

Abstract
Aldehyde dehydrogenases (ALDHs) belong to a superfamily of enzymes that play a key role in the metabolism of aldehydes of both endogenous and exogenous derivation. The human ALDH superfamily comprises 19 isozymes that possess important physiological and toxicological functions. The ALDH1A subfamily plays a pivotal role in embryogenesis and development by mediating retinoic acid signaling. ALDH2, as a key enzyme that oxidizes acetaldehyde, is crucial for alcohol metabolism. ALDH1A1 and ALDH3A1 are lens and corneal crystallins, which are essential elements of the cellular defense mechanism against ultraviolet radiation-induced damage in ocular tissues. Many ALDH isozymes are important in oxidizing reactive aldehydes derived from lipid peroxidation and thereby help maintain cellular homeostasis. Increased expression and activity of ALDH isozymes have been reported in various human cancers and are associated with cancer relapse. As a direct consequence of their significant physiological and toxicological roles, inhibitors of the ALDH enzymes have been developed to treat human diseases. This review summarizes known ALDH inhibitors, their mechanisms of action, isozyme selectivity, potency, and clinical uses. The purpose of this review is to 1) establish the current status of pharmacological inhibition of the ALDHs, 2) provide a rationale for the continued development of ALDH isozyme-selective inhibitors, and 3) identify the challenges and potential therapeutic rewards associated with the creation of such agents.
I. Introduction
The aldehyde dehydrogenases (ALDHs) have a surprisingly broad spectrum of biological activities. ALDH activity is crucial to the biosynthesis of retinoic acid, an important regulator of vertebrate development, and to the metabolism of the neurotransmitter GABA (Yoshida et al., 1992; Vasiliou et al., 2000; Sophos and Vasiliou, 2003; Vasiliou and Nebert, 2005; Marchitti et al., 2008; Niederreither and Dollé, 2008; Kim et al., 2011a). From the toxicological point of view, dehydrogenase enzymatic activity of ALDHs is important in alcohol metabolism through aldehyde detoxification and for cellular homeostasis by eliminating reactive aldehydes derived from lipid peroxidation (Vasiliou et al., 2000). Increased ALDH activity, however, has been found to interfere with certain chemotherapeutic treatments (Sládek, 1999; Jelski et al., 2007; Tanei et al., 2009; Deng et al., 2010). In addition, ALDHs can act as esterases (Blackwell et al., 1983; Yoshida et al., 1998) and also perform nonenzymatic functions, such as reducing osmotic stress in plants, binding to macromolecules (such as androgen and cholesterol) and protecting the mammalian cornea from UV exposure (Estey et al., 2007; Chen et al., 2009). Genetic polymorphisms of ALDH isozymes that result in diminished enzymatic activity are associated with several clinical disease states, including Sjögren-Larsson syndrome, Type 2 hyperprolinemia, pyridoxine-dependent seizures, hyperammonemia, γ-hydroxybutyric aciduria, and alcoholic liver disease (Marchitti et al., 2008).
In the past, the well established role of ALDH in alcohol metabolism has driven the research behind the discovery of ALDH inhibitors. Accumulation of acetaldehyde after ethanol consumption leads to the development of unpleasant physiological effects comprising facial flushing, nausea, and tachycardia. This condition, termed the alcohol flushing syndrome, commonly occurs in subjects possessing a genetic polymorphism that confers upon them reduced activity of ALDH2, the enzyme responsible for the efficient metabolism of acetaldehyde. This observation led to the initial development of selective ALDH2 inhibitors as antidipsotropic or alcohol-aversive agents (Keung, 2003). As our understanding of the roles played by the various ALDH isozymes in disease states continues to expand, the rationale for the development of selective inhibitors of the individual isozymes becomes more apparent. The availability of such inhibitors, at minimum, would permit verification of the putative roles of the isozymes. Optimally, such inhibitors would be used to treat disease states in which ALDH activity is implicated in their pathophysiology (Hiltbrand et al., 2009; Yao et al., 2010; Zhang et al., 2005, 2011). This review summarizes the current state of knowledge about inhibitors of the ALDH superfamily with respect to their selectivity, efficacy, and structure-activity relationships and their applications in treating disease states.
II. The Aldehyde Dehydrogenase Superfamily of Enzymes
The human ALDH superfamily comprises 19 known NAD(P+)-dependent enzymes that irreversibly catalyze the oxidation of both endogenously and exogenously produced aldehydes to their respective carboxylic acids (Fig. 1). In 1998, a gene nomenclature based on peptide sequence identity was instituted such that 1) families within the superfamily shared more than 40% sequence identity and 2) members of the same subfamily shared more than 60% sequence identity. ALDH activity is constitutively expressed in mammalian tissues, with the highest level in the liver, followed by the kidney, uterus, and brain (Alnouti and Klaassen, 2008). This activity is typically a composite of one or more of the ALDH isozymes, and some cells can also be induced to express ALDH isozymes under conditions of stress (Rice et al., 2008).
Fig. 1.

The ALDH superfamily of enzymes. Clustering dendrogram depicting the evolution of mammalian ALDHs from a single gene, their human chromosomal location, preferred substrate, and available X-ray crystal structures from the Research Collaboratory for Structural Bioinformatics (RCSB-PDB) database. *, substrate for hydrolase reaction of ALDH1L2 (Strickland et al., 2011). [Modified from Marchitti SA, Deitrich RA, and Vasiliou V (2007) Neurotoxicity and metabolism of the catecholamine-derived 3,4-dihydroxyphenylacetaldehyde and 3,4-dihydroxyphenylglycolaldehyde: the role of aldehyde dehydrogenase. Pharmacol Rev 59:125–150. Copyright © 2007 American Society for Pharmacology and Experimental Therapeutics. Used with permission.]
A. Enzymatic Functions of Aldehyde Dehydrogenase Isozymes
During embryogenesis, many developmental genes are transcriptionally regulated by retinoic acid (RA), the active metabolite of vitamin A and RA receptors (Niederreither and Dollé, 2008). The ALDH1A subfamily comprises the primary ALDHs (ALDH1A1/-1A2/-1A3) that synthesize RA from retinal and are therefore crucial in regulating RA signaling. These ALDH isozymes have a high affinity for the oxidation of both all-trans- and 9-cis-retinal, exhibiting a Km in the lower micromolar range (Yoshida et al., 1992; Chen et al., 2011a). In agreement with a developmental role, these isozymes are expressed in tissues during vertebrate development in a spatial-temporal fashion (Downes and Holmes, 1992; Grün et al., 2000). Some stem cells, such as those associated with hematopoiesis, possess higher ALDH activity than normal cells, a characteristic that can be exploited for the isolation of primitive stem cell populations (Storms et al., 1999; Storms et al., 2005). Such activity in hematopoietic stem cells mediates RA signaling and thereby serves to regulate the self-renewal and differentiation of these cells (Hess et al., 2006; Rice et al., 2008).
The ALDH2 isozyme is predominantly linked with acetaldehyde detoxification in the second step of alcohol metabolism. Other ALDHs, including ALDH1A1 and ALDH1B1, also contribute to acetaldehyde oxidation, albeit with lesser affinity than the ALDH2 isozyme (Wang et al., 2009; Stagos et al., 2010). A genetic polymorphism in the ALDH2 gene can result in a substantial decrease in its acetaldehyde metabolizing capacity. The alcohol flushing syndrome observed in East Asian populations is caused by acetaldehyde accumulation manifesting as a result of reduced ALDH2 activity associated with the expression of the ALDH2*2 in this population (Higuchi et al., 1995).
ALDH activity also supports cellular homeostasis through protection from reactive oxygen species generated under conditions of oxidative stress. In this context, ALDH1A1 and ALDH3A1 (also respectively referred to as lens and corneal crystallins) are the isozymes that defend ocular surface tissues from reactive oxygen species (Estey et al., 2007; Chen et al., 2009). In addition, they maintain the integrity of the lens and cornea through nonenzymatic activities, including direct absorption of harmful UV radiation or serving as chaperones to prevent aggregation of misfolded proteins (Estey et al., 2010). ALDH7A1 has been shown to protect cells against hyperosmotic stress by generating osmolytes, such as betaine, and simultaneously metabolizing toxic aldehydes derived from lipid peroxidation (Brocker et al., 2010; Brocker et al., 2011).
ALDHs have the capacity to influence neural function, particularly in dopaminergic nerves. Both ALDH1A1 and ALDH2 have been implicated in dopamine metabolism. Inhibition of these isozymes leads to the accumulation of neurotoxic dopamine metabolites, such as 3,4-dihydroxyphenylacetaldehyde, that are thought to contribute to the pathogenesis of Parkinson's disease (Burke et al., 2003; Marchitti et al., 2007; Allen et al., 2010). Dopaminergic pathways are involved in the reinforcing effects of cocaine addiction. Inhibition of ALDH2 suppresses cocaine-seeking behavior in conditioned rats through a 3,4-dihydroxyphenylacetaldehyde-dependent decrease in dopamine synthesis (Yao et al., 2010). Likewise, ethanol consumption by rodents has been shown to be reduced by ALDH inhibitor treatment through a mechanism independent of increases in systemic levels of acetaldehyde (Keung et al., 1995) and potentially involving suppression of central dopamine release (Arolfo et al., 2009). Such a central nervous system mechanism may contribute to the low incidence of alcohol consumption/addiction observed in subjects harboring the ALDH2*2 genetic variant that encodes the inactive ALDH2 enzyme. In addition, the isozyme ALDH5A1, also known as mitochondrial succinic semialdehyde dehydrogenase, is an important regulator of GABA metabolism (Kim et al., 2011a). It is believed that, through their metabolic functions, ALDHs have a significant involvement in neuroprotection (Marchitti et al., 2007).
Finally, several ALDH isozymes have been discovered to have both regulatory as well as metabolic roles in cancer. Cancer stem cell (CSC) populations have elevated ALDH activity (Tanei et al., 2009), which can confer resistance to selected anticancer agents by metabolic inactivation. Such interference with chemotherapy is speculated to be a cause of relapse in cancer patients (Honoki et al., 2010; Su et al., 2010; Kim et al., 2011b). Because of its high expression, ALDH activity is used as a specific marker for CSCs through the ALDEFLUOR assay (Alison et al., 2010); however, it is important to recognize that the high ALDH activity observed in CSCs are probably due to multiple or distinct ALDH isozymes, the contribution of each depending on the specific type of the tumor (Marcato et al., 2011). For example, ALDH7A1 was recently implicated in facilitation of prostate cancer metastasis (van den Hoogen et al., 2011) whereas ALDH1B1 was found to be highly induced in human colonic adenocarcinoma (Chen et al., 2011b).
B. Structure and Catalysis of Aldehyde Dehydrogenase
The ALDH enzymes share a wide range of common physiological functions and substrates. Nevertheless, each of the isozymes has evolved distinct substrate specificities (Wang et al., 2009). Exploiting differences in the catalytic active site architecture would then facilitate the design of new inhibitors selective for each isozyme.
Certain classes of the ALDH isozymes have been cocrystallized with their preferred substrates, a process that has facilitated characterization of the active site structure (Liu et al., 1997; Cobessi et al., 2000; Larson et al., 2005; D'Ambrosio et al., 2006; Lowe et al., 2008). Most isozymes share the same catalytic mechanism for both dehydrogenase and esterase activity, except for ALDH6A1, which uses CoA as a cofactor rather than NAD(P+) (Kazmi et al., 1992; Xiao et al., 2009). This feature of the ALDH superfamily probably relates to the following catalytic residues being highly conserved: Cys302, Lys192, and Glu268. Mammalian ALDHs function as either homotetramers or homodimers, with an active site in each monomer. The tetramers may display either full- or half-site reactivity with substrate, depending on the pH and the presence of divalent cations (Takahashi et al., 1981; Weiner and Takahashi, 1983; Rodriguez-Zavala and Weiner, 2002). In general, each of these monomers comprise 500 to 600 amino acids, although there are some classes, such as ALDH16A1, -18A1, -1L1, and -1L2, that have up to 800 or 900 amino acids (Min et al., 1988; Strickland et al., 2011). Crystallographic studies indicate that the ALDH monomer across all classes share high homology in three distinct functional regions: coenzyme (NAD) binding domain, catalytic domain, and oligomerization domain (Fig. 2, A and B) (Liu et al., 1997; Steinmetz et al., 1997; Colby et al., 1998; Cobessi et al., 2000; D'Ambrosio et al., 2006).
Fig. 2.

Molecular architecture of ALDH isozymes. A, the representative ALDH monomer visualized from Protein Data Bank code 1O01 (Perez-Miller and Hurley, 2003): human mitochondrial aldehyde dehydrogenase complexed with crotonaldehyde, NADH, and Mg2+. The functional regions are highlighted: catalytic domain (brown), NAD binding domain (purple), and oligomerization domain (green). B, top view of the monomer showing active site area. C, magnified view of B showing Cys302 (a), aldehyde substrate (b), ordered water molecule between Glu268 and Cys302 (c), Glu268 (d), Mg2+ ion (e; not drawn to scale), and NADH cofactor (f).
Site-directed mutagenesis experiments have revealed that catalysis occurs in five steps (illustrated in Fig. 3A) (Hempel et al., 1999): 1) activation of the catalytic Cys302 via a water-mediated proton abstraction by Glu268, 2) consequent nucleophilic attack on the electrophilic aldehyde by the thiolate group of Cys302, 3) formation of a tetrahedral thiohemiacetal intermediate (deacylation) with concomitant hydride transfer to the pyridine ring of NAD+, 4) hydrolysis of the resulting thioester intermediate, and 5) dissociation of the reduced cofactor and subsequent regeneration of the enzyme by NAD+ binding. Glu268 functions as a general base required to activate the Cys302 through deprotonation, leading to the thiohemiacetal formation preceding the deacylation step (Wang and Weiner, 1995; Sheikh et al., 1997). It was proposed that the first and the fourth steps require an ordered water molecule bound to the side chain of Glu268 (Fig. 2C) (i.e., to facilitate both the deprotonation of Cys302 and the subsequent hydrolysis of the thioester intermediate). The active site is located at the base of hydrophobic tunnel 12 Å from the surface of the enzyme, near the tetrameric interface and opposite the cofactor binding site (Fig. 4). Residues lining this tunnel are believed to confer the substrate specificity of each isozyme. The NAD+ cofactor is bound to the enzyme through 10 hydrogen bonds, four of which involve two residues, Lys192 and Glu399, that are highly conserved in all ALDH isozymes (Ni et al., 1997). Both residues make two hydrogen bonds each, one to the adenosine ribose and the other to the nicotinamide ribose of NAD+. Site-directed mutagenesis of these residues in ALDH2 changes the rate-limiting step of the reaction from the deacylation step to the hydride transfer step. Lys192 mutants alter substrate specificity from aliphatic to aromatic aldehydes in ALDH2 (Ni et al., 1997). The ALDH1, ALDH2, and ALDH3 classes of isozymes differ substantially in the rate-limiting step depending on the effect of cofactor binding, presence of divalent ions, and pH of the reaction (Takahashi et al., 1981; Perez-Miller and Hurley, 2003). Reduced cofactor release is the rate-limiting step for ALDH1A1, whereas for ALDH3A1, it is the hydride transfer.
Fig. 3.

A, mechanism of aldehyde oxidation. 1, activation of the catalytic Cys302 by the ordered water molecule and Glu268 through proton abstraction. 2, nucleophilic attack on the carbonyl carbon of the aldehyde by the thiolate group of the catalytic Cys302. 3, formation of the tetrahedral thiohemiacetal intermediate (deacylation) and hydride transfer from the tetrahedral thiohemiacetal intermediate to the pyridine ring of NAD+. 4, hydrolysis of the resulting thioester intermediate. 5, dissociation of the reduced cofactor and subsequent regeneration of the enzyme by NAD+ binding. B, proposed mechanism of ester hydrolysis. 1, catalytic cysteine residue is activated by hydrogen abstraction from a basic residue (Glu268) in catalytic site. 2, thiolate ion attacks the substrate p-nitrophenyl acetate leading to oxyanion intermediate formation. 3, the intermediate rearranges and nitrophenolic group leaves. 4, Glu268 residue abstracts hydrogen from nearby ordered water molecule, making it a nucleophile. It attacks the carbonyl in the thioacyl enzyme complex. 5, formation of the tetrahedral intermediate that again rearranges to release acetic acid and free enzyme.
Fig. 4.

Active site topography. The active site is located at the base of a hydrophobic tunnel located 12 Å from the surface of the enzyme. a, lipophilic surface potential rendering of the ALDH2 structure (1O01) (Perez-Miller and Hurley, 2003). The lipophilic potential is indicated by a color code (blue, hydrophilic; green/brown, hydrophobic). b, inset magnification of tunnel from a with the substrate in view. c, side view of the enzyme in a, with enzyme structure in blue ribbon and lipophilic surface in green and brown; the hydrophobic tunnel is shown in brackets with NADH, aldehyde substrate (ball and stick models), and magnesium ion (purple sphere) inside.
The proposed mechanism of the esterase activity of the ALDHs (shown in Fig. 3B) shares some common properties with the dehydrogenase activities. For example, it requires the activated Cys302 residue and Glu268 but does not require the presence of NAD(P)+. However, the impact that the presence of cofactor has on the reaction is variable and isozyme-dependent. For most ALDH1 and ALDH2 isozymes, addition of cofactor initiates the esterase reaction, an effect thought to occur through increased nucleophilicity of the active site Cys302 (Takahashi et al., 1981). However, recent studies on ALDH2 relate the effect of cofactor to that of Alda-1 (mentioned earlier as an activator of ALDH2 and ALDH2*2), where the presence of either cofactor or Alda-1 initiates the esterase reaction by increasing the frequency of productive encounters with the active site Cys302 (Perez-Miller et al., 2010). It is likely that the variable effects of cofactor, like the variable effects of mutations on conserved active site residues, are related to the differences among the ALDH isozyme in their rate-limiting steps.
The genetic polymorphism ALDH2*2 affects ≈50% of the Asian population and results in a diminution in ALDH2 activity (Goedde and Agarwal, 1987; Higuchi et al., 1995). This is a result of a substitution of Glu487 for Lys487. The failure of Lys487 to form hydrogen bonds with Arg264 in its own subunit (or with Arg475 in its dimer subunit) results in a weaker NAD+ binding site, a situation that ultimately affects the kinetic properties of the enzyme (Larson et al., 2007). Mechanistic studies have revealed differences between the ALDH2 and ALDH3 isozymes with respect to the rate-limiting step, the function of conserved Glu333 and Glu209 residues and the differential effect of NAD+ binding on the esterase activity (Mann and Weiner, 1999). Such isozyme-specific differences in interacting residues, along with differences in oligomeric state, inhibition mechanisms, and substrate specificity, could be exploited for the development of selective inhibitors (Batt and Crow, 1989; Yoshida et al., 1998).
III. Clinical Applications of Aldehyde Dehydrogenase Inhibitors
The current development of ALDH inhibitors is being driven by emerging clinical needs. It has to be noted, however, that pharmacological inhibitors have been developed for only 3 of the 19 ALDH isozymes. These are the enzymes involved in the metabolism of alcohol (ALDH2) and the anticancer oxazaphosphorine drugs (ALDH1A1 and ALDH3A1).
Alcohol-related disease is the third leading lifestyle-related cause of death in the United States, claiming approximately 23,000 lives in 2006 alone (Heron et al., 2009). In 2009, alcohol abuse claimed 2.3 million lives internationally (Mokdad et al., 2004). In the liver, ethanol is first metabolized to acetaldehyde by alcohol dehydrogenases and cytochrome P450 2E1 (Lieber, 1994). The acetaldehyde thus produced is oxidized mainly by mitochondrial ALDH2 (Weiner, 1979; Goedde and Agarwal, 1987; Batt and Crow, 1989; Soyka and Rösner, 2010). The unpleasant alcohol flushing syndrome experienced by persons with the ALDH2*2 polymorphism renders a large population of Asians intolerant of what might be considered “normal” levels of alcohol consumption. This observation led to the development of molecules that selectively inhibit ALDH2 with the idea of creating drugs that would render a person vulnerable to acetaldehyde-associated symptoms after alcohol consumption (e.g., alcohol-aversive drugs for alcoholics or antidipsotropic agents) (Mason and Heyser, 2010; Rösner et al., 2010; Soyka and Rösner, 2010). The capacity of ALDH to metabolize acetaldehyde is important from a toxicological standpoint because accumulation of acetaldehyde leads to formation of protein adducts (from lipid peroxidation) and subsequent loss of critical physiological functions. For example, in ethanol-induced cardiac toxicity, accumulation of acetaldehyde increases myocardial susceptibility to ischemia reperfusion injury (Wang et al., 2008; Chen et al., 2010).
An important clinical issue in cancer treatment is the development of chemotherapeutic drug resistance. Although most patients with advanced disease respond to initial chemotherapy, the majority of them has a relapse within 5 years of completing treatment; one third of patients are intrinsically resistant. It is now well accepted that CSCs, a small subset of cancer cells, can be resistant to chemotherapy and thereby promote cancer relapse by acting as a source of tumor cells (Honoki et al., 2010; Su et al., 2010). A key biochemical feature of CSCs is high ALDH activity, as illustrated in numerous recent studies documenting the profound tumorigenic potential of ALDH-expressing cells (Su et al., 2010; Chen et al., 2011b; Kim et al., 2011b). In addition, increased activity of ALDH1A1 and ALDH3A1 isozymes has been associated with the development of resistance of tumor cell populations to oxazaphosphorine anticancer drugs, such as cyclophosphamide and procarbazine (Sládek, 1999; Giorgianni et al., 2000; Zhang et al., 2005; Tanei et al., 2009; Honoki et al., 2010). The role of ALDH in the resistance mechanism is supported by the observation that the potency of these drugs are increased when ALDH activity is inhibited (Croker and Allan, 2011). Based on these observations, ALDH isozymes have been proposed as new pharmacological targets for the more effective treatment of cancer. In this regard, new agents are being developed and tested as ALDH inhibitors, particularly targeting the ALDH1A1 and ALDH3A1 isozymes (Parajuli et al., 2011).
Although inhibition of individual ALDH isozymes represents a potentially fruitful approach to treating disease, it is important to recognize that an accumulating body of literature suggests that activation of ALDH may produce beneficial effects in specific disease or therapeutic conditions. For example, by protecting ALDH2 from inactivation, Alda-1, a small-molecule activator of ALDH, prevents the tolerance that normally attends long-term nitroglycerin therapy (Perez-Miller et al., 2010). Such tolerance is a growing concern among physicians treating ischemic and congestive heart disease. In addition to playing a central role in the vasodilator actions of nitroglycerin, ALDH2 may be important in protecting ischemic myocardium. By preventing ALDH2 inactivation by ischemia-associated 4-hydroxy-2-nonenal, Alda-1 reduces the extent of infarction-induced injury (Budas et al., 2009, 2010; Chen et al., 2010). Patients suffering from Fanconi's anemia are more vulnerable to acetaldehyde-induced DNA damage (Alter et al., 2005; Joenje, 2011). Given the important role of ALDH2 in metabolizing toxic aldehydes, it is reasonable to speculate how the administration of an ALDH activator would be beneficial in these patients. The fact that activation or inhibition of ALDH enzymes can have beneficial effects (depending on the specific disease or treatment conditions) underscores the need for a greater understanding of the role played by each isozyme in specific diseases. Such knowledge will allow the prediction of beneficial and deleterious effects of any developed inhibitors or activators.
IV. Pharmacological Inhibitors of Aldehyde Dehydrogenase Enzymes
The following 14 ALDH inhibitors will be discussed in their alphabetic order. Their mechanisms of action, isozyme selectivity, inhibition potency, and other features are summarized in Table 1.
TABLE 1.
Known inhibitors of aldehyde dehydrogenases
| Inhibitor | Active Metabolite | Type of Inhibition | IC50 |
Other Features | References | ||
|---|---|---|---|---|---|---|---|
| hALDH1A1 | hALDH2 | hALDH3A1 | |||||
| μM | μM | μM | |||||
| Ampal | Thioampal | Irreversible | N.A. | N.A. | N.A. | In rat hepatoma cells (JM2), thioampal derivatives inhibited ALDH1 and ALDH3 isoenzymes significantly. Also noted were growth inhibition and anti-tumor activity by these derivatives. | Quemener et al., 1989; Canuto et al., 2001; Quash et al., 2008 |
| Benomyl | MBT | Irreversible | N.A. | N.A. | N.A. | MBT showed IC50 of 0.23 μM with mouse ALDH2, with activation by microsomes. The activation is inhibited by P450 inhibitor. | Axness and Fleeker, 1979; Staub et al., 1998, 1999 |
| Citrala | Neral and geranial | Reversible noncompetitive | N.A. | N.A. | N.A. | Geranial showed positive cooperativity with hALDH2; n ≈ 2–2.5; Citral Ki for Rat ALDH2 = 360 nM | Boyer and Petersen, 1991; Kikonyogo et al., 1999 |
| Chloral hydratea | Chloral hydrate | Reversible competitive | 1–10 | N.A. | N.A. | Also inhibits esterase activity noncompetitively (Ki = 19 μM) with NAD+ | Blackwell et al., 1983 |
| Chlorpropamide analogs NPI-1 API-1 | NPI-1 | Irreversible | N.A. | N.A. | 121 | All of the analogs tested were proven to be more active against human tumor isolated ALDH3A1 than against normal tissue ALDH3A1. | Lee et al., 1992; Nagasawa et al., 1995; Rekha et al., 1998 |
| API-1 | N.A. | N.A. | 223 | ||||
| Coprine | 1-Amino cyclopropanol | Irreversible | 50 | N.A. | N.A. | Inhibits the esterase activity. Displays mutagenicity and gonadotoxicity. | Marchner and Tottmar, 1983; Michelot, 1992 |
| Cyanamide | HNO | Irreversible | N.A. | 10 | N.A. | Requires activation by catalase. Also inhibits catalase activity. Liver injury reported with simultaneous consumption of alcohol. | Shirota et al., 1987; Nagasawa et al., 1990 |
| Daidzin | Daidzin | Reversible competitive | 28 | 0.29 | N.A. | Structure analogs are being developed as antidipsotropic agents. | Lowe et al., 2008; Arolfo, 2009 |
| CVT-10216 | CVT-10216 | 1.3 | 0.029 | N.A. | |||
| DEABa | DEAB | Reversible competitive | 0.55 | N.A. | N.A. | No activation required. Confers sensitivity to cyclophosphamide-resistant cells. | Croker and Allan, 2011 |
| DPABa | DPAB | <1 | N.A. | N.A. | |||
| Disulfiram | Disulfiram | Irreversible | 0.15 | 1.45 | N.A. | Inhibits dopamine β-hydroxylase; displays copper chelation activity; inhibits carboxylesterase and cholinesterase. | Lipsky et al., 2001; Lin et al., 2010; Schroeder et al., 2010; Wang et al., 2011 |
| DDTC-SO | 0.27 | 2.2 | N.A. | ||||
| DDTC-SO2 | 0.12 | 62 | N.A. | ||||
| DETC-SO | N.A. | 1.2 | N.A. | ||||
| DETC-SO2 | N.A. | 0.4 | N.A. | ||||
| Gossypol | Gossypol | Reversible noncompetitive | 75 | 45 | 7.5 (competitive) | Inhibits several other dehydrogenase enzymes. Interacts with alcohol in system to form a toxic metabolite, sufficient for aversion from drinking. Known for antitumor, antiviral, and antiparasitic activities. | Burgos et al., 1986; Rekha and Sladek, 1997; Pang et al., 2011 |
| Kynurenine Tryptophan metabolites | KA | N.A. | N.A. | N.A. | N.A. | Tryptophane metabolites in the kynurenine pathway seem to have the potential for ALDH2 specific inhibition. | Badawy and Morgan, 2007; Badawy et al., 2011 |
| 3-HK | N.A. | N.A. | N.A. | N.A. | |||
| 3-HAA | N.A. | N.A. | N.A. | N.A. | |||
| Molinate | Molinate | Irreversible | N.A. | >50 | N.A. | Molinate sulfoxide was found to inhibit ALDH activity in mouse striatal synaptosomes only, suggesting other isozymes as possible targets. | Allen et al., 2010 |
| Molinate sulfoxide | High | ||||||
| Molinate sulfone | <50 | ||||||
| Nitroglycerina | NO3− | Irreversible | N.A. | N.A. | N.A. | DTT but not GSH partially protected GTN-induced inhibition of dehydrogenase activity. ALDH2 inactivation has a role in impaired GTN reduction and nitrate tolerance | Chen, 2002; Beretta et al., 2008; Daiber et al., 2010; Wenzl et al., 2011 |
| Pargylinea | Propiolaldehyde | Reversible competitive | 1.6 | 1.8 | N.A. | Activated by CYP2E1 in liver microsomes; irreversible inhibitor of monoaminoxidase | DeMaster et al., 1980, 1986; Moridani et al., 2001 |
N.A., inhibition data not available for hALDHs, although data exists for inhibition of mouse and other mammalian isozymes; API-1, 4-chloro-N-ethyl-N-[(propylamino)carbonyl]benzenesulfonamide; NPI-1, (benzoyloxy)[(4-chlorophenyl)sulfonyl]carbamic acid 1,1-dimethylethyl ester; KA, kynurenic acid; 3-HK, 3-hydroxykynurenine; 3-HAA, 3-hydroxyanthranilic acid.
These compounds are not strict inhibitors but competitive substrates capable of inactivation through Michael-adduct formation in the active site.
A. 4-Amino-4-methyl-2-pentyne-1-al
4-Amino-4-methyl-2-pentyne-1-al (AMPAL) and 2-methyl-5-(methylsulfanyl)-5-oxopentan-2-aminium are two irreversible inhibitors of the ALDH1 and ALDH3 isozymes. AMPAL is also known for its antitumor activity and thus substantiates the relationship between resistant neoplasticity and high ALDH activity (Quemener et al., 1989). Rat JM2 hepatoma cells exhibit high ALDH1 and ALDH3 activity. Two analogs of AMPAL, 4-methyl-1-(methylsulfanyl)-4-(2,2,5,5-tetramethylpyrrolidin-1-yl) pent-2-yn-1-one) and 4-(dimethylamino)-4-methyl-1-(methylsulfanyl)pent-2-yn-1-one, inhibited the ALDH activity in these cells and elicited significant cell growth inhibition (Canuto et al., 2001). The analogs differ from each other in the substituent at the C4 position in AMPAL. This evidence establishes possible role of ALDHs in stalling growth in tumor cells.
B. Benomyl
Benomyl (methyl-[1-[(butylamino)carbonyl]-1H-benzimidazol-2-yl]carbamate) is a benzimidazole fungicide that is selectively toxic to microorganisms and invertebrates (Somerville, 1986). This agent is a potent inhibitor of mouse liver mitochondrial ALDH in vitro and in vivo (Staub et al., 1999). When administered to mice in vivo, benomyl specifically inhibits ALDH2 (IC50, 24 μmol/kg) (Staub et al., 1998). Such selectivity for ALDH2 is not apparent in vitro, suggesting that this benomyl is biotransformed in vivo to intermediates that are more potent inhibitors of ALDH2.
Insight into the mechanisms involved in the in vivo formation of mouse ALDH2 inhibitory metabolites originated from studies describing the presence of S-(N-butylcarbamoyl)GSH (GSBT) in bile and S-(N-butylcarbamoyl)cysteine (CysBT) and N-acetyl-S-(N-butylcarbamoyl)cysteine in urine (Axness and Fleeker, 1979). These metabolites primarily are derived from the butylcarbamyl moiety, which gives clues to the nature of metabolites mediating the inhibition. Identification of these metabolites suggest that GSBT and CysBT are activated by β-lyase to inhibitors (Fig. 5). S-Methyl-N-butylthiocarbamate (MBT) is one such metabolite identified in the liver as a transient inhibitory intermediate and found to be bioactivated to the final inhibitors by members of the microsomal cytochrome P450 family (Staub et al., 1998).
Fig. 5.

Benomyl and its active metabolites. Benomyl is metabolized to BIC. BIC is then conjugated with GSH to form GSBT. Under the action of γ-glutamyltranspeptidase, GSBT is transformed to Cys-BT. β-Lyase then converts Cys-BT to N-butylthiocarbamic acid, which is then converted to MBT. Under the action of P450s, MBT gives rise to various sulfoxide and N-hydroxyl products that inhibit the ALDH2 active site through carbomylation of the active cysteine. [Adapted from Staub RE, Quistad GB, and Casida JE (1998) Mechanism for benomyl action as a mitochondrial aldehyde dehydrogenase inhibitor in mice. Chem Res Toxicol 11:535–543. Copyright © 1998 American Chemical Society. Used with permission.]
On the basis of these observations, a six-step activation pathway for benomyl metabolism and ALDH inhibition has been postulated. The first step involves spontaneous release of butyl isocyanate (BIC), another potent inhibitor of mitochondrial ALDH2, from the parent compound. The other product, methylbenzimidazole-carbamate, did not inhibit the enzyme to the same extent as the parent compound and thus was not pursued. Once released, BIC conjugates with GSH to form GSBT, which is hydrolyzed by γ-glutamyltranspeptidase and cysteine to CysBT. This is subsequently converted to N-butylthiocarbamic acid by β-lyase. Methylation of N-butylthiocarbamic acid by S-methyltransferase forms the penultimate inhibitor MBT, which is then subject to P450-mediated N-hydroxylation or sulfoxidation, the products of which have the potential to carbamoylate the active site Cys302 of ALDH2 (Axness and Fleeker, 1979; Staub et al., 1998, 1999). The earlier metabolites, GSBT, NAcSBT, and Cys-BT, from the GSH pathway were all found to inhibit mitochondrial ALDH2 in vitro, suggesting that there are yet other metabolites of benomyl acting as inhibitors. This suggests the need for structure-activity relationship studies that need to be conducted on these metabolites.
C. Chloral
Trichloroacetaldehyde monohydrate (or chloral) was among the most potent hypnotic agents first synthesized. Early on, it was recognized that, when coingested with alcohol, chloral elicited a rapid induction of sleep, often referred to as the “Mickey Finn” effect. Chloral is a competitive substrate of ALDH with a slow kcat and IC50 values ranging from 1 to 10 μM (Poon et al., 2002). In vivo, its metabolites include trichloroacetic acid, proving that it functions as a substrate (Poon et al., 2000) there by being capable of inhibiting the metabolism of other aldehydic substrates of ALDHs. The mechanism by which it inhibits ALDH is thought to involve reversible thiohemiacetal formation with Cys302 in the active site. Because of the powerful electron-withdrawing properties of the Cl3C group, the hydride transfer step of the catalytic mechanism is likely to be impaired.
D. Chlorpropamide analogs
The potential of oral hypoglycemic agents, including chlorpropamide and tolbutamide, to impair the oxidation of acetaldehyde in patients ingesting alcohol-containing beverages was well documented 5 decades ago (Grün et al., 2000). Numerous reports thereafter described the occurrence of malaise and facial flushing in 15 to 23% of human subjects taking these oral hypoglycemic agents after alcohol ingestion (Yoshida et al., 1992). A study in mice suggested that coadministration of chlorpropamide or tolbutamide with alcohol resulted in inhibition of ALDH2, chlorpropamide being relatively more potent. That the inhibition was detected in liver homogenates of mice pretreated with these agents suggested that the nature of the ALDH2 inhibition was irreversible (Niederreither and Dollé, 2008). The inhibitory effect appeared to be selective for ALDH2 in that neither agent affected the ALDH1 or ALDH3 isozymes. Furthermore, the effect seemed to be mediated by their metabolites, nitroxyl and n-propyl isocyanate, because the parent compounds did not induce any inhibition of ALDH2 in vitro. It is predicted that the mode of inhibition would be through the nitroxyl attack of the thiol group of Cys302. Ester and alkyl analogs of chlorpropamide, designed to release nitroxyl and n-propyl isocyanate during esterase metabolism, have been developed as prodrug inhibitors of the dehydrogenase activity of ALDHs. Identification of nitroxyl as an inhibitor established a new class of compounds that could serve as ALDH inhibitors (Lee et al., 1992; Lee et al., 2001). The selectivity of some of these analogs (Fig. 6) for various isozymes has been examined by Rekha et al. (1998). Ester [(benzoyloxy)[(4-chlorophenyl)sulfonyl]carbamic acid 1,1-dimethylethyl ester] and alkyl [4-chloro-N-ethyl-N-[(propylamino)carbonyl]benzenesulfonamide] analogs showed the greatest inhibitory potency against recombinant ALDH2 and yeast ALDH, a substrate-nonspecific ALDH. The vulnerability of the other isozymes to inhibition (according to their IC50 values) was as follows: recombinant ALDH2 > ALDH3 (isolated from normal human tissues) = recombinant ALDH1. It is noteworthy that ALDH from human tumor tissue was more sensitive to inhibition by the two analogs tested than that obtained from its normal tissue counterpart (Sreerama and Sladek, 1993; Rekha and Sladek, 1997; Sládek, 1999).
Fig. 6.

Structural analogs of chlorpropamide. Ester analog of chlorpropamide, (benzoyloxy)[(4-chlorophenyl)sulfonyl]carbamic acid 1,1-dimethylethyl ester (NPI-1). Alkyl analog of chlorpropamide, 4-chloro-N-ethyl-N-[(propylamino)carbonyl]benzenesulfonamide (API-1). [Reprinted from Rekha GK, Devaraj VR, Sreerama L, Lee MJ, Nagasawa HT, and Sladek NE (1998) Inhibition of human class 3 aldehyde dehydrogenase, and sensitization of tumor cells that express significant amounts of this enzyme to oxazaphosphorines, by chlorpropamide analogues. Biochem Pharmacol 55:465–474. Copyright © 1998 Elsevier. Used with permission.]
E. Citral
Citral (3,7-dimethyl-2,6-octadienal) belongs to a class of volatile α,β-unsaturated aldehydes that occur naturally in herbs and citrus fruits. It can be found in synthetic products such as food additives, cosmetics, and detergents. Like other α,β-unsaturated aldehydes (e.g., acrolein, crotonaldehyde, and 4-hydroxynonenal), it is more of a slow, noncompetitive inhibitor than a strict inhibitor of ALDH1, ALDH2, and ALDH3 isozymes (Boyer and Petersen, 1991; Kikonyogo et al., 1999) Citral occurs as a 1:2 mixture of cis and trans isomers, termed neral and geranial, respectively; geranial is the preferred isomer for inhibition of the ALDH1 isozyme (Kikonyogo et al., 1999). For ALDH2, neral shows Michelis-Menten kinetics and geranial shows positive co-operativity. The Km for citral with ALDH3 is extremely low compared with ALDH1 and ALDH2, suggesting that it might be a specific inhibitor of this isozyme (Kikonyogo et al., 1999). Geranial is preferentially metabolized by ALDH1 because it has a very high Vmax/Km ratio, probably as a result of its structural resemblance to all-trans retinaldehyde (Kikonyogo et al., 1999), the natural substrate for ALDH1. Citral has been used to study vertebrate development as a result of its capacity to inhibit retinoic acid synthesis (Kronmiller et al., 1995; Tanaka et al., 1996).
F. Coprine
Coprine is the active ingredient of the mushroom Coprinopsis atramentaria. Minor poisoning incidents associated with ingestion of the mushrooms with alcohol, termed coprinus syndrome, led to the development of the active compound for use as an antidipsotropic agent. Coprine does not inhibit ALDH2 in vitro but does so in vivo as a consequence of its hydrolysis to its active metabolite, 1-amino cyclopropanol (Tottmar and Lindberg, 1977), which inhibits ALDH2 irreversibly through electrophilic attack and covalent modification of the Cys302 thiol (Marchner and Tottmar, 1983). In vitro, the rate of inactivation seemed to increase at higher concentrations of NAD+ in solution, suggesting that a conformational change in ALDH makes it more amenable for the inhibitor to interact with its active site. The esterase activity of ALDH is also inhibited, albeit to a lesser extent compared with the dehydrogenase activity (Marchner and Tottmar, 1983). Unlike disulfiram (see below), it does not inhibit dopamine β-hydroxylase (Carlsson et al., 1978; Tottmar and Hellstrom, 1979). Despite its potency at inhibiting ALDH activity, mutagenic and gonadotoxic side effects prevented further pursuit of this compound as a candidate for drug development (Michelot, 1992).
G. Cyanamide
Cyanamide (in the form of calcium carbimide citrate salt) is an ALDH inhibitor used therapeutically as an alcohol-aversive agent (Temposil) in Europe, Canada, and Japan. It is a prodrug that requires conversion to its active metabolite (Fig. 7). Bioactivation requires catalase and H2O2 to generate N-hydroxycyanamide, which spontaneously decomposes to cyanide (Shirota et al., 1987) (produced at much lower concentrations than that required to elicit toxicity) and nitroxyl, the latter serving as the inhibitor of ALDH (Nagasawa et al., 1990). The mode of inhibition was found to be both reversible (via intersubunit disulfide formation) and irreversible (via sulfinamide formation) depending on the pH of inactivation (DeMaster et al., 1998) via the formation of an N-hydroxysulfinamide intermediate. Given that nitroxyl reacts with thiol proteins, it may be predicted that nitroxyl treatment could lead to depletion of cellular GSH. However, contrary to expectations, extremely low concentrations of nitroxyl deactivate select thiol proteins, such as glyceraldehyde 3-phosphate dehydrogenase, without altering the cellular GSH/GSSG ratio (DeMaster et al., 1998; Paolocci et al., 2007). Pro-drugs of nitroxyl, such as derivatives of N,O-diacylated N-hydroxy-arylsulfonamide (Fukuto et al., 1992) and N-acyloxy-O-arenesulfonylated benzenecarboximidic acid (Lee et al., 2001), are being developed, albeit with more emphasis on their use as vasodilators (Nagasawa et al., 1995) that are bioactivated by the esterase activity of the ALDH enzyme. In the treatment of alcohol addiction, cyanamide is a second-line drug relative to disulfiram owing to its greater capacity to induce liver injury compared with disulfiram (Tamai et al., 2000).
Fig. 7.

Cyanamide inhibition of ALDH. Bioactivation of cyanamide releases nitroxyl as the active inhibitor, which inhibits irreversibly through sulfonamide formation (1) or reversibly through intersubunit disulfide formation (2).
H. Daidzin (antioxidant Isoflavone)
Daidzin, daidzein, and puerarin are phytochemicals derived from the Chinese edible vine Pueraria lobata. They have been used for cancer prevention and antidipsotropic treatments in ancient Chinese medicine (Keung and Vallee, 1993; McGregor, 2007). Daidzin (7-O-glucosyl-4′-hydroxyisoflavone) (Fig. 8), and its structural analogs are among the very few highly potent inhibitors of the ALDH2 isozyme (IC50, 80 nM) (Lowe et al., 2008). Structural requirements of these molecules for ALDH2 inhibition include a polar 4′-substituent with hydrogen binding capacity, such as −NH2 and −OH, and an isoflavone group that blocks the substrate binding site in the hydrophobic cleft (Lowe et al., 2008). Based on these observations, new analogs of daidzin have been developed as highly selective, reversible inhibitors of ALDH2. For example, CVT-10216 (Fig. 8) shows an IC50 of 29 nM for ALDH2 and 1300 nM for ALDH1 (Arolfo et al., 2009). An additional advantage of these inhibitors is their minimal effect on monoamine oxidase (Yao et al., 2010). CVT-10216 has also seen application in cocaine addiction, where inhibition of ALDH2 causes aberrant dopamine metabolism, leading to the production of a metabolite that suppresses dopamine synthesis and cocaine self-administration in animal models (Yao et al., 2010).
Fig. 8.
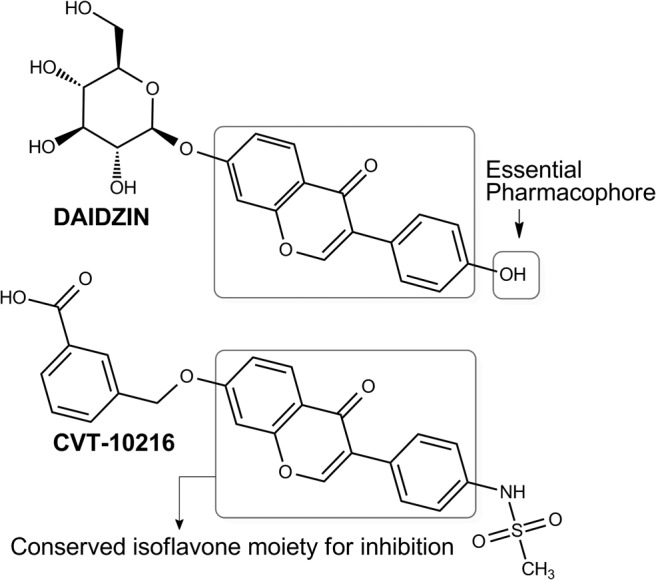
Antioxidant isoflavones. Inhibition of ALDH2 by daidzin and CVT-10216 is dependent on a conserved isoflavone moiety (box) and a 4′-substituent identified as the essential pharmacophore.
I. 4-(Diethylamino)benzaldehyde
4-(Diethylamino)benzaldehyde (DEAB) seems to be more of a slow competitive substrate (like citral and chloral) than a classic inhibitor of ALDH isozymes (Russo et al., 1988), although the precise mechanism of inhibition remains to be elucidated. It has been used as an ALDH1-selective inhibitor for repressing retinoic acid synthesis in studies investigating the role of retinoic acid in abnormal embryonic development in vertebrates (van den Hoogen et al., 2011). The DEAB derivative 4-(N,N-dipropylamino)benzaldehyde is also a competitive inhibitor of retinal oxidation (Russo et al., 2002). Despite the use of DEAB as a potent inhibitor of ALDH1, its specificity has not been completely ascertained. When administered in doses of 50 and 100 mg/kg to mice, DEAB increased the blood acetaldehyde concentration and inhibited the fraction of ethanol converted to acetate by 32.5 and 67.5%, respectively (Mahmoud et al., 1993). In recent years, a commercial Aldefluor assay has been developed and widely used to identify stem cells from normal and cancerous tissues on the basis of the observation that these cells exhibit high ALDH activity (Moreb et al., 2007). In this assay, the fluorescence of a cell membrane-permeable substrate is enhanced by ALDH activation of the molecule. In this assay, DEAB treatment is used as a control to define the contribution of ALDH activity to the fluorescence changes (Storms et al., 1999).
J. Disulfiram
Disulfiram (tetraethylthioperoxydicarbonic diamide), commercially known as Antabuse, has been used since 1948 as an alcohol-aversive agent in the treatment of alcoholism (Bell and Smith, 1949). Consumption of alcohol by patients given therapeutic doses of disulfiram results in acetaldehyde accumulation, the physiological consequences of which include low blood pressure, tachycardia, facial flushing, nausea, and vertigo (Bell and Smith, 1949). These symptoms, collectively called disulfiram ethanol reaction (Tottmar and Hellstrom, 1979; Marcato et al., 2011), serve as the means by which the individual is discouraged from indulging in alcohol intake. Disulfiram itself inhibits ALDH1A1 more potently that it does ALDH2 (Moore et al., 1998), which led to some early confusion as to the identity of the enzyme inhibited in vivo. This is attributed to the fact that the hydrophobic tunnel in the enzyme's architecture through which the substrate enters is larger in ALDH1 and therefore capable of accommodating disulfiram, a bulky molecule, more effectively (Moore et al., 1998). In the 1980s, cephalosporin antibiotics had been known to cause alcohol flushing reactions in a manner similar to that of disulfiram (Fromtling and Gadebusch, 1983). The disulfide bond in the 5,5′-dithiobis(1-methyltetrazole) side chain of these agents is shown to mediate the inactivation of the catalytic Cys302 of ALDH1A1 enzyme in vitro, giving clues to the mode of enzyme inhibition (Kitson, 1986).
The molecular mechanism of ALDH inhibition is mediated by metabolic products of disulfiram (Fig. 9). The first of these involves the reduction of the disulfide bond and concurrent liberation of diethyldithiocarbamate (DDTC), an inhibitor of ALDH in vivo but not in vitro (Deitrich and Erwin, 1971; Kitson, 1975; Vallari and Pietruszko, 1982; Kitson, 1983; Lipsky et al., 2001). DDTC is converted by hepatic thiol methyltransferases to S-methyl-N,N-diethyldithiocarbamate (DETC) and S-methyl-N,N-diethyldithiocarbamate (Me-DDTC), both potent inhibitors of mitochondrial ALDH2 in vivo. Subsequent P450-catalyzed oxidation metabolites of DETC and Me-DDTC, including DETC-sulfoxide (DETC-SO) and S-methyl-N,N-diethylthiocarbamate-sulfoxide (Me-DDTC-SO) and -sulfone (Me-DDTC-SO2), also inhibit mitochondrial ALDH (Hart and Faiman, 1992; Mays et al., 1996, 1998) (Fig. 9). The inhibition induced by all of these compounds is irreversible in nature because of carbamylation of the catalytic Cys302 residue (Staub et al., 1999; Lipsky et al., 2001; Shen et al., 2001). Glutathione blunts the inhibition caused by DETC-SO and DETC-SO2 but does not affect inhibition induced by Me-DDTC-SO, which is relatively more potent (Lam et al., 1997; Mays et al., 1998). This gives rise to the intriguing possibility that, if intracellular levels of GSH are sufficient to mitigate the inhibitory action of the metabolites, Me-DDTC-SO may perhaps be a better candidate for specific inhibition of ALDH2, given its ability to overcome GSH scavenging (Mays et al., 1998).
Fig. 9.

Disulfiram and its metabolites. DDTC is liberated from disulfiram by disulfide exchange with the catalytic cysteine of ALDH. Hepatic thiomethyl transferases subsequently give rise to all of the other metabolites. [Adapted from Pike MG, Mays DC, Macomber DW, and Lipsky JJ (2001) Metabolism of a disulfiram metabolite, S-methylN,N-diethyldithiocarbamate, by flavin monooxygenase in human renal microsomes. Drug Metab Dispos 29:127–132. Copyright © 2001 American Society for Pharmacology and Experimental Therapeutics. Used with permission.]
In addition to being used as an antidipsotropic agent, disulfiram has found a new therapeutic use in treating cocaine addiction via an ALDH-independent mechanism. This is thought to involve disulfiram inhibition of dopamine β-hydroxylase (Barth and Malcolm, 2010; Schroeder et al., 2010), the copper-dependent enzyme required for metabolism of dopamine to norepinephrine in noradrenergic neurons. The resultant disulfiram-induced decrease in norepinephrine release from noradrenergic nerves impinging on midbrain dopaminergic neurons is thought to reduce excitation of these dopaminergic neurons and thereby attenuate the cocaine-induced euphoria (Gaval-Cruz and Weinshenker, 2009). These inhibitory effects are attributed to the copper-chelating activity of the disulfiram metabolite DDTC. Disulfiram inhibits other copper-dependent enzymes, such as carboxylesterase and cholinesterase (Zemaitis and Greene, 1976). This same copper-chelating action is thought to induce proteasome inhibition and subsequent cancer cell apoptosis (Wang et al., 2011), leading to the proposal that disulfiram could serve as an anticancer therapy (Lin and Lin, 2011). The observed inhibition by disulfiram of prostate cancer cell growth, together with its ability to act as a DNA-demethylating agent (Lin et al., 2011) support this proposed use. Clinical trials are currently under way that evaluate its ability to repress cancer growth (Kona et al., 2011; Lin and Lin, 2011).
K. Gossypol
Gossypol, a terpenoid aldehyde extracted from cottonseed, was traditionally used in Chinese medicine as a male contraceptive. More recently, ongoing investigations have focused on its antitumor activity (Cengiz et al., 2010) as a result of documented antineoplastic activity in human breast adenocarcinoma (Van Poznak et al., 2001; Van Poznak and Seidman, 2002; Pang et al., 2011) and metastatic prostate cancer (Huang et al., 2009). Inhibition of the apoptosis regulatory proteins B-cell lymphoma-extra-large and B-cell lymphoma 2 was thought to underlie gossypol's beneficial effects in cancer treatment (Huang et al., 2010). However, this compound inhibits ALDH and other NAD(P+)-catalyzed oxidations, such as aldose reductases, lactate dehydrogenases, and malate dehydrogenases (Burgos et al., 1986). With respect to ALDH-induced oxidation, gossypol acts as a noncompetitive inhibitor and shows relatively more selectivity for the ALDH3 isozyme than the ALDH1 and ALDH2 isozymes (Rekha and Sladek, 1997). It is noteworthy that the nature of inhibition was always competitive with respect to the cofactor involved but not the aldehyde substrate (Rekha and Sladek, 1997). This observation suggests that gossypol may interact with the cofactor binding site, which may underlie the commonality of inhibition of dehydrogenases. Gossypol does not inhibit the esterase activity of the ALDHs (Rekha and Sladek, 1997).
L. Kynurenine Metabolites
High tryptophan levels in patients with chronic fatigue syndrome seemed to induce alcohol aversion (Castell et al., 1999). This anecdotal evidence led researchers to look for possible ALDH inhibitors in tryptophan metabolites. Four tryptophan metabolites in the kynurenine pathway (3-hydroxykynurenine, 3-hydroxyanthranilic acid, kynurenic acid, and indol-3-ylpyruvic acid), have been shown to inhibit rat liver mitochondrial ALDH2 (Badawy and Morgan, 2007). These agents are being developed as potential antidipsotropic treatments (Badawy et al., 2011).
M. Molinate
Molinate is a thiocarbamate derivative used as a rice pesticide. It was phased out because of its neurological and reproductive toxicity in rodents (Chen et al., 2011b). The mechanism of molinate-induced testicular toxicity is thought to be due to the binding of a sulfoxide metabolite of molinate to the protein hydrolase A, a carboxyl esterase found in Leydig cells (Jewell and Miller, 1998). Studies in human and rat liver microsomes and slices have shown that the sulfoxide metabolite and its sulfone are detoxified via glutathione conjugation to a far greater extent in humans than in rodents (Jewell and Miller, 1999). ALDH inhibitory activity of the thiocarbamate pesticides was surmised from the observation that farmers working with these pesticides were rendered alcohol-sensitive (Hart and Faiman, 1995). Molinate and its sulfoxide and sulfone metabolites have been shown to inhibit human recombinant ALDH2 in vitro (Allen et al., 2010). This occurs through an irreversible modification of the active site cysteine residue (Cys302) and thiocarbamate formation (Fig. 10) (Hjelle et al., 1981). Other thiocarbamate herbicides, such as pebulate, vernolate, butylate, triallate, and cycloate (and their metabolites) also inhibit ALDH activity (Quistad et al., 1994).
Fig. 10.

Molinate and its active metabolites. Molinate is metabolized to molinate sulfoxide and finally to molinate sulfone. [Reprinted from Allen EM, Anderson DG, Florang VR, Khanna M, Hurley TD, and Doorn JA (2010) Relative inhibitory potency of molinate and metabolites with aldehyde dehydrogenase 2: implications for the mechanism of enzyme inhibition. Chem Res Toxicol 23:1843–1850. Copyright © 2010 American Chemical Society. Used with permission.]
N. Nitroglycerin
Nitroglycerin or glyceryl trinitrate (GTN) belongs to a class of potent organic nitrates used for almost a century to treat myocardial ischemia. GTN, along with other lower potency nitrates such as isosorbide dinitrate (ISDN), induces coronary vasodilation by a nitric oxide (NO)-dependent mechanism (Daiber et al., 2008). ALDH2 plays a significant role in the bioactivation of nitroglycerin to NO (Chen et al., 2002; Beretta et al., 2008) through a mechanism involving its esterase activity (Fig. 11). NO2 is cleaved at position 3 of GTN to produce 1,2-glyceryl dinitrate and nitrite (NO2−), whereas the enzyme itself gets inactivated through disulfide bridge formation with an adjacent cysteine. The NO2− undergoes subsequent bioactivation by respiratory chain cytochrome oxidase to yield NO, which then induces vasodilation via a cGMP-dependent mechanism (Chen and Stamler, 2006; Daiber and Münzel, 2010). The vasodilatory effects of GTN decrease with long-term administration, a phenomenon called nitrate tolerance (Münzel et al., 2005; Daiber et al., 2010). NO produced from GTN can react with cellular superoxide to form peroxynitrite, which then oxidizes the catalytic thiol of ALDH2. Such inhibition of mitochondrial ALDH2 dehydrogenase activity also abolishes the esterase activity needed for activation of GTN, thus mitigating GTN-induced vasodilation.
Fig. 11.

Nitroglycerin interactions with ALDH. GTN is hydrolyzed by ALDH to form 1,2-glyceryl dinitrate (1,2 GDN) and a thionitrate intermediate bound to the ALDH active site cysteine (Cys302). Two proposed pathways can then be followed: in pathway A, an adjacent cysteine in the active site (Cys301) attacks the thiol of Cys302, forming a disulfide bond and releasing nitrite ion. The nitrite ion gets further metabolized to release NO, which induces vasodilation, whereas the ALDH enzyme is oxidized. In pathway B, the thionitrate intermediate rearranges to form sulfenyl nitrite, which leads to the formation of either a sulfinyl radical (C) and NO or an inactivated sulfinate (D) and HNO. [Adapted from Wenzl MV, Beretta M, Griesberger M, Russwurm M, Koesling D, Schmidt K, Mayer B, and Gorren AC (2011) Site-directed mutagenesis of aldehyde dehydrogenase-2 suggests three distinct pathways of nitroglycerin biotransformation. Mol Pharmacol 80:258–266. Copyright © 2011 American Society for Pharmacology and Experimental Therapeutics. Used with permission.]
Nitrate ester-based vasodilators such as ISDN are very attractive for the treatment of angina pectoris until the issue of GTN tolerance. The pathway responsible for the bioactivation of low-potency nitrates such as ISDN is through activation by P450 enzyme(s) in the endoplasmic reticulum (Münzel et al., 2005). It has been established that isosorbide esters also irreversibly inhibit the dehydrogenase and esterase activities of ALDH1 and ALDH2 (Mukerjee and Pietruszko, 1994; Pietruszko et al., 1995). Both of these isozymes are protected from inhibition in the presence of NAD+ and chloral (the slow competitive substrate for ALDHs), suggesting that the isosorbide esters act as substrates (Mukerjee and Pietruszko, 1994). The restoration of ISDN vasodilator activity by strong reducing agents, such as DTT (but not cysteine or reduced GSH) consolidates the involvement of disulfide bond formation in its inhibitory action on ALDH (Mukerjee and Pietruszko, 1994; Daiber et al., 2004). However, Murphy et al. (2005) reported that ISDN does not inhibit ALDH2 and only selectively inhibits the ALDH3 isozyme (contradicting the above study), which, in turn, is capable of being reactivated by DTT. GTN was found to inhibit both isozymes, but only ALDH3 was reactivated by DTT (Murphy et al., 2005). This suggests that ALDH3 may be subject to a unique mode of inhibition by GTN that differs from the inhibition of ALDH2. A recent series of studies involving site-directed mutagenesis experiments led Wenzl et al. (2011) to propose three distinct pathways for GTN biotransformation by ALDH2. The dominant pathway involves the formation of nitrite and active site Cys (301 and 303) disulfide formation (Fig. 11). The second pathway involves irreversible inactivation of Cys302, promoted by Glu268. A third pathway was suggested by the fact that an ALDH2 triple mutant (E268Q/C301S/C303S) produced superstoichiometric amounts of NO directly while forming a DTT-reducible species that proved relatively resistant to inactivation, although there is a need for further experimental validation. The experimental validation of the third pathway may open up possibilities of developing new vasodilators that can bypass the tolerance induced by GTN-mediated ALDH2 inactivation.
O. Pargyline
Pargyline (N-benzyl-N-methylprop-2-yn-1-amine) is an irreversible monoamine oxidase inhibitor used in the past as an antihypertensive agent. A clue that it affected ALDH was the manifestation of disulfiram-like reactions when patients taking pargyline ingested alcohol. Pargyline is activated by CYP2E1 in liver microsomes (Fig. 12) (Moridani et al., 2001) via N-depropargylation and N-demethylation (DeMaster et al., 1980) to yield propiolaldehyde, a highly reactive α,β-acetylenic aldehyde that irreversibly inactivates ALDH2 (DeMaster and Nagasawa, 1978). Other minor pathways of P450-mediated N-debenzylation and N-oxidation have been suggested (Fig. 12). Whatever the pathway, propiolaldehyde and propargylamine seem to be the final effectors of ALDH inactivation. The mechanism of inactivation is thought to involve the irreversible modification of the catalytic cysteine (Cys302) via either carbamoylation or enone formation. The highly reactive propiolaldehyde certainly can react with cellular thiols, including those in ALDH enzymes, and cause depletion of GSH and subsequent lipid peroxidation (Moridani et al., 2001). Other in vivo metabolites of pargyline, such as benzylamine and propargylamine, also inhibit rat ALDH2 activity but do so indirectly via GSH depletion (DeMaster et al., 1986). Although propiolaldehyde has been found to be preferentially metabolized by ALDH2 (relative to ALDH1) (Ferencz-Biro and Pietruszko, 1984; Lebsack et al., 1977), both isozymes are protected from inhibition by propiolaldehyde by incubation with NAD+.
Fig. 12.

Pargyline metabolism. CYP 2E1-mediated activation of pargyline via N-depropargylation (A), N-demethylation (B), N-debenzylation (C), and N-oxidation (D). N-methylpropargylamine and N-benzylpropargylamine are metabolized further by P450s (E) to yield propioaldehyde as the final effector of ALDH inactivation. Major and minor pathways are depicted by solid and dashed lines, respectively. [Adapted from DeMaster EG, Shirota FN, and Nagasawa HT (1986) Role of propiolaldehyde and other metabolites in the pargyline inhibition of rat liver aldehyde dehydrogenase. Biochem Pharmacol 35:1481–1489. Copyright © 1986 Elsevier. Used with permission.]
V. Concluding Remarks
ALDHs are a group of structurally related proteins. The majority of ALDHs facilitate the oxidation of aldehyde substrates to their corresponding carboxylic acids. Some ALDH isozymes exhibit esterase activity and functions unrelated to catalytic activity. In so doing, ALDH isozymes play diverse but physiologically and toxicologically critical roles. Increased or suppressed ALDH activity has been implicated in a variety of diseases, including cancer. Therefore, the application of pharmacological inhibitors or activators of ALDHs represents a rational approach for the treatment of these pathological conditions. Of the 19 known human ALDH enzymes, only a few of them have been characterized biochemically, specifically ALDH1A1, ALDH1B1, ALDH2, ALDH3A1, ALDH3B1, and ALDH7A1. Although these ALDH isozymes exhibit distinct substrate specificity, they also show an overlapping spectrum for substrates, making it difficult to precisely delineate isozyme-specific effects. Only three ALDH isozymes, ALDH1A1, ALDH2, and ALDH3A1, have been studied for vulnerability to pharmacological inhibition. No antagonists have been developed that inhibit each ALDH isozyme without affecting the others. This lack of selectivity of available antagonists for specific ALDH isozymes has not prevented clinical usage. Under these circumstances, their effects on non-ALDH enzymes limit their therapeutic utility. Nevertheless, the absence of selective pharmacological tools prevents verification of the putative roles of the ALDHs in human diseases or treatments. The emergence of ALDH isozymes as potentially important therapeutic targets serves as an impetus for the need for the development of selective inhibitors. Information derived from proteomic and structure-activity relationship studies are providing a rational basis for the development of ALDH isozyme-specific inhibitors. It is hoped that the content provided in this review serves to motivate and advance this process.
Acknowledgments
This work was supported by the National Institutes of Health National Eye Institute [Grants EY017963, EY11490 (both to V.V.)]; and the National Institutes of Health National Institute on Alcohol Abuse and Alcoholism [Grants R21-AA017754 (to V.V.), R37-AA00930 (to D.R.P.), AA018123, AA19746 (to T.D.H.)]. We thank Dr. Philip Reigan for his help with molecular modeling and visualization of the ALDH protein in Figs. 2 and 4.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Koppaka, Thompson, Chen, Ellermann, Nicolaou, Juvonen, Petersen, Deitrich, Hurley, and Vasiliou.
This article is available online at http://pharmrev.aspetjournals.org.
- ALDH
- aldehyde dehydrogenase
- AMPAL
- 4-amino-4-methyl-2-pentyne-1-al
- BIC
- butyl isocyanate
- CSC
- cancer stem cell
- CysBT
- S-(N-butylcarbamoyl)cysteine
- DDTC
- diethyldithiocarbamate
- DEAB
- 4-(diethylamino)benzaldehyde
- DETC
- S-methyl-N,N-diethyldithiocarbamate
- DETC-SO
- S-methyl-N,N-diethyldithiocarbamate sulfoxide
- DTT
- dithiothreitol
- GSBT
- S-(N-butylcarbamoyl)GSH
- GTN
- glyceryl trinitrate
- ISDN
- isosorbide dinitrate
- MBT
- S-methyl-N-butylthiocarbamate
- Me-DDTC
- S-methyl-N,N-diethyldithiocarbamate
- Me-DDTC-SO
- S-methyl-N,N-diethylthiocarbamate-sulfoxide
- Me-DDTC-SO2
- S-methyl-N,N-diethylthiocarbamate-sulfone
- NAcSBT
- N-acetyl-S-(N-butylcarbamoyl)cysteine
- P450
- cytochrome P450
- RA
- retinoic acid.
References
- Alison MR, Guppy NJ, Lim SM, Nicholson LJ. (2010) Finding cancer stem cells: are aldehyde dehydrogenases fit for purpose? J Pathol 222:335–344 [DOI] [PubMed] [Google Scholar]
- Allen EM, Anderson DG, Florang VR, Khanna M, Hurley TD, Doorn JA. (2010) Relative inhibitory potency of molinate and metabolites with aldehyde dehydrogenase 2: implications for the mechanism of enzyme inhibition. Chem Res Toxicol 23:1843–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alnouti Y, Klaassen CD. (2008) Tissue distribution, ontogeny, and regulation of aldehyde dehydrogenase (Aldh) enzymes mRNA by prototypical microsomal enzyme inducers in mice. Toxicol Sci 101:51–64 [DOI] [PubMed] [Google Scholar]
- Alter BP, Joenje H, Oostra AB, Pals G. (2005) Fanconi anemia: adult head and neck cancer and hematopoietic mosaicism. Arch Otolaryngol Head Neck Surg 131:635–639 [DOI] [PubMed] [Google Scholar]
- Arolfo MP, Overstreet DH, Yao L, Fan P, Lawrence AJ, Tao G, Keung WM, Vallee BL, Olive MF, Gass JT, et al. (2009) Suppression of heavy drinking and alcohol seeking by a selective ALDH-2 inhibitor. Alcohol Clin Exp Res 33:1935–1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axness ME, Fleeker JR. (1979) Metabolism of the butylcarbamoyl moiety of benomyl in rat. Pestic Biochem Physiol 11:1–12 [Google Scholar]
- Badawy AA, Bano S, Steptoe A. (2011) Tryptophan in alcoholism treatment I: kynurenine metabolites inhibit the rat liver mitochondrial low Km aldehyde dehydrogenase activity, elevate blood acetaldehyde concentration and induce aversion to alcohol. Alcohol Alcohol 46:651–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badawy AA, Morgan CJ. (2007) Tryptophan metabolites as potent inhibitors of aldehyde dehydrogenase activity and potential alcoholism-aversion therapeutic agents. Int Congr Ser 1304:344–351 [Google Scholar]
- Barth KS, Malcolm RJ. (2010) Disulfiram: an old therapeutic with new applications. CNS Neurol Disord Drug Targets 9:5–12 [DOI] [PubMed] [Google Scholar]
- Batt RD, Crow KE. (1989) Human Metabolism of Alcohol: Regulation, Enzymology and Metabolism of Ethanol, CRC Press, Boca Raton, FL [Google Scholar]
- Bell RG, Smith HW. (1949) Preliminary report on clinical trials of antabuse. Can Med Assoc J 60:286–288 [PMC free article] [PubMed] [Google Scholar]
- Beretta M, Gruber K, Kollau A, Russwurm M, Koesling D, Goessler W, Keung WM, Schmidt K, Mayer B. (2008) Bioactivation of nitroglycerin by purified mitochondrial and cytosolic aldehyde dehydrogenases. J Biol Chem 283:17873–17880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell LF, Bennett AF, Buckley PD. (1983) Relationship between the mechanisms of the esterase and dehydrogenase activities of the cytoplasmic aldehyde dehydrogenase from sheep liver. An alternative view. Biochemistry 22:3784–3791 [DOI] [PubMed] [Google Scholar]
- Boyer CS, Petersen DR. (1991) The metabolism of 3,7-dimethyl-2,6-octadienal (citral) in rat hepatic mitochondrial and cytosolic fractions. Interactions with aldehyde and alcohol dehydrogenases. Drug Metab Dispos 19:81–86 [PubMed] [Google Scholar]
- Brocker C, Cantore M, Failli P, Vasiliou V. (2011) Aldehyde dehydrogenase 7A1 (ALDH7A1) attenuates reactive aldehyde and oxidative stress induced cytotoxicity. Chem Biol Interact 191:269–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocker C, Lassen N, Estey T, Pappa A, Cantore M, Orlova VV, Chavakis T, Kavanagh KL, Oppermann U, Vasiliou V. (2010) Aldehyde dehydrogenase 7A1 (ALDH7A1) is a novel enzyme involved in cellular defense against hyperosmotic stress. J Biol Chem 285:18452–18463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budas GR, Disatnik MH, Chen CH, Mochly-Rosen D. (2010) Activation of aldehyde dehydrogenase 2 (ALDH2) confers cardioprotection in protein kinase C epsilon (PKCε) knockout mice. J Mol Cell Cardiol 48:757–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budas GR, Disatnik MH, Mochly-Rosen D. (2009) Aldehyde dehydrogenase 2 in cardiac protection: a new therapeutic target? Trends Cardiovasc Med 19:158–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos C, Gerez de Burgos NM, Rovai LE, Blanco A. (1986) In vitro inhibition by gossypol of oxidoreductases from human tissues. Biochem Pharmacol 35:801–804 [DOI] [PubMed] [Google Scholar]
- Burke WJ, Li SW, Williams EA, Nonneman R, Zahm DS. (2003) 3,4-Dihydroxyphenylacetaldehyde is the toxic dopamine metabolite in vivo: implications for Parkinson's disease pathogenesis. Brain Res 989:205–213 [DOI] [PubMed] [Google Scholar]
- Canuto RA, Muzio G, Salvo RA, Maggiora M, Trombetta A, Chantepie J, Fournet G, Reichert U, Quash G. (2001) The effect of a novel irreversible inhibitor of aldehyde dehydrogenases 1 and 3 on tumour cell growth and death. Chem Biol Interact 130–132:209–218 [DOI] [PubMed] [Google Scholar]
- Carlsson A, Henning M, Lindberg P, Martinson P, Trolin G, Waldeck B, Wickberg B. (1978) On the disulfiram-like effect of coprine, the pharmacologically active principle of Coprinus atramentarius. Acta Pharmacol Toxicol (Copenh) 42:292–297 [DOI] [PubMed] [Google Scholar]
- Castell LM, Yamamoto T, Phoenix J, Newsholme EA. (1999) The role of tryptophan in fatigue in different conditions of stress. Adv Exp Med Biol 467:697–704 [DOI] [PubMed] [Google Scholar]
- Cengiz E, Karaca B, Kucukzeybek Y, Gorumlu G, Gul MK, Erten C, Atmaca H, Uzunoglu S, Karabulut B, Sanli UA, et al. (2010) Overcoming drug resistance in hormone- and drug-refractory prostate cancer cell line, PC-3 by docetaxel and gossypol combination. Mol Biol Rep 37:1269–1277 [DOI] [PubMed] [Google Scholar]
- Chen CH, Sun L, Mochly-Rosen D. (2010) Mitochondrial aldehyde dehydrogenase and cardiac diseases. Cardiovasc Res 88:51–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Koppaka V, Thompson DC, Vasiliou V. (2011a) Focus on molecules: ALDH1A1: from lens and corneal crystallin to stem cell marker. Exp Eye Res. http://dx.doi.org/10.1016/j.exer.2011.04.008 [DOI] [PMC free article] [PubMed]
- Chen Y, Mehta G, Vasiliou V. (2009) Antioxidant defenses in the ocular surface. Ocul Surf 7:176–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Orlicky DJ, Matsumoto A, Singh S, Thompson DC, Vasiliou V. (2011b) Aldehyde dehydrogenase 1B1 (ALDH1B1) is a potential biomarker for human colon cancer. Biochem Biophys Res Commun 405:173–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Stamler JS. (2006) Bioactivation of nitroglycerin by the mitochondrial aldehyde dehydrogenase. Trends Cardiovasc Med 16:259–265 [DOI] [PubMed] [Google Scholar]
- Chen Z, Zhang J, Stamler JS. (2002) Identification of the enzymatic mechanism of nitroglycerin bioactivation Proc Natl Acad Sci USA 99:8306–8311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobessi D, Tête-Favier F, Marchal S, Branlant G, Aubry A. (2000) Structural and biochemical investigations of the catalytic mechanism of an NADP-dependent aldehyde dehydrogenase from Streptococcus mutans. J Mol Biol 300:141–152 [DOI] [PubMed] [Google Scholar]
- Colby TD, Bahnson BJ, Chin JK, Klinman JP, Goldstein BM. (1998) Active site modifications in a double mutant of liver alcohol dehydrogenase: structural studies of two enzyme-ligand complexes. Biochemistry 37:9295–9304 [DOI] [PubMed] [Google Scholar]
- Croker AK, Allan AL. (2011) Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDH(hi)CD44 (+) human breast cancer cells. Breast Cancer Res Treat http://dx.doi.org/10.1007/s10549-011-1692-y [DOI] [PubMed]
- D'Ambrosio K, Pailot A, Talfournier F, Didierjean C, Benedetti E, Aubry A, Branlant G, Corbier C. (2006) The first crystal structure of a thioacylenzyme intermediate in the ALDH family: new coenzyme conformation and relevance to catalysis. Biochemistry 45:2978–2986 [DOI] [PubMed] [Google Scholar]
- Daiber A, Harrison DG, Münzel T.(2008) Doubt about an essential role for constitutive nitric oxide synthase in nitroglycerin-mediated vasodilation Proc Natl Acad Sci USA 105:E92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiber A, Münzel T. (2010) Nitrate reductase activity of mitochondrial aldehyde dehydrogenase (ALDH-2) as a redox sensor for cardiovascular oxidative stress. Methods Mol Biol 594:43–55 [DOI] [PubMed] [Google Scholar]
- Daiber A, Münzel T, Gori T. (2010) Organic nitrates and nitrate tolerance–state of the art and future developments. Adv Pharmacol 60:177–227 [DOI] [PubMed] [Google Scholar]
- Daiber A, Oelze M, Coldewey M, Bachschmid M, Wenzel P, Sydow K, Wendt M, Kleschyov AL, Stalleicken D, Ullrich V, et al. (2004) Oxidative stress and mitochondrial aldehyde dehydrogenase activity: a comparison of pentaerythritol tetranitrate with other organic nitrates. Mol Pharmacol 66:1372–1382 [DOI] [PubMed] [Google Scholar]
- Deitrich RA, Erwin VG. (1971) Mechanism of the inhibition of aldehyde dehydrogenase in vivo by disulfiram and diethyldithiocarbamate. Mol Pharmacol 7:301–307 [PubMed] [Google Scholar]
- DeMaster EG, Nagasawa HT. (1978) Inhibition of aldehyde dehydrogenase by propiolaldehyde, a possible metabolite of pargyline. Res Commun Chem Pathol Pharmacol 21:497–505 [PubMed] [Google Scholar]
- DeMaster EG, Redfern B, Nagasawa HT. (1998) Mechanisms of inhibition of aldehyde dehydrogenase by nitroxyl, the active metabolite of the alcohol deterrent agent cyanamide. Biochem Pharmacol 55:2007–2015 [DOI] [PubMed] [Google Scholar]
- DeMaster EG, Shirota FN, Nagasawa HT. (1980) Microsomal N-depropargylation of pargyline to propiolaldehyde, an irreversible inhibitor of mitochondrial aldehyde dehydrogenase. Adv Exp Med Biol 132:219–228 [DOI] [PubMed] [Google Scholar]
- DeMaster EG, Shirota FN, Nagasawa HT. (1986) Role of propiolaldehyde and other metabolites in the pargyline inhibition of rat liver aldehyde dehydrogenase. Biochem Pharmacol 35:1481–1489 [DOI] [PubMed] [Google Scholar]
- Deng S, Yang X, Lassus H, Liang S, Kaur S, Ye Q, Li C, Wang LP, Roby KF, Orsulic S, et al. (2010) Distinct expression levels and patterns of stem cell marker, aldehyde dehydrogenase isoform 1 (ALDH1), in human epithelial cancers. PLoS One 5:e10277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downes J, Holmes R. (1992) Development of aldehyde dehydrogenase and alcohol dehydrogenase in mouse eye: evidence for light-induced changes. Biol Neonate 61:118–123 [DOI] [PubMed] [Google Scholar]
- Estey T, Cantore M, Weston PA, Carpenter JF, Petrash JM, Vasiliou V. (2007) Mechanisms involved in the protection of UV-induced protein inactivation by the corneal crystallin ALDH3A1. J Biol Chem 282:4382–4392 [DOI] [PubMed] [Google Scholar]
- Estey T, Chen Y, Carpenter JF, Vasiliou V. (2010) Structural and functional modifications of corneal crystallin ALDH3A1 by UVB light. PLoS One 5:e15218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferencz-Biro K, Pietruszko R. (1984) Inhibition of human aldehyde dehydrogenase isozymes by propiolaldehyde. Alcohol Clin Exp Res 8:302–307 [DOI] [PubMed] [Google Scholar]
- Fromtling RA, Gadebusch HH. (1983) Ethanol-cephalosporin antibiotic interactions: an animal model for the detection of disulfiram (Antabuse)-like effects. Methods Find Exp Clin Pharmacol 5:595–600 [PubMed] [Google Scholar]
- Fukuto JM, Hszieh R, Gulati P, Chiang KT, Nagasawa HT. (1992) N,O-diacylated-N-hydroxyarylsulfonamides: nitroxyl precursors with potent smooth muscle relaxant properties. Biochem Biophys Res Commun 187:1367–1373 [DOI] [PubMed] [Google Scholar]
- Gaval-Cruz M, Weinshenker D. (2009) mechanisms of disulfiram-induced cocaine abstinence: antabuse and cocaine relapse. Mol Interv 9:175–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgianni F, Bridson PK, Sorrentino BP, Pohl J, Blakley RL. (2000) Inactivation of aldophosphamide by human aldehyde dehydrogenase isozyme 3. Biochem Pharmacol 60:325–338 [DOI] [PubMed] [Google Scholar]
- Goedde HW, Agarwal DP. (1987) Polymorphism of aldehyde dehydrogenase and alcohol sensitivity. Enzyme 37:29–44 [DOI] [PubMed] [Google Scholar]
- Grün F, Hirose Y, Kawauchi S, Ogura T, Umesono K. (2000) Aldehyde dehydrogenase 6, a cytosolic retinaldehyde dehydrogenase prominently expressed in sensory neuroepithelia during development. J Biol Chem 275:41210–41218 [DOI] [PubMed] [Google Scholar]
- Hart BW, Faiman MD. (1992) In vitro and in vivo inhibition of rat liver aldehyde dehydrogenase by S-methyl N,N-diethylthiolcarbamate sulfoxide, a new metabolite of disulfiram. Biochem Pharmacol 43:403–406 [DOI] [PubMed] [Google Scholar]
- Hart BW, Faiman MD. (1995) Inhibition of rat liver low Km aldehyde dehydrogenase by thiocarbamate herbicides. Occupational implications. Biochem Pharmacol 49:157–163 [DOI] [PubMed] [Google Scholar]
- Hempel J, Perozich J, Chapman T, Rose J, Boesch JS, Liu ZJ, Lindahl R, Wang BC. (1999) Aldehyde dehydrogenase catalytic mechanism. A proposal. Adv Exp Med Biol 463:53–59 [DOI] [PubMed] [Google Scholar]
- Heron M, Hoyert DL, Murphy SL, Xu J, Kochanek KD, Tejada-Vera B. (2009) Deaths: final data for 2006. Natl Vital Stat Rep 57:1–134 [PubMed] [Google Scholar]
- Hess DA, Wirthlin L, Craft TP, Herrbrich PE, Hohm SA, Lahey R, Eades WC, Creer MH, Nolta JA. (2006) Selection based on CD133 and high aldehyde dehydrogenase activity isolates long-term reconstituting human hematopoietic stem cells. Blood 107:2162–2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi S, Matsushita S, Murayama M, Takagi S, Hayashida M. (1995) Alcohol and aldehyde dehydrogenase polymorphisms and the risk for alcoholism. Am J Psychiatry 152:1219–1221 [DOI] [PubMed] [Google Scholar]
- Hiltbrand E, Fournet G, Giliberto JP, Paret MJ, Chantepie J, Morel D, Goré J, Reichert U, Mehier H, Rochedix ME, et al. (2009) Synergetic bitherapy in mice with xenografts of human prostate cancer using a methional mimic (METLICO) and an aldehyde dehydrogenase 3 inhibitor (MATE): systemic intraperitoneal (IP) and targeted intra-tumoral (IT) administration. Curr Med Chem 16:1184–1191 [DOI] [PubMed] [Google Scholar]
- Hjelle JJ, Grubbs JH, Beer DG, Petersen DR. (1981) Inhibition of rat liver aldehyde dehydrogenase by carbon tetrachloride. J Pharmacol Exp Ther 219:821–826 [PubMed] [Google Scholar]
- Honoki K, Fujii H, Kubo A, Kido A, Mori T, Tanaka Y, Tsujiuchi T. (2010) Possible involvement of stem-like populations with elevated ALDH1 in sarcomas for chemotherapeutic drug resistance. Oncol Rep 24:501–505 [DOI] [PubMed] [Google Scholar]
- Huang LH, Hu JQ, Tao WQ, Li YH, Li GM, Xie PY, Liu XS, Jiang J. (2010) Gossypol inhibits phosphorylation of Bcl-2 in human leukemia HL-60 cells. Eur J Pharmacol 645:9–13 [DOI] [PubMed] [Google Scholar]
- Huang YW, Wang LS, Dowd MK, Wan PJ, Lin YC. (2009) (−)-Gossypol reduces invasiveness in metastatic prostate cancer cells. Anticancer Res 29:2179–2188 [PubMed] [Google Scholar]
- Jelski W, Chrostek L, Szmitkowski M. (2007) The activity of class I, III, and IV of alcohol dehydrogenase isoenzymes and aldehyde dehydrogenase in gastric cancer. Dig Dis Sci 52:531–535 [DOI] [PubMed] [Google Scholar]
- Jewell WT, Miller MG. (1998) Identification of a carboxylesterase as the major protein bound by molinate. Toxicol Appl Pharmacol 149:226–234 [DOI] [PubMed] [Google Scholar]
- Jewell WT, Miller MG. (1999) Comparison of human and rat metabolism of molinate in liver microsomes and slices. Drug Metab Dispos 27:842–847 [PubMed] [Google Scholar]
- Joenje H. (2011) Metabolism: alcohol, DNA and disease. Nature 475:45–46 [DOI] [PubMed] [Google Scholar]
- Kazmi SM, Li D, Koop K, Conant J, Lau CY.(1992) Role of aldehyde dehydrogenase in the biological activity of spermine dialdehyde, a novel immunosuppressive/purging agent. Pharmacol Res 25:383–392 [DOI] [PubMed] [Google Scholar]
- Keung WM. (2003) Anti-dipsotropic isoflavones: the potential therapeutic agents for alcohol dependence. Med Res Rev 23:669–696 [DOI] [PubMed] [Google Scholar]
- Keung WM, Lazo O, Kunze L, Vallee BL. (1995) Daidzin suppresses ethanol consumption by Syrian golden hamsters without blocking acetaldehyde metabolism. Proc Natl Acad Sci USA 92:8990–8993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keung WM, Vallee BL. (1993) Daidzin and daidzein suppress free-choice ethanol intake by Syrian golden hamsters. Proc Natl Acad Sci USA 90:10008–10012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikonyogo A, Abriola DP, Dryjanski M, Pietruszko R. (1999) Mechanism of inhibition of aldehyde dehydrogenase by citral, a retinoid antagonist. Eur J Biochem 262:704–712 [DOI] [PubMed] [Google Scholar]
- Kim KJ, Pearl PL, Jensen K, Snead OC, Malaspina P, Jakobs C, Gibson KM. (2011a) Succinic semialdehyde dehydrogenase: biochemical-molecular-clinical disease mechanisms, redox regulation, and functional significance. Antioxid Redox Signal 15:691–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MP, Fleming JB, Wang H, Abbruzzese JL, Choi W, Kopetz S, McConkey DJ, Evans DB, Gallick GE. (2011b) ALDH activity selectively defines an enhanced tumor-initiating cell population relative to CD133 expression in human pancreatic adenocarcinoma. PLoS One 6:e20636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitson TM. (1983) Mechanism of inactivation of sheep liver cytoplasmic aldehyde dehydrogenase by disulfiram. Biochem J 213:551–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitson TM. (1975) The effect of disulfiram on the aldehyde dehydrogenases of sheep liver. Biochem J 151:407–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitson TM. (1986) The effect of 5,5′-dithiobis(1-methyltetrazole) on cytoplasmic aldehyde dehydrogenase and its implications for cephalosporin-alcohol reactions. Alcohol Clin Exp Res 10:27–32 [DOI] [PubMed] [Google Scholar]
- Kronmiller JE, Beeman CS, Nguyen T, Berndt W. (1995) Blockade of the initiation of murine odontogenesis in vitro by citral, an inhibitor of endogenous retinoic acid synthesis. Arch Oral Biol 40:645–652 [DOI] [PubMed] [Google Scholar]
- Lam JP, Mays DC, Lipsky JJ. (1997) Inhibition of recombinant human mitochondrial and cytosolic aldehyde dehydrogenases by two candidates for the active metabolites of disulfiram. Biochemistry 36:13748–13754 [DOI] [PubMed] [Google Scholar]
- Larson HN, Weiner H, Hurley TD. (2005) Disruption of the coenzyme binding site and dimer interface revealed in the crystal structure of mitochondrial aldehyde dehydrogenase “Asian” variant. J Biol Chem 280:30550–30556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson HN, Zhou J, Chen Z, Stamler JS, Weiner H, Hurley TD. (2007) Structural and functional consequences of coenzyme binding to the inactive asian variant of mitochondrial aldehyde dehydrogenase: roles of residues 475 and 487. J Biol Chem 282:12940–12950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebsack ME, Petersen DR, Collins AC, Anderson AD. (1977) Preferential inhibition of the low Km aldehyde dehydrogenase activity by pargyline. Biochem Pharmacol 26:1151–1154 [DOI] [PubMed] [Google Scholar]
- Lee MJ, Nagasawa HT, Elberling JA, DeMaster EG. (1992) Prodrugs of nitroxyl as inhibitors of aldehyde dehydrogenase. J Med Chem 35:3648–3652 [DOI] [PubMed] [Google Scholar]
- Lee MJ, Shoeman DW, Goon DJ, Nagasawa HT. (2001) N-hydroxybenzenecarboximidic acid derivatives: a new class of nitroxyl-generating prodrugs. Nitric Oxide 5:278–287 [DOI] [PubMed] [Google Scholar]
- Lieber CS. (1994) Hepatic and metabolic effects of ethanol: pathogenesis and prevention. Ann Med 26:325–330 [DOI] [PubMed] [Google Scholar]
- Lin J, Haffner MC, Zhang Y, Lee BH, Brennen WN, Britton J, Kachhap SK, Shim JS, Liu JO, Nelson WG, et al. (2011) Disulfiram is a DNA demethylating agent and inhibits prostate cancer cell growth. Prostate 71:333–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin LZ, Lin J. (2011) Antabuse (disulfiram) as an affordable and promising anticancer drug. Int J Cancer 129:1285–1286; author reply 1286–1287 [DOI] [PubMed] [Google Scholar]
- Lipsky JJ, Shen ML, Naylor S. (2001) In vivo inhibition of aldehyde dehydrogenase by disulfiram. Chem Biol Interact 130–132:93–102 [DOI] [PubMed] [Google Scholar]
- Liu ZJ, Hempel J, Sun J, Rose J, Hsiao D, Chang WR, Chung YJ, Kuo I, Lindahl R, Wang BC. (1997) Crystal structure of a class 3 aldehyde dehydrogenase at 2.6 A resolution. Adv Exp Med Biol 414:1–7 [DOI] [PubMed] [Google Scholar]
- Lowe ED, Gao GY, Johnson LN, Keung WM. (2008) Structure of daidzin, a naturally occurring anti-alcohol-addiction agent, in complex with human mitochondrial aldehyde dehydrogenase. J Med Chem 51:4482–4487 [DOI] [PubMed] [Google Scholar]
- Mahmoud MI, Potter JJ, Colvin OM, Hilton J, Mezey E. (1993) Effect of 4-(diethylamino)benzaldehyde on ethanol metabolism in mice. Alcohol Clin Exp Res 17:1223–1227 [DOI] [PubMed] [Google Scholar]
- Mann CJ, Weiner H. (1999) Differences in the roles of conserved glutamic acid residues in the active site of human class 3 and class 2 aldehyde dehydrogenases. Protein Sci 8:1922–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcato P, Dean CA, Giacomantonio CA, Lee PW. (2011) Aldehyde dehydrogenase: its role as a cancer stem cell marker comes down to the specific isoform. Cell Cycle 10:1378–1384 [DOI] [PubMed] [Google Scholar]
- Marchitti SA, Brocker C, Stagos D, Vasiliou V. (2008) Non-P450 aldehyde oxidizing enzymes: the aldehyde dehydrogenase superfamily. Expert Opin Drug Metab Toxicol 4:697–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchitti SA, Deitrich RA, Vasiliou V. (2007) Neurotoxicity and metabolism of the catecholamine-derived 3,4-dihydroxyphenylacetaldehyde and 3,4-dihydroxyphenylglycolaldehyde: the role of aldehyde dehydrogenase. Pharmacol Rev 59:125–150 [DOI] [PubMed] [Google Scholar]
- Marchner H, Tottmar O. (1983) Studies in vitro on the inactivation of mitochondrial rat-liver aldehyde dehydrogenase by the alcohol-sensitizing compounds cyanamide, 1-aminocyclopropanol and disulfiram. Biochem Pharmacol 32:2181–2188 [DOI] [PubMed] [Google Scholar]
- Mason BJ, Heyser CJ. (2010) The neurobiology, clinical efficacy and safety of acamprosate in the treatment of alcohol dependence. Expert Opin Drug Saf 9:177–188 [DOI] [PubMed] [Google Scholar]
- Mays DC, Nelson AN, Lam-Holt J, Fauq AH, Lipsky JJ. (1996) S-methyl-N,N-diethylthiocarbamate sulfoxide and S-methyl-N,N-diethylthiocarbamate sulfone, two candidates for the active metabolite of disulfiram. Alcohol Clin Exp Res 20:595–600 [DOI] [PubMed] [Google Scholar]
- Mays DC, Ortiz-Bermudez P, Lam JP, Tong IH, Fauq AH, Lipsky JJ. (1998) Inhibition of recombinant human mitochondrial aldehyde dehydrogenase by two intermediate metabolites of disulfiram. Biochem Pharmacol 55:1099–1103 [DOI] [PubMed] [Google Scholar]
- McGregor NR. (2007) Pueraria lobata (Kudzu root) hangover remedies and acetaldehyde-associated neoplasm risk. Alcohol 41:469–478 [DOI] [PubMed] [Google Scholar]
- Michelot D. (1992) Poisoning by Coprinus atramentarius. Nat Toxins 1:73–80 [DOI] [PubMed] [Google Scholar]
- Min H, Shane B, Stokstad EL. (1988) Identification of 10-formyltetrahydrofolate dehydrogenase-hydrolase as a major folate binding protein in liver cytosol. Biochim Biophys Acta 967:348–353 [DOI] [PubMed] [Google Scholar]
- Mokdad AH, Marks JS, Stroup DF, Gerberding JL. (2004) Actual causes of death in the United States, 2000. JAMA 291:1238–1245 [DOI] [PubMed] [Google Scholar]
- Moore SA, Baker HM, Blythe TJ, Kitson KE, Kitson TM, Baker EN. (1998) Sheep liver cytosolic aldehyde dehydrogenase: the structure reveals the basis for the retinal specificity of class 1 aldehyde dehydrogenases. Structure 6:1541–1551 [DOI] [PubMed] [Google Scholar]
- Moreb JS, Zucali JR, Ostmark B, Benson NA. (2007) Heterogeneity of aldehyde dehydrogenase expression in lung cancer cell lines is revealed by Aldefluor flow cytometry-based assay. Cytometry B Clin Cytom 72:281–289 [DOI] [PubMed] [Google Scholar]
- Moridani MY, Khan S, Chan T, Teng S, Beard K, O'Brien PJ. (2001) Cytochrome P450 2E1 metabolically activates propargyl alcohol: propiolaldehyde-induced hepatocyte cytotoxicity. Chem Biol Interact 130–132:931–942 [DOI] [PubMed] [Google Scholar]
- Mukerjee N, Pietruszko R. (1994) Inactivation of human aldehyde dehydrogenase by isosorbide dinitrate. J Biol Chem 269:21664–21669 [PubMed] [Google Scholar]
- Münzel T, Daiber A, Mülsch A. (2005) Explaining the phenomenon of nitrate tolerance. Circ Res 97:618–628 [DOI] [PubMed] [Google Scholar]
- Murphy TC, Arntzen R, Picklo MJ., Sr (2005) Nitrate-based vasodilators inhibit multiple vascular aldehyde dehydrogenases. Cardiovasc Toxicol 5:321–332 [DOI] [PubMed] [Google Scholar]
- Nagasawa HT, DeMaster EG, Redfern B, Shirota FN, Goon DJ. (1990) Evidence for nitroxyl in the catalase-mediated bioactivation of the alcohol deterrent agent cyanamide J Med Chem 33:3120–3122 [DOI] [PubMed] [Google Scholar]
- Nagasawa HT, Kawle SP, Elberling JA, DeMaster EG, Fukuto JM. (1995) Prodrugs of nitroxyl as potential aldehyde dehydrogenase inhibitors vis-a-vis vascular smooth muscle relaxants. J Med Chem 38:1865–1871 [DOI] [PubMed] [Google Scholar]
- Ni L, Sheikh S, Weiner H. (1997) Involvement of glutamate 399 and lysine 192 in the mechanism of human liver mitochondrial aldehyde dehydrogenase. J Biol Chem 272:18823–18826 [DOI] [PubMed] [Google Scholar]
- Niederreither K, Dollé P. (2008) Retinoic acid in development: towards an integrated view. Nat Rev Genet 9:541–553 [DOI] [PubMed] [Google Scholar]
- Pang X, Wu Y, Wu Y, Lu B, Chen J, Wang J, Yi Z, Qu W, Liu M. (2011) (−)-Gossypol suppresses the growth of human prostate cancer xenografts via modulating VEGF signaling-mediated angiogenesis. Mol Cancer Ther 10:795–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolocci N, Jackson MI, Lopez BE, Miranda K, Tocchetti CG, Wink DA, Hobbs AJ, Fukuto JM. (2007) The pharmacology of nitroxyl (HNO) and its therapeutic potential: Not just the janus face of NO. Pharmacol Ther 113:442–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parajuli B, Kimble-Hill AC, Khanna M, Ivanova Y, Meroueh S, Hurley TD. (2011) Discovery of novel regulators of aldehyde dehydrogenase isoenzymes. Chem Biol Interact 191:153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Miller S, Younus H, Vanam R, Chen CH, Mochly-Rosen D, Hurley TD. (2010) Alda-1 is an agonist and chemical chaperone for the common human aldehyde dehydrogenase 2 variant. Nat Struct Mol Biol 17:159–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Miller SJ, Hurley TD. (2003) Coenzyme isomerization is integral to catalysis in aldehyde dehydrogenase. Biochemistry 42:7100–7109 [DOI] [PubMed] [Google Scholar]
- Pietruszko R, Mukerjee N, Blatter EE, Lehmann T. (1995) Nitrate esters as inhibitors and substrates of aldehyde dehydrogenase. Adv Exp Med Biol 372:25–34 [DOI] [PubMed] [Google Scholar]
- Poon R, Nadeau B, Chu I. (2000) Biochemical effects of chloral hydrate on male rats following 7-day drinking water exposure. J Appl Toxicol 20:455–461 [DOI] [PubMed] [Google Scholar]
- Poon R, Nakai J, Yagminas A, Benoit F, Moir D, Chu I, Valli VE. (2002) Subchronic toxicity of chloral hydrate on rats: a drinking water study. J Appl Toxicol 22:227–236 [DOI] [PubMed] [Google Scholar]
- Quash G, Fournet G, Courvoisier C, Martinez RM, Chantepie J, Paret MJ, Pharaboz J, Joly-Pharaboz MO, Goré J, André J, et al. (2008) Aldehyde dehydrogenase inhibitors: α,β-acetylenic N-substituted aminothiolesters are reversible growth inhibitors of normal epithelial but irreversible apoptogens for cancer epithelial cells from human prostate in culture. Eur J Med Chem 43:906–916 [DOI] [PubMed] [Google Scholar]
- Quemener V, Quash G, Moulinoux JP, Penlap V, Ripoll H, Havouis R, Doutheau A, Goré J. (1989) In vivo antitumor activity of 4-amino 4-methyl 2-pentyne 1-al, an inhibitor of aldehyde dehydrogenase. In Vivo 3:325–330 [PubMed] [Google Scholar]
- Quistad GB, Sparks SE, Casida JE. (1994) Aldehyde dehydrogenase of mice inhibited by thiocarbamate herbicides. Life Sci 55:1537–1544 [DOI] [PubMed] [Google Scholar]
- Rekha GK, Devaraj VR, Sreerama L, Lee MJ, Nagasawa HT, Sladek NE. (1998) Inhibition of human class 3 aldehyde dehydrogenase, and sensitization of tumor cells that express significant amounts of this enzyme to oxazaphosphorines, by chlorpropamide analogues. Biochem Pharmacol 55:465–474 [DOI] [PubMed] [Google Scholar]
- Rekha GK, Sladek NE. (1997) Inhibition of human class 3 aldehyde dehydrogenase, and sensitization of tumor cells that express significant amounts of this enzyme to oxazaphosphorines, by the naturally occurring compound gossypol. Adv Exp Med Biol 414:133–146 [DOI] [PubMed] [Google Scholar]
- Rice KL, Izon DJ, Ford J, Boodhoo A, Kees UR, Greene WK. (2008) Overexpression of stem cell associated ALDH1A1, a target of the leukemogenic transcription factor TLX1/HOX11, inhibits lymphopoiesis and promotes myelopoiesis in murine hematopoietic progenitors. Leuk Res 32:873–883 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Zavala JS, Weiner H. (2002) Structural aspects of aldehyde dehydrogenase that influence dimer-tetramer formation. Biochemistry 41:8229–8237 [DOI] [PubMed] [Google Scholar]
- Rösner S, Hackl-Herrwerth A, Leucht S, Vecchi S, Srisurapanont M, Soyka M. (2010) Opioid antagonists for alcohol dependence. Cochrane Database Syst Rev 12:CD001867. [DOI] [PubMed] [Google Scholar]
- Russo J, Barnes A, Berger K, Desgrosellier J, Henderson J, Kanters A, Merkov L. (2002) 4-(N,N-dipropylamino)benzaldehyde inhibits the oxidation of all-trans retinal to all-trans retinoic acid by ALDH1A1, but not the differentiation of HL-60 promyelocytic leukemia cells exposed to all-trans retinal. BMC Pharmacol 2:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo JE, Hauguitz D, Hilton J. (1988) ) Inhibition of mouse cytosolic aldehyde dehydrogenase by 4-(diethylamino)benzaldehyde. Biochem Pharmacol 37:1639–1642 [DOI] [PubMed] [Google Scholar]
- Schroeder JP, Cooper DA, Schank JR, Lyle MA, Gaval-Cruz M, Ogbonmwan YE, Pozdeyev N, Freeman KG, Iuvone PM, Edwards GL, et al. (2010) Disulfiram attenuates drug-primed reinstatement of cocaine seeking via inhibition of dopamine beta-hydroxylase. Neuropsychopharmacology 35:2440–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh S, Ni L, Hurley TD, Weiner H. (1997) The potential roles of the conserved amino acids in human liver mitochondrial aldehyde dehydrogenase. J Biol Chem 272:18817–18822 [DOI] [PubMed] [Google Scholar]
- Shen ML, Johnson KL, Mays DC, Lipsky JJ, Naylor S. (2001) Determination of in vivo adducts of disulfiram with mitochondrial aldehyde dehydrogenase. Biochem Pharmacol 61:537–545 [DOI] [PubMed] [Google Scholar]
- Shirota FN, DeMaster EG, Nagasawa HT. (1987) Cyanide is a product of the catalase-mediated oxidation of the alcohol deterrent agent, cyanamide. Toxicol Lett 37:7–12 [DOI] [PubMed] [Google Scholar]
- Sládek NE. (1999) Aldehyde dehydrogenase-mediated cellular relative insensitivity to the oxazaphosphorines. Curr Pharm Des 5:607–625 [PubMed] [Google Scholar]
- Somerville L. (1986) The metabolism of fungicides. Xenobiotica 16:1017–1030 [DOI] [PubMed] [Google Scholar]
- Sophos NA, Vasiliou V. (2003) Aldehyde dehydrogenase gene superfamily: the 2002 update. Chem Biol Interact 143–144:5–22 [DOI] [PubMed] [Google Scholar]
- Soyka M, Rösner S. (2010) Emerging drugs to treat alcoholism. Expert Opin Emerg Drugs 15:695–711 [DOI] [PubMed] [Google Scholar]
- Sreerama L, Sladek NE. (1993) Identification and characterization of a novel class 3 aldehyde dehydrogenase overexpressed in a human breast adenocarcinoma cell line exhibiting oxazaphosphorine-specific acquired resistance. Biochem Pharmacol 45:2487–2505 [DOI] [PubMed] [Google Scholar]
- Stagos D, Chen Y, Brocker C, Donald E, Jackson BC, Orlicky DJ, Thompson DC, Vasiliou V. (2010) Aldehyde dehydrogenase 1B1: molecular cloning and characterization of a novel mitochondrial acetaldehyde-metabolizing enzyme. Drug Metab Dispos 38:1679–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staub RE, Quistad GB, Casida JE. (1998) Mechanism for benomyl action as a mitochondrial aldehyde dehydrogenase inhibitor in mice. Chem Res Toxicol 11:535–543 [DOI] [PubMed] [Google Scholar]
- Staub RE, Quistad GB, Casida JE. (1999) S-methyl N-butylthiocarbamate sulfoxide: selective carbamoylating agent for mouse mitochondrial aldehyde dehydrogenase. Biochem Pharmacol 58:1467–1473 [DOI] [PubMed] [Google Scholar]
- Steinmetz CG, Xie P, Weiner H, Hurley TD. (1997) Structure of mitochondrial aldehyde dehydrogenase: the genetic component of ethanol aversion. Structure 5:701–711 [DOI] [PubMed] [Google Scholar]
- Storms RW, Green PD, Safford KM, Niedzwiecki D, Cogle CR, Colvin OM, Chao NJ, Rice HE, Smith CA. (2005) Distinct hematopoietic progenitor compartments are delineated by the expression of aldehyde dehydrogenase and CD34. Blood 106:95–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storms RW, Trujillo AP, Springer JB, Shah L, Colvin OM, Ludeman SM, Smith C. (1999) Isolation of primitive human hematopoietic progenitors on the basis of aldehyde dehydrogenase activity. Proc Natl Acad Sci USA 96:9118–9123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland KC, Krupenko NI, Dubard ME, Hu CJ, Tsybovsky Y, Krupenko SA. (2011) Enzymatic properties of ALDH1L2, a mitochondrial 10-formyltetrahydrofolate dehydrogenase. Chem Biol Interact 191:129–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Qiu Q, Zhang X, Jiang Z, Leng Q, Liu Z, Stass SA, Jiang F. (2010) Aldehyde dehydrogenase 1 A1-positive cell population is enriched in tumor-initiating cells and associated with progression of bladder cancer. Cancer Epidemiol Biomarkers Prev 19:327–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Weiner H, Filmer DL. (1981) Effects of pH on horse liver aldehyde dehydrogenase: alterations in metal ion activation, number of functioning active sites, and hydrolysis of the acyl intermediate. Biochemistry 20:6225–6230 [DOI] [PubMed] [Google Scholar]
- Tamai H, Yokoyama A, Okuyama K, Takahashi H, Maruyama K, Suzuki Y, Ishii H. (2000) Comparison of cyanamide and disulfiram in effects on liver function. Alcohol Clin Exp Res 24:97S–99S [PubMed] [Google Scholar]
- Tanaka M, Tamura K, Ide H. (1996) Citral, an inhibitor of retinoic acid synthesis, modifies chick limb development. Dev Biol 175:239–247 [DOI] [PubMed] [Google Scholar]
- Tanei T, Morimoto K, Shimazu K, Kim SJ, Tanji Y, Taguchi T, Tamaki Y, Noguchi S. (2009) Association of breast cancer stem cells identified by aldehyde dehydrogenase 1 expression with resistance to sequential Paclitaxel and epirubicin-based chemotherapy for breast cancers. Clin Cancer Res 15:4234–4241 [DOI] [PubMed] [Google Scholar]
- Tottmar O, Hellstrom E. (1979) Blood pressure response to ethanol in relation to acetaldehyde levels and dopamine-beta-hydroxylase activity in rats pretreated with disulfiram, cyanamide and coprine. Acta Pharmacol Toxicol (Copenh) 45:272–281 [DOI] [PubMed] [Google Scholar]
- Tottmar O, Lindberg P. (1977) Effects on rat liver acetaldehyde dehydrogenases in vitro and in vivo by coprine, the disulfiram-like constituent of Coprinus atramentarius. Acta Pharmacol Toxicol (Copenh) 40:476–481 [PubMed] [Google Scholar]
- Vallari RC, Pietruszko R. (1982) Human aldehyde dehydrogenase: mechanism of inhibition of disulfiram. Science 216:637–639 [DOI] [PubMed] [Google Scholar]
- van den Hoogen C, van der Horst G, Cheung H, Buijs JT, Pelger RC, van der Pluijm G. (2011) The aldehyde dehydrogenase enzyme 7A1 is functionally involved in prostate cancer bone metastasis. Clin Exp Metastasis 28:615–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Poznak C, Seidman AD. (2002) Critical review of current treatment strategies for advanced hormone insensitive breast cancer. Cancer Invest 20 (Suppl 2):1–14 [DOI] [PubMed] [Google Scholar]
- Van Poznak C, Seidman AD, Reidenberg MM, Moasser MM, Sklarin N, Van Zee K, Borgen P, Gollub M, Bacotti D, Yao TJ, et al. (2001) Oral gossypol in the treatment of patients with refractory metastatic breast cancer: a phase I/II clinical trial. Breast Cancer Res Treat 66:239–248 [DOI] [PubMed] [Google Scholar]
- Vasiliou V, Nebert DW. (2005) Analysis and update of the human aldehyde dehydrogenase (ALDH) gene family. Hum Genomics 2:138–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiliou V, Pappa A, Petersen DR. (2000) Role of aldehyde dehydrogenases in endogenous and xenobiotic metabolism. Chem Biol Interact 129:1–19 [DOI] [PubMed] [Google Scholar]
- Wang F, Zhai S, Liu X, Li L, Wu S, Dou QP, Yan B. (2011) A novel dithiocarbamate analogue with potentially decreased ALDH inhibition has copper-dependent proteasome-inhibitory and apoptosis-inducing activity in human breast cancer cells. Cancer Lett 300:87–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GW, Guo Y, Vondriska TM, Zhang J, Zhang S, Tsai LL, Zong NC, Bolli R, Bhatnagar A, Prabhu SD. (2008) Acrolein consumption exacerbates myocardial ischemic injury and blocks nitric oxide-induced PKCepsilon signaling and cardioprotection. J Mol Cell Cardiol 44:1016–1022 [DOI] [PubMed] [Google Scholar]
- Wang MF, Han CL, Yin SJ. (2009) Substrate specificity of human and yeast aldehyde dehydrogenases. Chem Biol Interact 178:36–39 [DOI] [PubMed] [Google Scholar]
- Wang X, Weiner H. (1995) Involvement of glutamate 268 in the active site of human liver mitochondrial (class 2) aldehyde dehydrogenase as probed by site-directed mutagenesis. Biochemistry 34:237–243 [DOI] [PubMed] [Google Scholar]
- Weiner H. (1979) Alcohol dehydrogenase, in Biochemistry and Pharmacology of Ethanol (Majchrowicz E, Noble EP. eds) pp 125–144, Plenum Press, New York [Google Scholar]
- Weiner H, Takahashi K. (1983) Effects of magnesium and calcium on mitochondrial and cytosolic liver aldehyde dehydrogenases. Pharmacol Biochem Behav 18 (Suppl 1):109–112 [DOI] [PubMed] [Google Scholar]
- Wenzl MV, Beretta M, Griesberger M, Russwurm M, Koesling D, Schmidt K, Mayer B, Gorren AC. (2011) Site-directed mutagenesis of aldehyde dehydrogenase-2 suggests three distinct pathways of nitroglycerin biotransformation. Mol Pharmacol 80:258–266 [DOI] [PubMed] [Google Scholar]
- Xiao T, Shoeb M, Siddiqui MS, Zhang M, Ramana KV, Srivastava SK, Vasiliou V, Ansari NH. (2009) Molecular cloning and oxidative modification of human lens ALDH1A1: implication in impaired detoxification of lipid aldehydes. J Toxicol Environ Health A 72:577–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L, Fan P, Arolfo M, Jiang Z, Olive MF, Zablocki J, Sun HL, Chu N, Lee J, Kim HY, et al. (2010) Inhibition of aldehyde dehydrogenase-2 suppresses cocaine seeking by generating THP, a cocaine use-dependent inhibitor of dopamine synthesis. Nat Med 16:1024–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida A, Hsu LC, Davé V. (1992) Retinal oxidation activity and biological role of human cytosolic aldehyde dehydrogenase. Enzyme 46:239–244 [DOI] [PubMed] [Google Scholar]
- Yoshida A, Rzhetsky A, Hsu LC, Chang C. (1998) Human aldehyde dehydrogenase gene family. Eur J Biochem 251:549–557 [DOI] [PubMed] [Google Scholar]
- Zemaitis MA, Greene FE. (1976) Impairment of hepatic microsomal and plasma esterases of the rat by disulfiram and diethyldithiocarbamate. Biochem Pharmacol 25:453–459 [DOI] [PubMed] [Google Scholar]
- Zhang J, Tian Q, Chan SY, Duan W, Zhou S. (2005) Insights into oxazaphosphorine resistance and possible approaches to its circumvention. Drug Resist Updat 8:271–297 [DOI] [PubMed] [Google Scholar]
- Zhang P, Xu D, Wang S, Fu H, Wang K, Zou Y, Sun A, Ge J. (2011) Inhibition of aldehyde dehydrogenase 2 activity enhances antimycin-induced rat cardiomyocytes apoptosis through activation of MAPK signaling pathway. Biomed Pharmacother 65:590–593 [DOI] [PubMed] [Google Scholar]