In addition to constitutive activity, each receptor can be further stimulated by either α-MSH (Fig. 4A) or the small-molecule agonist BMS-470539 (Fig. 4B) in a concentration-dependent manner. Although there is a trend toward lower agonist potency at each of the three variants versus the wild-type receptor, only the F196L polymorphism leads to a significant shift in the EC50, in response to both αMSH and BMS-470539 (Table 2).
Fig. 3.

The MC1R is constitutively active with receptor variants showing decreased basal activity. HEK293 cells were transiently transfected with the indicated concentrations of receptor-encoding cDNA and a CRE6x-Luc reporter gene construct. Twenty-four hours after transfection, luciferase activity was determined as described under Materials and Methods. Luciferase activity is expressed as a percentage of basal activity relative to values obtained after transfection of 8 ng of wild-type receptor cDNA (= 100%). Data points represent the mean ± S.E.M from at least three independent experiments, each performed in triplicate. Comparison of wild-type versus variant receptor activity after transfection with 8 ng of corresponding cDNA: **, p < 0.01; ***, p < 0.001.
Fig. 4.

All MC1R variants respond to stimulation with the peptide agonist αMSH (A) and the small-molecule agonist BMS-470539 (B). HEK293 cells were transiently transfected with receptor-encoding cDNA and a CRE6x-Luc reporter gene construct. Twenty-four hours after transfection, cells were incubated for 4 h with either no ligand or increasing concentrations of αMSH or BMS-470539. After stimulation, luciferase activity was determined as described under Materials and Methods and normalized relative to maximal stimulation at the wild-type MC1R. Data points represent the mean ± S.E.M from at least three independent experiments, each performed in triplicate.
TABLE 2.
Agonist potency at the wild-type versus variant receptors
Half-maximal effective concentrations (EC50 values) were calculated, and corresponding pEC50 values for wild-type versus variant receptors were compared.
| Receptor | αMSH |
BMS-470539 |
||
|---|---|---|---|---|
| EC50 | pEC50 | EC50 | pEC50 | |
| nM | nM | |||
| WT | 0.2 | 9.72 ± 0.17 | 2.3 | 8.59 ± 0.14 |
| V60L | 0.5 | 9.34 ± 0.06 | 6.1 | 8.27 ± 0.13 |
| R163Q | 0.3 | 9.55 ± 0.18 | 4.6 | 8.35 ± 0.11 |
| F196L | 1.1 | 8.99 ± 0.17* | 45.0 | 7.36 ± 0.06*** |
, p < 0.05;
, p < 0.001.
The Surface Expression of MC1R Variants V60L and F196L Are Decreased Relative to Wild Type.
To determine whether the decrease in basal signaling observed with MC1R variants correlates with a change in MC1R expression, ELISAs were performed on HA-tagged receptors. Assays were done by using either permeabilized or unpermeabilized cells to assess total (Fig. 5A) and surface (Fig. 5B) expression, respectively. Although there were no changes in total expression, a significant decrease in surface expression of the V60L and F196L variants was observed. It is of note that basal activity and the EC50 for agonist-induced signaling of the HA-tagged receptors were similar to corresponding values observed for respective nontagged MC1Rs (data not shown).
Fig. 5.

V60L and F196L variants have decreased cell-surface expression with no change in total expression. HEK293 cells were transfected with 8 ng of cDNA encoding either the wild-type or mutant HA-tagged MC1Rs. After 24 h, expression levels were measured by ELISA as described under Materials and Methods. Data are graphed as a percentage of wild-type receptor expression. Data points represent the mean ± S.E.M from at least three independent experiments, each performed with six replicates. Comparison of wild-type versus variant receptor expression: *, p < 0.05; **, p < 0.01.
MC1R Variants Do Not Exhibit a Change in Binding Affinity.
To assess agonist affinity, radioligand binding assays were performed by using a fixed concentration of [125I]NDP-MSH as the tracer and increasing concentrations of αMSH as the unlabeled competitor. No significant differences in αMSH affinity were observed when comparing wild-type and variant receptors (Fig. 6; Table 3).
Fig. 6.

MC1R mutations have no effect on NDP-MSH binding affinity. HEK293 cells in 24-well plates were transiently transfected with wild-type or variant receptors. Forty-eight hours later, [125I]NDP-MSH radioligand binding with increasing concentrations of unlabeled α-MSH was evaluated. Cells were incubated for 2 h at room temperature in the absence or presence of unlabeled α-MSH at the indicated concentrations. Data points represent the mean ± S.E.M from at least four independent experiments, each performed in triplicate. Data are expressed as a percentage of radioligand binding to the wild-type receptor in the absence of cold α-MSH.
TABLE 3.
Ki values for NDP-MSH binding experiments
No significant differences were observed between corresponding pKi values for the wild-type versus variant receptors (p > 0.05).
| Receptor | Ki | pKi |
|---|---|---|
| nM | ||
| WT | 3.9 | 8.46 ± 0.15 |
| V60L | 2.2 | 8.70 ± 0.12 |
| R163Q | 2.8 | 8.56 ± 0.07 |
| F196L | 4.2 | 8.35 ± 0.11 |
Selected MC1R Polymorphisms Attenuate the Ability of Agonists to Trigger Receptor-Mediated MAPK Activation.
In HEK293 cells expressing wild-type MC1R, stimulation with both αMSH (Fig. 7, A and B) and BMS-470539 (Fig. 7, C and D) resulted in a transient, concentration-dependent increase in phospho-p44/42 ERK levels. Although a strong phospho-p44/42 ERK signal (indicative of MAPK activation) was observed after 5 min of ligand exposure, this effect was markedly diminished after 20 min of incubation. Compared with the wild-type MC1R, all tested variants showed similar time patterns and maximal levels of ligand-induced MAPK activation at saturating concentrations of either αMSH (10−6 M) or BMS-470529 (10−5 M). The MAPK activation induced by lower concentrations of α-MSH at either the R163Q or F196L variants was significantly less than that observed with the stimulation of wild-type receptor at their respective agonist concentrations (Fig. 8, A and B). With the F196L variant, MAPK activation by lower concentrations of the small-molecule agonist BMS-470539 was also significantly decreased, whereas the effect of this ligand at the R163Q or V60L variants was comparable with wild type (Fig. 8, C and D).
Fig. 7.

Cells expressing the wild-type MC1R show concentration-dependent transient increases in MAPK activation in response to αMSH and BMS-470539. HEK293 cells were transfected with cDNA encoding the wild-type MC1R. Forty-eight hours after transfection, the cells were stimulated with the indicated concentrations of either αMSH or BMS-470539 for 5 or 20 min, or cells were not treated with ligand (0 min). After stimulation, cells were lysed and levels of phospho-ERK (pERK) and total ERK (tERK) measured by Western blot analysis as described under Materials and Methods. A, a representative Western blot illustrates the concentration- and time-dependent effects of αMSH on phospho-ERK levels. B, quantitation of the Western blot signal 5 min poststimulation. Data are expressed as a percentage of the effect induced by 10−6 M αMSH. Comparisons were made relative to this maximal value: **, p < 0.01; ***, p < 0.001. Each bar represents the mean ± S.E.M from at least four independent experiments. C, a representative Western blot illustrates the concentration- and time-dependent effects of BMS-470539 on phospho-ERK levels. D, quantitation of the Western blot signal 5 min poststimulation. Data are expressed as a percentage of the effect induced by 10−5 M BMS-470539. Comparisons were made relative to this maximal value: *, p < 0.05; **, p < 0.01. Each bar represents the mean ± S.E.M from at least four independent experiments.
Fig. 8.

MC1R polymorphisms can decrease MAPK activity induced by αMSH and/or BMS-470539 relative to the wild-type control. Forty-eight hours after transfection of MC1R encoding cDNA, HEK293 cells were stimulated with the indicated concentrations of either αMSH or BMS-470539 for 5 or 20 min, or cells were not treated with ligand (0 min). After stimulation, cells were lysed, and levels of phospho- and total ERK were measured by Western blot analysis as described under Materials and Methods. A, a representative Western blot illustrates the concentration- and time-dependent effects of αMSH on phospho-ERK levels in cells expressing wild-type versus variant receptors. B, quantitation of Western blot signals at the 5-min point. Data are expressed as a percentage of the αMSH-induced effect at the corresponding ligand concentration in cells expressing the wild-type MC1R. Bars represent the mean ± S.E.M from at least three independent experiments. Comparison of ligand effects at a given concentration in cells expressing wild-type versus variant receptors: *, p < 0.05; **, p < 0.01; ***, p < 0.001. C, a representative Western blot illustrates the concentration- and time-dependent effects of BMS-470539 on phospho-ERK levels in cells expressing wild-type versus variant receptors. D, quantitation of Western blot data at the 5-min point. Data are expressed as a percentage of the BMS-470539 induced effect at the corresponding ligand concentration in cells expressing the wild-type MC1R. Bars represent the mean ± S.E.M from at least three independent experiments. Comparison of ligand effects at a given concentration in cells expressing wild-type versus variant receptors: *, p < 0.05; **, p < 0.01.
MC1R Polymorphisms Do Not Affect BMS-470539-Induced Inhibition of NFκB Activity.
MC1R activation inhibits the NFκB signaling pathway in cellular models and may thereby contribute to the resolution of inflammatory processes (Wikberg et al., 2000; Getting, 2002). To explore this MC1R function, we compared the ability of BMS-470539 to inhibit TNFα-stimulated activation of a NFκB-luciferase construct in cells expressing wild-type versus variant receptors (Kang et al., 2006). These experiments were performed in the human type II alveolar epithelial cell line,A549, an established in vitro model of inflammation that is readily transfectable and suitable for studying the modulation of inflammation by melanocortin peptides (Bonnans et al., 2006; Getting et al., 2008). In A549 cells expressing either the wild-type or variant MC1R constructs (V60L and R163Q), TNFα stimulates NFκB activity to a comparable extent. In contrast, TNFα-induced stimulation of NFκB activity is significantly higher when the F196L variant is expressed (Fig. 9). BMS-470539 inhibits TNFα-stimulated NFκB luciferase activity to a similar extent in cells expressing either wild-type or variant MC1Rs (Fig. 9). Using the same assay, controls transfected with the empty pcDNA1.1 expression vector showed no response (data not shown).
Fig. 9.

BMS-470539 inhibits TNFα-induced NFκβ activity in cells expressing wild-type or variant MC1Rs. In the presence of TNFα alone, the F196L variant shows an elevated level of NFκB-luciferase activity relative to wild type. A549 cells were transiently transfected with cDNA encoding the indicated receptors together with an NFκB-luciferase reporter gene construct. Twenty-four hours after transfection, cells were preincubated with or without BMS-470539 for 30 min, followed by further incubation in the presence of TNFα (0.5 ng/ml) for another 4 h. Luciferase activity was then determined as described under Materials and Methods. Data are expressed as a percentage of TNFα-stimulated NFκB-luciferase activity observed in cells expressing wild-type MC1R, in the absence of BMS-470539. Bars represent the mean ± S.E.M from at least four independent experiments. In the absence of TNFα stimulation, no significant differences were observed between wild-type and variant receptors (p > 0.05). Comparison of TNFα-stimulated NFκB activity versus wild-type receptor control in the absence of BMS-470539: #, p < 0.05. Comparison of NFκB activity at each receptor in the presence of increasing concentrations of BMS-470539: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Discussion
Most pharmacological studies of the highly polymorphic MC1R have focused on missense variants associated with decreased pigmentation/red hair phenotypes. Here, we focus on the F196L isoform that is present in 11% of a sub-Saharan African cohort. We have examined how this previously uncharacterized SNP influences cAMP, MAPK, and NFκB signaling under basal conditions as well as in response to pharmacological stimulation with peptide (α-MSH) or small-molecule (BMS-470539) agonists. As a comparison, we also studied the V60L and R163Q variants, which are found with high frequency in white and Asian populations, respectively. Although these two variants have previously been reported to reduce cAMP production (Beaumont et al., 2005, 2007), their role in MAPK signaling and pathways associated with inflammation remained unexplored.
Assessment of these three naturally occurring mutations in the MC1R revealed that each showed a significant decrease in basal signaling; this loss of constitutive activity was most pronounced with expression of the F196L variant. The observed change in basal activity of the V60L and F196L variants may in part be explained by a decrease in cell surface expression. Because total expression of these variants was unchanged, it is possible that these missense variants lead to misfolding or structural instability of the receptor, resulting in intracellular retention, but not degradation (Conn and Ulloa-Aguirre, 2010). In contrast, the R163Q variant displayed a decrease in basal activity with no change in surface expression. The reduced level of constitutive signaling with this variant may be related to altered G-protein coupling given the location of this SNP within the second intracellular loop, a well established site of GPCR-G protein interaction. Further supporting the premise that this region is a determinant of MC1R basal activity, a decrease in cAMP signaling has been observed with substitution of an adjacent receptor residue, R162P (Jiménez-Cervantes et al., 2001).
In addition to the changes in basal activity, investigation of the F196L variant revealed a significant decrease in potency of αMSH, the endogenous peptide agonist. The shift in EC50 observed with receptor variants may be explained by either an alteration of the hormone binding site and/or defective transitioning from the inactive to the active receptor state (Lefkowitz et al., 1993; Beinborn et al., 2004). Given the location of this substitution within the fifth transmembrane domain (Schiöth et al., 1998) and the normal affinity of the F196L variant for radiolabeled NDP-MSH, it is unlikely that the hormone binding site is directly altered by this residue change. It is more plausible that the decrease in basal activity reflects a polymorphism-induced transition to a less active receptor state with an accompanying decrease in agonist potency as predicted by the extended ternary model (Lefkowitz et al., 1993). The V60L and R163Q polymorphisms displayed agonist potency and affinity similar to the wild-type receptor, consistent with previous reports (Schiöth et al., 1995; Ringholm et al., 2004; Beaumont et al., 2005, 2007).
Recent efforts have led to the development of a small-molecule agonist, BMS-470539, that specifically stimulates the MC1R versus other melanocortin receptor subtypes. As candidate therapeutics, advantages of small-molecule versus peptide ligands include resistance to proteolytic degradation and enhanced stability in vivo as well as the potential for oral application. Our results suggest that select SNPs may lead to either parallel or divergent changes in the function of endogenous peptide versus synthetic small-molecule ligands. This is best illustrated by comparing MAPK signaling between the F196L and R163Q variants. The F196L substitution leads to a generalized decrease in ligand induced effect (i.e., as with α-MSH, the potency of BMS-470539 is significantly attenuated). In contrast, the R163Q variant, while leading to reduced α-MSH potency, does not affect the response to BMS-470539. Results with the R163Q variant suggest a potential clinical utility of small molecules as drugs for rescuing defects in endogenous ligand activity (e.g., αMSH-induced ERK phosphorylation). In light of the variability in MC1R polymorphism-induced signaling, it will be important to assess both the αMSH and small-molecule agonist activity on each receptor variant to determine whether 1) endogenous signaling is compromised and 2) pharmacological rescue is possible.
Our data also highlight that MC1R SNP-induced changes in endogenous agonist signaling may either selectively alter one specific pathway or simultaneously affect multiple receptor-mediated functions. With the F196L variant, both αMSH-induced MAPK and cAMP signaling are attenuated. In contrast, the R163Q SNP shows a selective decrease in MAPK activation, whereas ligand-stimulated cAMP signaling is not affected. A prior study on MC1R SNPs identified mutants with an opposite phenotype; the MC1R variants R151C, R160W, and D294H show reduced cAMP production while maintaining normal MAPK signaling (Herraiz et al., 2009, 2011). Each of these latter variants is highly associated with red hair color and melanoma risk. In the future, it will be of interest to determine whether an opposite pattern of alterations (i.e., normal cAMP and compromised MAPK, as observed with the R163Q SNP) or a broader attenuation of signaling (found with the F196L variant, where both pathways are affected), may also predispose to abnormal physiology or disease.
The expression of polymorphic MC1Rs on macrophages, monocytes, dendritic cells, and mast cells raises the possibility that pathways linked to inflammation may be altered by missense variants in this receptor. To examine this question, we used A549 cells as an in vitro model system. TNF-α-induced, NFκB-mediated luciferase activity was quantified as an index of inflammatory response. In cells expressing either the wild-type MC1R or any of the three polymorphic receptors BMS-470539 decreased signaling via this pathway to a similar extent. The BMS-470539-induced effect was not altered by MC1R polymorphisms, even ones that have been shown to attenuate cAMP and MAPK signaling (as is the case with the F196L variant). It thus seems that MC1R small-molecule agonists have the potential to promote anti-inflammatory effects even when ligand-induced canonical signaling (e.g., cAMP production, MAPK activation) is compromised. Therefore, continued development and testing of MC1R agonists in models of inflammation may yield drug candidates with promising clinical efficacy even in individuals harboring MC1R mutations.
As described above, MC1R-induced inhibition of TNFα-stimulated NFκB signaling is sensitive to BMS-470539 stimulation. At the same time, our results suggest that this pathway it is also influenced by basal receptor activity. This effect is best illustrated by comparing the wild-type receptor with the F196L variant, where the F196L polymorphism shows reduced basal inhibitory activity. It thus seems that expression of the wild-type MC1R, even in the absence of ligand, is sufficient to dampen the basal inflammatory state. This assertion is further supported by experiments involving immunoneutralization of the MC1R in a human monocytic THP-1 cell line (Taherzadeh et al., 1999). After neutralization with an anti-MC1R-specific antibody, production of TNFα in resting THP-1 cells is increased, suggesting that the presence of the MC1R may negatively modulate inflammation. These data raise the possibility that individuals carrying loss-of-function MC1Rs may be at higher risk for inflammatory disease, a concern that should be further explored.
In conclusion, our studies demonstrate that loss-of-function SNPs in the MC1R can alter multiple intracellular signaling pathways. Affected pathways include production of cAMP, MAPK activation, and NFκB-mediated transcription. In this study, we illustrate novel pharmacological properties of the previously uncharacterized MC1R variant F196L. We also provide evidence to suggest that MC1R SNPs can bias the signaling of the endogenous agonist α-MSH, leading to a selective loss of one or more pathways. At the same time we highlight the ability of a small-molecule ligand, BMS-470539, to attenuate signaling linked to inflammation in the presence of MC1R variants that compromise other ligand-mediated activities. Taken together, these findings set the stage for future studies aimed at investigating the association between MC1R SNPs and clinical markers of inflammation. In addition, further efforts to identify and characterize small-molecule ligands of the MC1R (Catania et al., 2004) may help to define the potential of such compounds as anti-inflammatory drugs in normal individuals and in carriers of MC1R loss-of-function mutations.
Acknowledgments
We thank Kenneth Carlson and Bristol-Myers Squibb for the BMS-470539 compound; Bruce Levy and Jennifer Colby for the A549 cells; Ci Chen for excellent technical assistance; and Charles Contant for advice with statistical analysis.
This work was supported by the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant R01-DK072497] and the National Institutes of Health National Heart Lung and Blood Institute [Grant T32 HL069770].
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
- MC1R
- melanocortin 1 receptor
- GPCR
- G protein-coupled receptor
- MAPK
- mitogen-activated protein kinase
- SNP
- single-nucleotide polymorphism
- α-MSH
- α-melanocyte-stimulating hormone
- TNFα
- tumor necrosis factor-α
- HA
- hemagglutinin
- CRE
- cAMP-response element
- Luc
- luciferase
- ELISA
- enzyme-linked immunosorbent assay
- HEK
- human embryonic kidney
- PBS
- phosphate-buffered saline
- ERK
- extracellular signal-regulated kinase
- NDP
- Nle4-d-Phe7
- NFκB
- nuclear factor κB
- TBST
- Tris-buffered saline/Tween 20
- WT
- wild type
- BMS-470539
- 1-[1-(3-methyl-l-histidyl-O-methyl-d-tyrosyl)-4-phenyl-4-piperidinyl]-1-butanone.
Authorship Contributions
Participated in research design: Doyle, Fortin, Beinborn, and Kopin.
Conducted experiments: Doyle.
Performed data analysis: Doyle.
Wrote or contributed to the writing of the manuscript: Doyle, Fortin, Beinborn, and Kopin.
References
- Al-Fulaij MA, Ren Y, Beinborn M, Kopin AS. (2007) Identification of amino acid determinants of dopamine 2 receptor synthetic agonist function. J Pharmacol Exp Ther 321:298–307 [DOI] [PubMed] [Google Scholar]
- Al-Fulaij MA, Ren Y, Beinborn M, Kopin AS. (2008) Pharmacological analysis of human D1 AND D2 dopamine receptor missense variants. J Mol Neurosci 34:211–223 [DOI] [PubMed] [Google Scholar]
- Beaumont KA, Liu YY, Sturm RA. (2009) The melanocortin-1 receptor gene polymorphism and association with human skin cancer. Prog Mol Biol Transl Sci 88:85–153 [DOI] [PubMed] [Google Scholar]
- Beaumont KA, Newton RA, Smit DJ, Leonard JH, Stow JL, Sturm RA. (2005) Altered cell surface expression of human MC1R variant receptor alleles associated with red hair and skin cancer risk. Hum Mol Genet 14:2145–2154 [DOI] [PubMed] [Google Scholar]
- Beaumont KA, Shekar SN, Shekar SL, Newton RA, James MR, Stow JL, Duffy DL, Sturm RA. (2007) Receptor function, dominant negative activity and phenotype correlations for MC1R variant alleles. Hum Mol Genet 16:2249–2260 [DOI] [PubMed] [Google Scholar]
- Beinborn M, Ren Y, Bläker M, Chen C, Kopin AS. (2004) Ligand function at constitutively active receptor mutants is affected by two distinct yet interacting mechanisms. Mol Pharmacol 65:753–760 [DOI] [PubMed] [Google Scholar]
- Bonnans C, Fukunaga K, Levy MA, Levy BD. (2006) Lipoxin A4 regulates bronchial epithelial cell responses to acid injury. Am J Pathol 168:1064–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussif O, Lezoualc'h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. (1995) A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci U S A 92:7297–7301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscà R, Abbe P, Mantoux F, Aberdam E, Peyssonnaux C, Eychène A, Ortonne JP, Ballotti R. (2000) Ras mediates the cAMP-dependent activation of extracellular signal-regulated kinases (ERKs) in melanocytes. EMBO J 19:2900–2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catania A, Gatti S, Colombo G, Lipton JM. (2004) Targeting melanocortin receptors as a novel strategy to control inflammation. Pharmacol Rev 56:1–29 [DOI] [PubMed] [Google Scholar]
- Cohen C, Zavala-Pompa A, Sequeira JH, Shoji M, Sexton DG, Cotsonis G, Cerimele F, Govindarajan B, Macaron N, Arbiser JL. (2002) Mitogen-activated protein kinase activation is an early event in melanoma progression. Clin Cancer Res 8:3728–3733 12473582 [PubMed] [Google Scholar]
- Conn PM, Ulloa-Aguirre A. (2010) Trafficking of G-protein-coupled receptors to the plasma membrane: insights for pharmacoperone drugs. Trends Endocrinol Metab 21:190–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrhardt C, Schmolke M, Matzke A, Knoblauch A, Will C, Wixler V, Ludwig S. (2006) Polyethylenimine, a cost-effective transfection agent. Signal Transduction 6:179–184 [Google Scholar]
- Flanagan N, Healy E, Ray A, Philips S, Todd C, Jackson IJ, Birch-Machin MA, Rees JL. (2000) Pleiotropic effects of the melanocortin 1 receptor (MC1R) gene on human pigmentation. Hum Mol Genet 9:2531–2537 [DOI] [PubMed] [Google Scholar]
- Fortin JP, Schroeder JC, Zhu Y, Beinborn M, Kopin AS. (2010) Pharmacological characterization of human incretin receptor missense variants. J Pharmacol Exp Ther 332:274–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin JP, Zhu Y, Choi C, Beinborn M, Nitabach MN, Kopin AS. (2009) Membrane-tethered ligands are effective probes for exploring class B1 G protein-coupled receptor function. Proc Natl Acad Sci U S A 106:8049–8054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Borrón JC, Sánchez-Laorden BL, Jiménez-Cervantes C. (2005) Melanocortin-1 receptor structure and functional regulation. Pigment Cell Res 18:393–410 [DOI] [PubMed] [Google Scholar]
- Getting SJ. (2002) Melanocortin peptides and their receptors: new targets for anti-inflammatory therapy. Trends Pharmacol Sci 23:447–449 [DOI] [PubMed] [Google Scholar]
- Getting SJ. (2006) Targeting melanocortin receptors as potential novel therapeutics. Pharmacol Ther 111:1–15 [DOI] [PubMed] [Google Scholar]
- Getting S, Kaneva M, Kerrigan M. (2008) A role for melanocortin peptides in modulating oxidative stress induced inflammation in A549 cells. Proc Br Pharmacol Soc 6:C020 [Google Scholar]
- Herpin TF, Yu G, Carlson KE, Morton GC, Wu X, Kang L, Tuerdi H, Khanna A, Tokarski JS, Lawrence RM, et al. (2003) Discovery of tyrosine-based potent and selective melanocortin-1 receptor small-molecule agonists with anti-inflammatory properties. J Med Chem 46:1123–1126 [DOI] [PubMed] [Google Scholar]
- Herraiz C, Jiménez-Cervantes C, Zanna P, García-Borrón JC. (2009) Melanocortin 1 receptor mutations impact differentially on signalling to the cAMP and the ERK mitogen-activated protein kinase pathways. FEBS Lett 583:3269–3274 [DOI] [PubMed] [Google Scholar]
- Herraiz C, Journé F, Abdel-Malek Z, Ghanem G, Jiménez-Cervantes C, García-Borrón JC. (2011) Signaling from the human melanocortin 1 receptor to ERK1 and ERK2 mitogen-activated protein kinases involves transactivation of cKIT. Mol Endocrinol 25:138–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Cervantes C, Olivares C, González P, Morandini R, Ghanem G, García-Borrón JC. (2001) The Pro162 variant is a loss-of-function mutation of the human melanocortin 1 receptor gene. J Invest Dermatol 117:156–158 [DOI] [PubMed] [Google Scholar]
- John PR, Makova K, Li WH, Jenkins T, Ramsay M. (2003) DNA polymorphism and selection at the melanocortin-1 receptor gene in normally pigmented southern African individuals. Ann NY Acad Sci 994:299–306 [DOI] [PubMed] [Google Scholar]
- Kang L, McIntyre KW, Gillooly KM, Yang Y, Haycock J, Roberts S, Khanna A, Herpin TF, Yu G, Wu X, et al. (2006) A selective small molecule agonist of the melanocortin-1 receptor inhibits lipopolysaccharide-induced cytokine accumulation and leukocyte infiltration in mice. J Leukoc Biol 80:897–904 [DOI] [PubMed] [Google Scholar]
- Kazius J, Wurdinger K, van Iterson M, Kok J, Bäck T, Ijzerman AP. (2008) GPCR NaVa database: natural variants in human G protein-coupled receptors. Hum Mutat 29:39–44 [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Cotecchia S, Samama P, Costa T. (1993) Constitutive activity of receptors coupled to guanine nucleotide regulatory proteins. Trends Pharmacol Sci 14:303–307 [DOI] [PubMed] [Google Scholar]
- Manna SK, Aggarwal BB. (1998) α-Melanocyte-stimulating hormone inhibits the nuclear transcription factor NF-κB activation induced by various inflammatory agents. J Immunol 161:2873–2880 [PubMed] [Google Scholar]
- Rana BK, Hewett-Emmett D, Jin L, Chang BH, Sambuughin N, Lin M, Watkins S, Bamshad M, Jorde LB, Ramsay M, et al. (1999) High polymorphism at the human melanocortin 1 receptor locus. Genetics 151:1547–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringholm A, Klovins J, Rudzish R, Phillips S, Rees JL, Schiöth HB. (2004) Pharmacological characterization of loss of function mutations of the human melanocortin 1 receptor that are associated with red hair. J Invest Dermatol 123:917–923 [DOI] [PubMed] [Google Scholar]
- Roberts DW, Newton RA, Beaumont KA, Helen Leonard J, Sturm RA. (2006) Quantitative analysis of MC1R gene expression in human skin cell cultures. Pigment Cell Res 19:76–89 [DOI] [PubMed] [Google Scholar]
- Robinson SJ, Healy E. (2002) Human melanocortin 1 receptor (MC1R) gene variants alter melanoma cell growth and adhesion to extracellular matrix. Oncogene 21:8037–8046 [DOI] [PubMed] [Google Scholar]
- Sánchez-Más J, Hahmann C, Gerritsen I, García-Borrón JC, Jiménez-Cervantes C. (2004) Agonist-independent, high constitutive activity of the human melanocortin 1 receptor. Pigment Cell Res 17:386–395 [DOI] [PubMed] [Google Scholar]
- Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel MH, Heward CB, Burnett JB, Hadley ME. (1980) 4-Norleucine, 7-d-phenylalanine-α-melanocyte-stimulating hormone: a highly potent α-melanotropin with ultralong biological activity. Proc Natl Acad Sci U S A 77:5754–5758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiöth HB, Muceniece R, Wikberg JE, Chhajlani V. (1995) Characterisation of melanocortin receptor subtypes by radioligand binding analysis. Eur J Pharmacol 288:311–317 [DOI] [PubMed] [Google Scholar]
- Schiöth HB, Yook P, Muceniece R, Wikberg JE, Szardenings M. (1998) Chimeric melanocortin MC1 and MC3 receptors: identification of domains participating in binding of melanocyte-stimulating hormone peptides. Mol Pharmacol 54:154–161 [DOI] [PubMed] [Google Scholar]
- Taherzadeh S, Sharma S, Chhajlani V, Gantz I, Rajora N, Demitri MT, Kelly L, Zhao H, Ichiyama T, Catania A, et al. (1999) α-MSH and its receptors in regulation of tumor necrosis factor-α production by human monocyte/macrophages. Am J Physiol Regul Integr Comp Physiol 276:R1289–R1294 [DOI] [PubMed] [Google Scholar]
- Wikberg JE, Muceniece R, Mandrika I, Prusis P, Lindblom J, Post C, Skottner A. (2000) New aspects on the melanocortins and their receptors. Pharmacol Res 42:393–420 [DOI] [PubMed] [Google Scholar]
- Yoon SW, Goh SH, Chun JS, Cho EW, Lee MK, Kim KL, Kim JJ, Kim CJ, Poo H. (2003) α-Melanocyte-stimulating hormone inhibits lipopolysaccharide-induced tumor necrosis factor-α production in leukocytes by modulating protein kinase A, p38 kinase, and nuclear factor κB signaling pathways. J Biol Chem 278:32914–32920 [DOI] [PubMed] [Google Scholar]