

Abstract
Overdose of γ-hydroxybutyrate (GHB) frequently causes respiratory depression, occasionally resulting in death; however, little is known about the dose-response relationship or effects of potential overdose treatment strategies on GHB-induced respiratory depression. In these studies, the parameters of respiratory rate, tidal volume, and minute volume were measured using whole-body plethysmography in rats administered GHB. Intravenous doses of 200, 600, and 1500 mg/kg were administered to assess the dose-dependent effects of GHB on respiration. To determine the receptors involved in GHB-induced respiratory depression, a specific GABAB receptor antagonist, (2S)-(+)-5,5-dimethyl-2-morpholineacetic acid (SCH50911), and a specific GABAA receptor antagonist, bicuculline, were administered before GHB. The potential therapeutic strategies of receptor inhibition and monocarboxylate transporter (MCT) inhibition were assessed by inhibitor administration 5 min after GHB. The primary effect of GHB on respiration was a dose-dependent decrease in respiratory rate, accompanied by an increase in tidal volume, resulting in little change in minute volume. Pretreatment with 150 mg/kg SCH50911 completely prevented the decrease in respiratory rate, indicating agonism at GABAB receptors to be primarily responsible for GHB-induced respiratory depression. Administration of 50 mg/kg SCH50911 after GHB completely reversed the decrease in respiratory rate; lower doses had partial effects. Administration of the MCT inhibitor l-lactate increased GHB renal and total clearance, also improving respiratory rate. Administration of 5 mg/kg SCH50911 plus l-lactate further improved respiratory rate compared with the same dose of either agent alone, indicating that GABAB and MCT inhibitors, alone and in combination, represent potential treatment options for GHB-induced respiratory depression.
Introduction
γ-Hydroxybutyate (GHB) is a short-chain fatty acid present endogenously in many human tissues, resulting from production via GABA metabolism (Maitre, 1997). GHB has also recently been identified as a useful therapeutic agent for the treatment of narcolepsy and excessive daytime sleepiness in the form of sodium oxybate (Xyrem; Jazz Pharmaceuticals, Palo Alto, CA). However, GHB has become more popularly known as a drug of abuse. According to reports from the Drug Abuse Warning Network, there have consistently been 1000 to 2000 GHB-related emergency department visits reported annually in the United States over the past several years (Substance Abuse and Mental Health Services Administration, 2011). GHB overdose can result in manifestations including sedation, coma, hypothermia, bradycardia, respiratory depression, and death (Li et al., 1998; Sporer et al., 2003; Caldicott et al., 2004; Galicia et al., 2011). In a recent report of known GHB-associated fatalities, the most common cause of mortality was cardiorespiratory arrest (Zvosec et al., 2011). Respiratory depression with the need for mechanical ventilation is also frequently reported in nonfatal cases of GHB intoxication (Li et al., 1998; Mason and Kerns, 2002; Liechti and Kupferschmidt, 2004).
Although respiratory depression is a common symptom of GHB overdose, neither the dose-dependent effects of GHB on this measure nor the neurotransmitter receptors involved in GHB-induced respiratory depression have been investigated. There are several proposed actions of GHB, including 1) direct action at GABAB receptors (Bernasconi et al., 1992), 2) direct action at its own putative GHB receptor (Maitre, 1997), and 3) indirect action at GABA receptors via GABA production/release (Hechler et al., 1997; Gobaille et al., 1999). Although evidence exists for each of these mechanisms in vitro and/or in vivo, many of the toxicological effects of GHB, including sedation, hypothermia, and fatality, can be attributed to agonism at GABAB receptors (Carai et al., 2001, 2005; Kaupmann et al., 2003).
Along with a complex pharmacologic profile, the pharmacokinetics of GHB are also notably complicated. In humans, GHB exhibits dose-dependent pharmacokinetics, even at therapeutic concentrations (Palatini et al., 1993). Rats similarly display nonlinear pharmacokinetics, due to several concentration-dependent processes including saturable oral absorption, saturable metabolism, and saturable renal reabsorption (Lettieri and Fung, 1979; Morris et al., 2005). In both humans and rats, GHB metabolism is the predominant route of elimination at low doses, and renal excretion of unchanged drug is minimal (Lettieri and Fung, 1976; Brenneisen et al., 2004). Although limited information exists for supratherapeutic GHB doses in humans, it has been well documented in rats that renal clearance becomes an increasingly important route of elimination as GHB doses are increased (Morris et al., 2005). This nonlinear renal clearance can be attributed to a concentration-dependent transport process, leading to saturable renal reabsorption, demonstrated in our laboratory to involve the group of transporters known as monocarboxylate transporters (MCTs) (Morris et al., 2005; Wang et al., 2006). MCTs are proton-dependent transporters expressed throughout the body, and GHB is an identified substrate for MCTs 1, 2, and 4 (Wang et al., 2006; Wang and Morris, 2007). The ubiquitous expression of these transporters includes that in the intestine, kidney, and brain, regions of interest regarding GHB pharmacokinetics. Because of their role in the renal reabsorption of GHB, inhibition of these transporters represents a potential therapeutic strategy for GHB overdose. This strategy has been demonstrated to translate to in vivo effects on GHB disposition, and administration of MCT inhibitors increases the renal and total clearance in animal models of GHB overdose (Morris et al., 2005; Wang et al., 2008a,b). Likewise, the administration of the MCT inhibitor l-lactate, in combination with osmotic diuresis, increases the renal clearance of GHB in humans, as demonstrated in our pilot clinical study (Morris et al., 2011).
The first aim of this research was to investigate the dose-response relationship of GHB-induced respiratory depression, including the primary neurotransmitter receptors involved in eliciting this effect. The second was to assess potential treatment strategies, including MCT and receptor inhibition, for improving GHB-induced respiratory depression, because the application of these strategies for treating this pharmacodynamic endpoint have not been evaluated previously.
Materials and Methods
Chemicals and Reagents.
Sodium GHB used in these studies was provided by the National Institute on Drug Abuse. Deuterated GHB (GHB-d6) was purchased from Cerilliant Corporation (Round Rock, TX). Sodium l-lactate and bicuculline methiodide were purchased from Sigma-Aldrich (St. Louis, MO). (2S)-(+)-5,5-Dimethyl-2-morpholineacetic acid (SCH50911) was purchased from Tocris Bioscience (Ellisville, MO). High-performance liquid chromatography-grade acetonitrile and acetic acid were purchased from Honeywell Burdick & Jackson (Muskegon, MI).
Animals and Animal Surgery.
Male Sprague-Dawley rats (Harlan, Indianapolis, IN) weighing 270 to 330 g were used for all experiments. Animals were housed under controlled temperature and humidity with an artificial 12-h light/dark cycle, and food was available ad libitum. All animal protocols were approved by the Institutional Animal Care and Use Committee at the University at Buffalo. Animals were allowed to acclimate to their environment for a minimum of 1 week before surgical implantation of jugular and femoral vein cannulae under anesthesia with ketamine-xylazine. Cannulae were flushed daily with 40 IU/ml heparinized saline to maintain patency. Animals were allowed a minimum of 72 h for recovery from surgery before drug administration.
Plethysmography.
Measurement of respiration in these studies was performed using a whole-body plethysmograph (model PLY4213; Buxco Research Systems, Wilmington, NC). Plethysmography equipment included unrestrained plethysmography chambers consisting of a main (animal) chamber and reference chamber for buffering changes in atmospheric pressure. The plethysmography chambers were connected to a Rodent Bias Flow Supply (BFL0250) to draw expired CO2 out of the chambers and provide a smoothed flow of room air at a flow rate of 2.5 l/min per chamber. The plethysmography chambers included ports to which a pressure sensor was connected and led to the MAX 1500 preamplifier. Signals were collected, visualized, and quantitated using BioSystem XA software. Two additional ports were included in the chambers for the insertion of jugular and femoral vein cannulae, allowing for drug administration and blood sampling. Urine was collected at the base of the chamber at intervals by opening an additional port at the base. Calibration of chamber pressure was performed before every experiment by injection of 5 ml of air through the base port. At each recording, signals were collected for six intervals of 10 s each and averaged to represent 1 min of recording. Measurements for the parameters of respiratory frequency (rate), tidal volume, and minute volume (rate · tidal volume) were quantitated for each recording.
Pharmacokinetic/Pharmacodynamic Studies.
Rats were placed in plethysmography chambers 1 h before drug administration and allowed to acclimate to the chambers for 45 min before five baseline measurements of 1 min each were collected over 15 min. In all studies, GHB administration was considered time 0, and respiration measurements were recorded at 2.5, 5, 7.5, 10, 15, 20, 25, and 30 min and every 15 min thereafter for 480 min. Blood samples were collected, and collection times were optimized for each GHB dose according to previous studies (Felmlee et al., 2010b, 2011). Urine was collected at intervals up to 480 min. For overlapping pharmacokinetic/pharmacodynamic time points, blood and urine samples were taken directly after the recording of respiratory measurements.
Dose-Dependent Effects of GHB on Respiration.
To assess the dose-response relationship of GHB-induced respiratory depression, rats were administered GHB intravenously in doses of 200, 600, and 1500 mg/kg (four to six animals per dose). GHB was injected over 1 to 2 min as a 300 mg/ml solution in sterile water via the jugular vein cannula. A placebo control group received a 5 ml/kg saline bolus.
Neurotransmitter Receptors Involved in GHB-Induced Respiratory Depression.
To determine the primary receptors involved in GHB-induced respiratory depression, rats were pretreated with specific receptor antagonists. Bicuculline methiodide (5 mg/kg) was administered for inhibition of GABAA receptors and SCH50911 (150 mg/kg) for inhibition of GABAB receptors (three to four animals per group). Inhibitors were administered immediately after the collection of baseline respiratory measurements and 1500 mg/kg GHB was administered 5 min later. Data from dose-dependent experiments were used as the control. Bicuculline methiodide was administered as a 5 mg/ml solution in saline and SCH50911 as a 50 mg/ml solution in saline via the jugular vein cannula.
Potential Treatment Strategies.
To assess the effect of potential treatment strategies on GHB-induced respiratory depression, treatments were administered intravenously 5 min after 1500 mg/kg GHB. Treatment strategies included SCH50911 (2.5, 5, 10, and 50 mg/kg), the MCT inhibitor l-lactate (66 mg/kg bolus followed by a 302.5 mg/kg/h infusion for 8 h), and combination therapy of 5 mg/kg SCH50911 plus the same dose of l-lactate. Treatment groups included three to five animals per group, and were compared with the 1500 mg/kg control group from dose-dependent experiments to determine the effects of treatment on GHB-induced respiratory depression. The same l-lactate dose was also administered alone at time 0 to assess potential effects of this agent on respiration. In these experiments, SCH50911 was administered as a 2.5, 5, 10, or 50 mg/ml solution in saline via the jugular vein cannula and l-lactate as a 40 mg/ml solution in sterile water via the femoral vein cannula.
Plasma and Urine Sample Analysis.
GHB plasma concentrations were determined using an LC-MS/MS method, similar to those published previously (Fung et al., 2008; Felmlee et al., 2010a). Plasma samples were prepared by adding 5 μl of GHB-d6 (125 μg/ml) to 50 μl of sample. Plasma standards and quality controls were prepared by adding 5 μl of GHB-d6 and 5 μl of GHB stock solution to 45 μl of blank plasma, and 800 μl of 0.1% formic acid in acetonitrile was added to precipitate the plasma proteins. The samples were vortexed, followed by centrifugation at 10,000g for 20 min at 4°C. Then 750 μl of the supernatant was aspirated and evaporated under a stream of nitrogen gas. The samples were reconstituted in 250 μl of aqueous mobile phase.
The LC-MS/MS assay was performed on an Agilent 1100 series high-performance liquid chromatography system with binary pump and autosampler (Agilent Technologies, Santa Clara, CA) connected to a PerkinElmer Sciex API 3000 triple quadrupole tandem mass spectrometer with a TurboIonSpray (Applied Biosystems, Foster City, CA). Chromatographic separation was achieved by injecting 7 μl of sample on an Xterra MS C18 column (250 × 2.1 mm i.d., 5-μm particle size; Waters, Milford, MA). Mobile phase A consisted of 5:95 acetonitrile-water with 0.1% acetic acid and mobile phase B consisted of 95:5 acetonitrile-water with 0.1% acetic acid. The flow rate was 200 μl/min with the following gradient elution profile: 100 to 68% A over 7 min; 68 to 10% A over 3 min; and 10 to 100% over 5 min for a total run time of 15 min. The mass spectrometer was operated in a positive ionization mode with multiple reaction monitoring. Q1/Q3 m/z ratios for the parent/product ions of GHB and GHB-d6 were 105.2/87.2 and 111.1/93.2, respectively. The mass spectrometer parameters were optimized at a declustering potential of 18 V, focusing potential of 100 V, collision energy of 20 V, entrance potential of 10 V, and collision cell exit potential of 5 V. The ion spray voltage was set at 5500 V with temperature at 350°C. Nebulizer and curtain gas flow were set at 10 and 8 ml/min, respectively. The retention time for GHB was 4.15 min. The data were analyzed using Analyst software version 1.4.2 (Applied Biosystems).
Regression analysis of peak area ratios of GHB/GHB-d6 to GHB concentrations was used to assess linearity of the curve. The intraday and interday precision and accuracy were determined using quality control (QC) samples at 10 μg/ml (low QC), 125 μg/ml (medium QC), and 400 μg/ml (high QC). For determination of the intraday precision and accuracy, quality control samples were analyzed in triplicate on each day, whereas for the interday precision and accuracy, quality control samples were analyzed on three different days. A calibration curve was run on each analysis day along with the quality controls. The precision was determined by the coefficient of variation, and accuracy was measured by comparing the calculated concentration with the known concentration.
Urine samples were prepared and analyzed for GHB using a previously described LC-MS/MS method (Felmlee et al., 2010b). Plasma lactate concentrations were determined using a YSI 1500 Sport Lactate Analyzer (YSI, Inc., Yellow Springs, OH).
Data and Statistical Analysis.
Pharmacokinetic parameters were determined via noncompartmental analysis using WinNonlin 5.2 (Pharsight, Mountain View, CA). The area below the plasma concentration-time curve (AUC) was determined using the trapezoidal method. Total clearance (Cl) was determined as dose/AUC. Renal clearance (ClR) was determined as Ae/AUC, where Ae represents the amount excreted in the urine. Percentage of urinary excretion was calculated as Ae/dose. Metabolic or nonrenal clearance (Clm) was calculated as Cl − ClR. The pharmacodynamic descriptors of area below the effect curve (ABEC), maximum effect (Emax), time of maximum effect (Tmax), and duration of effect (Td) were used to determine the effects of inhibitor administration on GHB-induced respiratory depression. ABEC was calculated using WinNonlin. Td was determined for each animal as the time to return to its individual baseline respiratory frequency. Statistical analysis was performed using SigmaPlot 10.0 (Systat Software, Inc., San Jose, CA). Differences were considered significant when p < 0.05. One-way analysis of variance followed by Dunnett's or Tukey's post hoc tests was used to determine statistically significant differences in mean pharmacokinetic and pharmacodynamic parameters between groups. Paired t tests were used to determine statistically significant changes in respiratory parameters compared with baseline. In determining the effects of l-lactate alone on respiration, the average of the last hour of respiratory measurements was compared with the individual average baseline values. Mean steady-state lactate plasma concentrations were calculated as the average of hourly values beginning at 60 min.
Results
Plasma GHB LC-MS/MS Assay.
The lower limit of quantification for GHB in plasma was found to be 5 μg/ml with acceptable error in precision and accuracy of less than 20%. The endogenous concentrations of GHB in plasma are negligible compared with GHB concentrations obtained after administration of the lowest dose in our studies (Fung et al., 2004); therefore, the endogenous concentrations were not included in the calculation of GHB concentrations in plasma. The standard curve for GHB ranged from 5 to 500 μg/ml based on regression analysis of peak area ratios of GHB/GHB-d6 to GHB concentrations with a correlation coefficient (r2 >0.999). The intraday and interday precision and accuracy of the quality control samples are summarized in Table 1.
TABLE 1.
Intraday and interday accuracy and precision for GHB in rat plasma
Each measured concentration is the mean of triplicate measurements. The analysis was performed over 3 days.
| Nominal Concentration | Measured Concentration | S.D. | Precision | Accuracy | |
|---|---|---|---|---|---|
| μg/ml | CV% | % | |||
| Intraday | 10 | 10.8 | 0.12 | 1.07 | 107.7 |
| 125 | 121 | 4.36 | 3.60 | 96.8 | |
| 400 | 375 | 6.93 | 1.85 | 93.7 | |
| Interday | 10 | 10.5 | 0.32 | 3.05 | 105.2 |
| 125 | 118 | 2.96 | 2.50 | 94.9 | |
| 400 | 368 | 6.38 | 1.73 | 92.0 | |
CV, coefficient of variation.
Dose Dependence of GHB Pharmacokinetics/Pharmacodynamics.
GHB administration in increasing intravenous doses displayed nonlinear pharmacokinetics, as shown in Table 2, similar to previous reports (Lettieri and Fung, 1979; Morris et al., 2005). Renal clearance and the urinary excretion of GHB was almost negligible at the lowest dose of 200 mg/kg but represented the predominant route of elimination at the highest dose of 1500 mg/kg. The pharmacodynamic results of this experiment are shown in Fig. 1. Increasing doses of GHB resulted in a dose-dependent decrease in the parameter of respiratory rate, which was accompanied by a dose-dependent increase in tidal volume. Minute volume was unchanged with the 200 and 600 mg/kg doses but was significantly decreased with the 1500 mg/kg dose (95 ± 18 ml/min at baseline versus Emax of 54 ± 24 ml/min; mean ± S.D., p < 0.05). Raw plethysmography traces displaying the change in respiratory pattern with GHB administration are shown in Fig. 2. As a result of this experiment, respiratory rate was considered the primary parameter of interest for assessment of receptors involved and potential treatment strategies. It was also determined in this experiment that 1500 mg/kg GHB was the maximal dose that could be administered without causing death; therefore, this dose was used for further investigation.
TABLE 2.
Nonlinear pharmacokinetics of GHB
GHB was administered intravenously. Data are presented as mean (S.D.); n = 4 to 6. One-way analysis of variance followed by Tukey's post hoc test was used to determine statistically significant differences in pharmacokinetic parameters.
| 200 mg/kg | 600 mg/kg | 1500 mg/kg | |
|---|---|---|---|
| Cl, ml · kg−1 · min−1 | 7.60 (0.29) | 6.00 (0.74)a | 5.16 (0.70)a |
| ClR, ml · kg−1 · min−1 | 0.444 (0.20) | 1.68 (0.75)a | 3.18 (0.66)a,b |
| Urinary excretion, % | 6.0 (3) | 26.7 (11)a | 60.1 (7)a,b |
Significantly different from 200 mg/kg GHB (P < 0.05).
Significantly different from 200 and 600 mg/kg GHB (P < 0.05).
Fig. 1.

Dose-dependent effects of GHB on measures of respiration. GHB was administered intravenously at time 0. Data are presented as mean ± S.D.; n = 4 to 6.
Fig. 2.

Effect of GHB administration on respiratory pattern. Displayed are sample 10-s interval plethysmography traces obtained at baseline (A) and 30 min after administration of GHB 1500 mg/kg i.v. (B).
Neurotransmitter Receptors Involved in GHB-Induced Respiratory Depression.
Effects of pretreatment with specific receptor antagonists are given in Table 3. Administration of the GABAB inhibitor, SCH50911 (150 mg/kg), before GHB, resulted in no significant decrease in respiratory rate nor a change in tidal volume compared with baseline, as displayed in Fig. 3. This inhibitor also increased the nonrenal clearance of GHB, but not the total clearance at this dose. Administration of the GABAA inhibitor, bicuculline methiodide (5 mg/kg), before GHB, resulted in no change in the respiratory effects compared with those for GHB alone and had no significant effects on GHB pharmacokinetics.
TABLE 3.
Effects of specific receptor antagonists on the pharmacokinetics/pharmacodynamics of GHB (1500 mg/kg i.v.)
SCH50911 (150 mg/kg) and bicuculline methiodide (5 mg/kg) were administered intravenously 5 min before GHB. Data are presented as mean (S.D.); n = 3 to 5. One-way analysis of variance followed by Dunnett's post hoc test was used to determine statistically significant differences in mean pharmacokinetic and pharmacodynamic parameters with inhibitor administration compared with those with GHB alone.
| GHB | GHB + SCH50911 | GHB + Bicuculline | |
|---|---|---|---|
| Cl, ml · kg−1 · min−1 | 5.16 (0.70) | 6.07 (0.47) | 5.02 (0.14) |
| ClR, ml · kg−1 · min−1 | 3.18 (0.66) | 2.84 (0.32) | 2.72 (0.78) |
| Clm, ml · kg−1 · min−1 | 1.99 (0.17) | 3.23 (0.78)* | 2.30 (0.63) |
| Frequency ABEC, breaths | 10,500 (2700) | —a | 10,900 (1300) |
| Frequency Emax, breaths/min | 17 (7) | — | 15 (9) |
| Frequency Tmax, min | 53.0 (19) | — | 67.5 (13) |
Significantly different from GHB alone (P < 0.05).
—, no ABEC, Emax, or Tmax values could be calculated because respiration is similar to the baseline values; SCH50911 completely prevented any significant decrease in frequency compared with baseline.
Fig. 3.

Effect of specific receptor inhibitors on GHB-induced respiratory depression. GHB (1500 mg/kg) was administered intravenously, alone and after pretreatment with the GABAB receptor antagonist SCH50911 (150 mg/kg) and the GABAA receptor antagonist bicuculline methiodide (5 mg/kg). Inhibitors were administered intravenously 5 min before GHB. Data are presented as mean ± S.D.; n = 3 to 5.
Potential Treatment Strategies.
Effects of potential treatment strategies on GHB-induced respiratory depression are given in Table 4. Administration of 50 mg/kg SCH50911 5 min after GHB completely reversed the GHB-induced decrease in respiratory rate, as shown in Fig. 4; there was no significant decrease in respiratory rate compared with baseline after the administration of SCH50911. In fact, a slight, but significant, increase in respiratory rate was observed at early time points in SCH50911-treated animals. Lower doses of 2.5, 5, and 10 mg/kg SCH50911 did not completely reverse GHB-induced respiratory depression, and significant decreases in respiratory rate were still observed after antagonist administration. Administration of 10 mg/kg SCH50911 significantly improved all pharmacodynamic parameters, whereas 5 mg/kg improved only the ABEC and Emax and 2.5 mg/kg had no significant effect on any pharmacodynamic parameter compared with that of GHB alone. Administration of 50 mg/kg SCH50911 also increased the nonrenal clearance of GHB, similar to the higher dose of this receptor antagonist, but this effect was not observed at lower doses. Administration of the MCT inhibitor l-lactate significantly increased GHB renal and total clearance and resulted in significant decreases in the frequency ABEC and Td but did not improve the Emax. The combined administration of 5 mg/kg SCH50911 plus l-lactate improved all pharmacodynamic endpoints compared with those for GHB alone. When l-lactate was administered alone at the same dose as that given for GHB, the plasma lactate concentrations obtained with this dose were much lower in the absence of GHB, indicating an effect of GHB on lactate pharmacokinetics. A higher dose of 66 mg/kg plus 605 mg/kg/h l-lactate was then administered alone to achieve lactate concentrations similar to those obtained with GHB administration; ∼1.5 mM mean increases in plasma lactate concentrations were obtained with this higher dose and with the lower dose administered concomitantly with GHB, as shown in Fig. 5. As displayed in Fig. 6, this higher dose of l-lactate had no significant effect on respiratory rate, tidal volume, or minute volume.
TABLE 4.
Effects of potential treatment strategies on the pharmacokinetics/pharmacodynamics of GHB (1500 mg/kg i.v.)
Data are presented as mean (SD); n = 3 to 5. Control = administration of GHB 1500 mg/kg intravenously. SCH50911 and l-lactate were administered intravenously 5 min after GHB. l-Lactate was administered as a 66 mg/kg bolus followed by a 302.5 mg/kg/h infusion for 8 h. One-way analysis of variance followed by Tukey's post hoc test was used to detect statistically significant differences in mean pharmacokinetic and pharmacodynamic parameters.
| Control |
SCH50911 |
l-Lactate | 5 mg/kg SCH50911 + l-Lactate | ||||
|---|---|---|---|---|---|---|---|
| 50 mg/kg | 10 mg/kg | 5 mg/kg | 2.5 mg/kg | ||||
| Cl, ml · kg−1 · min−1 | 5.16 (0.70) | 6.17 (0.41) | 6.13 (0.22) | 6.13 (0.23) | 6.05 (0.69) | 6.40 (0.62)a | 7.61 (0.062)a,b,c |
| ClR, ml · kg−1 · min−1 | 3.18 (0.66) | 3.37 (0.038) | 4.03 (0.21) | 3.78 (0.36) | 3.56 (0.50) | 4.22 (0.63)a | 5.28 (0.42)a,b |
| Clm, ml · kg−1 · min−1 | 1.99 (0.17) | 2.80 (0.52)a | 2.09 (0.37) | 2.35 (0.17) | 2.50 (0.19) | 2.19 (0.55) | 2.33 (0.44) |
| Frequency ABEC, breaths | 10500 (2700) | —d | 3690 (1440)a | 5500 (1440)a | 8720 (513) | 5470 (1550)a | 3170 (957)a |
| Frequency Emax, breaths/min | 17 (7) | — | 51 (3)a | 44 (6)a | 33 (2) | 24 (5) | 45 (6)a |
| Td, h | 4.35 (1.3) | — | 2.50 (0.20)a | 3.15 (0.28) | 4.62 (1.2) | 2.45 (0.62)a | 2.17 (0.14)a |
Significantly different from control (P < 0.05).
Significantly different from 5 mg/kg SCH50911 alone (P < 0.05).
Significantly different from l-lactate alone (P < 0.05).
—, no ABEC, Emax, or Td values could be calculated because respiration is similar to the baseline values; no significant decrease in frequency compared with baseline was observed after administration of 50 mg/kg SCH50911.
Fig. 4.

Effect of potential treatment strategies on respiratory rate after GHB administration. A, dose-dependent effects of SCH50911. B, effects of l-lactate and l-lactate and SCH50911 combination therapy. All treatments were administered intravenously 5 min after GHB (1500 mg/kg i.v.). Data are presented as mean ± S.D.; n = 3 to 5.
Fig. 5.

Plasma lactate concentrations after administration of l-lactate alone and with GHB. Data are presented as mean ± S.D.; n = 4 to 5. l-Lactate low-dose (LD) = 66 mg/kg + 302.5 mg/kg/h. l-Lactate high-dose (HD) = 66 mg/kg + 605 mg/kg/h. l-Lactate LD and HD were administered alone at time 0. l-Lactate LD was administered 5 min after GHB when administered concomitantly.
Fig. 6.
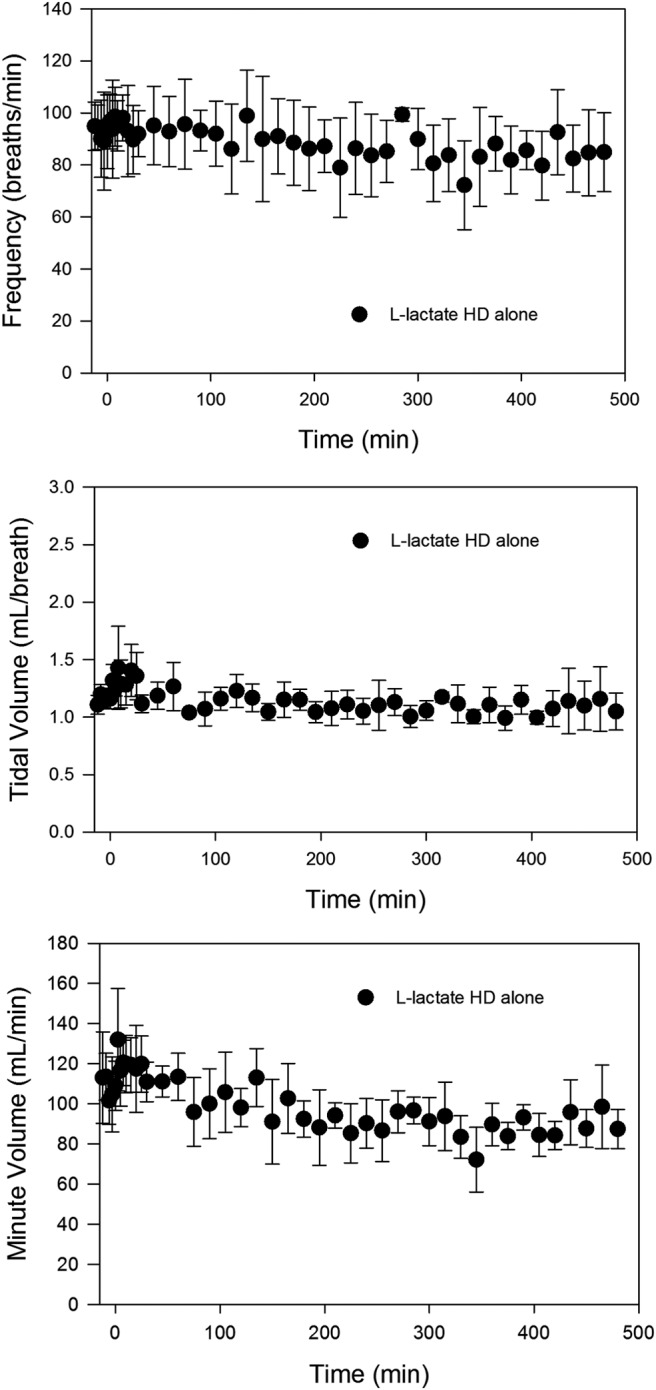
Effect of l-lactate on respiration. Data are presented as mean ± S.D.; n = 4. l-Lactate high dose (HD) = 66 mg/kg + 605 mg/kg/h, administered at time 0.
Discussion
Although abuse of GHB and its precursors has been recognized, no pharmacologic treatment for GHB overdose exists, and current treatment consists only of supportive care. Because of the high rates of respiratory depression reported in both fatal and nonfatal cases of GHB overdose, this pharmacodynamic endpoint serves as a clinically relevant marker for the evaluation of potential GHB overdose treatment strategies. The current data indicate that the primary effect of GHB on respiration is a decrease in respiratory rate, accompanied by a compensatory increase in tidal volume, allowing minute volume to remain constant until doses approach lethality. This respiratory pattern is similar to that noted in some clinical cases of GHB overdose (Mason and Kerns, 2002), indicating that our rat model is relevant for studying this endpoint. When the respiratory rate decreases substantially and tidal volume reaches a physiologic limit, minute volume decreases steeply with increases in GHB concentrations, resulting in fatality. Although we observed no fatalities at doses of 1500 mg/kg i.v. and lower, we observed fatality in approximately 50% of animals administered 1750 mg/kg, consistent with previous reports indicating that the LD50 of GHB administered intravenously in rats is 1700 mg/kg (Laborit, 1964). These results indicate that minute volume is an insensitive measure of GHB intoxication, because there is little change in this measure before death, and the primary parameter of interest in GHB-induced respiratory depression is respiratory rate.
Our data indicate that the decrease in respiratory rate after GHB administration is mediated primarily by action at GABAB receptors, because of complete inhibition of the effect of GHB on this parameter with pretreatment of a GABAB receptor antagonist, SCH50911. This inhibitor also exhibited a surprising effect on GHB pharmacokinetics, significantly increasing its nonrenal clearance. This effect was probably not translated to increased total clearance because nonrenal clearance is a minor route of GHB elimination at 1500 mg/kg but may be responsible for some of the effects of this inhibitor observed on GHB toxicodynamics in studies at lower GHB doses. Although the primary mechanism of GHB-induced respiratory depression was evident from GABAB receptor antagonism, we sought to assess any contribution of GABA to the effects of GHB by administration of bicuculline, a GABAA receptor antagonist. Bicuculline pretreatment did not significantly affect any of the pharmacodynamic descriptors for frequency or tidal volume, compared with GHB alone, indicating a negligible effect of GABA agonism at GABAA receptors on GHB-induced respiratory depression. We did not assess the effects of GHB receptor antagonism in these studies, because previous publications indicated minimal or even protective effects of GHB at this receptor with regard to toxicodynamic endpoints (Carai et al., 2001, 2005). Because of results indicating the role of GABAB receptors in GHB-induced respiratory depression, SCH50911 was also administered after GHB to assess the potential of GABAB inhibition as a treatment strategy for reversing GHB-induced respiratory depression. Similar to other reports (Carai et al., 2001, 2005), complete reversal of this toxicodynamic endpoint required a high dose of this inhibitor (50 mg/kg), although significant effects were observed at doses as low as 5 mg/kg. Previous studies have assessed the effect of SCH50911 alone on respiration, reporting no effect at doses up to 100 mg/kg (Bolser et al., 1995).
Although GABAB antagonists have the capability of completely preventing/reversing GHB-induced respiratory depression, these agents are not currently available for human use. In addition, complete reversal of the toxicological effects of GHB requires large doses of these inhibitors. In contrast, MCT inhibition with l-lactate represents a practical treatment strategy because of its clinical availability (lactated Ringer's solution and Lactate for Injection USP) and demonstrated safety and efficacy for increasing GHB renal clearance in humans at relevant clinical dosages (Morris et al., 2011). The dose of l-lactate administered in this study was given to target a 1 to 2 mM increase in plasma lactate concentrations, the same range targeted in our clinical study. Higher l-lactate regimens were also administered in the current study but with no greater effect. When administered at 5 min after GHB, l-lactate significantly increased GHB total and renal clearances, resulting in significant improvement of the frequency ABEC and Td. A significant decrease in respiratory rate was still observed, however, with no improvement in the Emax. It was hypothesized that further improvement in respiratory rate may be obtained by combination therapy including a low-dose GABAB antagonist and l-lactate, because of the differing mechanisms and partial, but temporally distinct, effects on GHB-induced respiratory depression. We administered SCH50911 at the lowest effective dose of 5 mg/kg with l-lactate, and, as the results of these experiments demonstrate, this combination therapy resulted in significant improvement in all pharmacodynamic parameters, which was not achieved with the same doses of these inhibitors alone. A pertinent effect of l-lactate, with and without SCH50911, is the improvement in the duration of respiratory depression, Td. A need for intubation with mechanical ventilation is frequently reported in cases of overdose of GHB, even when respiratory depression does not result in fatality, and improvement in Td probably indicates improvement in the duration of intubation. This parameter may be of particular concern after oral overdose of GHB and its precursors, because of the prolonged high GHB plasma concentrations observed after oral ingestion of these agents at high doses (Lettieri and Fung, 1978, 1979; Fung et al., 2008). Another interesting effect of the SCH50911-l-lactate combination was increased GHB clearance compared with that for l-lactate alone. Along with the improvement in pharmacodynamic parameters, this increased clearance of GHB may represent an added benefit of combination therapy compared with treatment with either drug as a single agent. Taken together, these data suggest that l-lactate administration is effective for treating GHB-induced respiratory depression, with and without GABAB antagonists. Further studies should include the assessment of treatment strategies in oral GHB overdose and the efficacy of treatments administered at various times after the GHB overdose. Also of importance is the assessment of respiratory depression in GHB overdose when it is concomitantly administered with other drugs of abuse, particularly ethanol and opiates, which themselves can affect respiration.
Because of reports indicating both stimulatory and inhibitory effects of l-lactate on respiration (Gorman et al., 1988; Tappy et al., 1996; Olsson et al., 2002), the dose of l-lactate administered concomitantly with GHB in these studies was administered alone to determine the effects of our targeted lactate plasma concentrations on respiration. Although an effect of l-lactate on GHB pharmacokinetics has been well described (Morris et al., 2005; Wang et al., 2006, 2008a), an interesting observation in this study was an apparent effect of GHB on l-lactate pharmacokinetics. Much lower plasma lactate concentrations were observed when the same dose of l-lactate was administered alone compared with those achieved with GHB coadministration, indicating inhibition of lactate clearance by GHB. Urinary recovery of lactate was increased with GHB administration, compared with that with l-lactate alone, probably as a result of inhibition of renal MCT-mediated reabsorption (0.055 ± 0.015% versus 1.4 ± 0.41% total dose; mean ± S.D., p < 0.01). This effect was not translated in the plasma profiles because of the negligible role of renal clearance in total lactate elimination at this dose. The decrease in lactate clearance, therefore, indicates a decrease in l-lactate metabolism with GHB administration. This effect is not entirely unexpected considering that hepatic lactate metabolism has been demonstrated to be uptake rate-limited (Metcalfe et al., 1986). Administration of β-hydroxybutyrate, an MCT substrate with affinity similar to that of GHB, significantly inhibited lactate uptake and metabolism in hepatocytes and a rat liver perfusion model at a concentration of 10 mM (Metcalfe et al., 1986). Concentrations of GHB in the current studies reach 40 mM with the 1500 mg/kg dose and are therefore probably high enough to significantly inhibit lactate transport and metabolism. Because of the observed pharmacokinetic interaction, a higher dose of l-lactate was administered to achieve the same targeted 1 to 2 mM increase in plasma concentrations. This dose resulted in no significant change in any respiratory parameter, although an insignificant 16% mean decrease in minute volume was observed. The lack of significant effect in our study is probably due to lower plasma lactate concentrations obtained compared with those in other studies that reported effects of l-lactate on respiration and indicates no adverse effects of our targeted concentrations.
In summary, GHB overdose leads to respiratory depression due to a decrease in respiratory rate, and, similar to other toxicological effects of GHB, this effect is mediated primarily by agonism at GABAB receptors. Increasing GHB clearance via MCT inhibition with l-lactate improves GHB-induced respiratory depression, and the clinical availability of this treatment option makes this a practical therapeutic strategy. A novel treatment strategy including combination therapy of a low dose of a GABAB antagonist and l-lactate may represent a safe and effective GHB overdose treatment strategy, pending the availability of GABAB receptor antagonists for clinical use. Further studies are necessary to assess the efficacy of treatment strategies including l-lactate after an oral overdose of GHB and its precursors, alone and with other drugs of abuse.
Acknowledgments
We thank Donna Ruszaj for her assistance in developing the current LC-MS/MS method.
This work was supported by the National Institutes of Health National Institute on Drug Abuse [Grant DA023223] and Pfizer Global Research and Development.
This work was previously presented in part as an abstract: Morse B, Uhlander J, and Morris M. Respiratory depression in γ-hydroxybutyrate overdose: interaction with ethanol and treatment using monocarboxylate transporter inhibition, AAPS Annual Meeting and Exposition; 2011 Oct 23–27; Washington DC. American Association of Pharmaceutical Sciences, Arlington, VA.
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
- GHB
- γ-hydroxybutyrate
- MCT
- monocarboxylate transporter
- SCH50911
- (2S)-(+)-5,5-dimethyl-2-morpholineacetic acid
- LC
- liquid chromatography
- MS/MS
- mass spectrometry
- Emax
- maximum effect
- Td
- duration of effect
- AUC
- area under the plasma concentration-time curve
- ABEC
- area below the effect curve.
Authorship Contributions
Participated in research design: Morse and Morris.
Conducted experiments: Morse.
Contributed new reagents or analytic tools: Morse, Vijay, and Morris.
Performed data analysis: Morse.
Wrote or contributed to the writing of the manuscript: Morse, Vijay, and Morris.
References
- Bernasconi R, Lauber J, Marescaux C, Vergnes M, Martin P, Rubio V, Leonhardt T, Reymann N, Bittiger H. (1992) Experimental absence seizures: potential role of γ-hydroxybutyric acid and GABAB receptors. J Neural Transm Suppl 35:155–177 [DOI] [PubMed] [Google Scholar]
- Bolser DC, Blythin DJ, Chapman RW, Egan RW, Hey JA, Rizzo C, Kuo SC, Kreutner W. (1995) The pharmacology of SCH 50911: a novel, orally-active GABA-β receptor antagonist. J Pharmacol Exp Ther 274:1393–1398 [PubMed] [Google Scholar]
- Brenneisen R, Elsohly MA, Murphy TP, Passarelli J, Russmann S, Salamone SJ, Watson DE. (2004) Pharmacokinetics and excretion of γ-hydroxybutyrate (GHB) in healthy subjects. J Anal Toxicol 28:625–630 [DOI] [PubMed] [Google Scholar]
- Caldicott DG, Chow FY, Burns BJ, Felgate PD, Byard RW. (2004) Fatalities associated with the use of γ-hydroxybutyrate and its analogues in Australasia. Med J Aust 181:310–313 [DOI] [PubMed] [Google Scholar]
- Carai MA, Colombo G, Brunetti G, Melis S, Serra S, Vacca G, Mastinu S, Pistuddi AM, Solinas C, Cignarella G, et al. (2001) Role of GABAB receptors in the sedative/hypnotic effect of γ-hydroxybutyric acid. Eur J Pharmacol 428:315–321 [DOI] [PubMed] [Google Scholar]
- Carai MA, Colombo G, Gessa GL. (2005) Resuscitative effect of a γ-aminobutyric acid B receptor antagonist on γ-hydroxybutyric acid mortality in mice. Ann Emerg Med 45:614–619 [DOI] [PubMed] [Google Scholar]
- Felmlee MA, Krzyzanski W, Morse BL, Morris ME. (2011) Use of a local sensitivity analysis to inform study design based on a mechanistic toxicokinetic model for γ-hydroxybutyric acid. AAPS J 13:240–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felmlee MA, Roiko SA, Morse BL, Morris ME. (2010a) Concentration-effect relationships for the drug of abuse gamma-hydroxybutyric acid. J Pharmacol Exp Ther 333:764–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felmlee MA, Wang Q, Cui D, Roiko SA, Morris ME. (2010b) Mechanistic toxicokinetic model for γ-hydroxybutyric acid: inhibition of active renal reabsorption as a potential therapeutic strategy. AAPS J 12:407–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung HL, Haas E, Raybon J, Xu J, Fung SM. (2004) Liquid chromatographic-mass spectrometric determination of endogenous gamma-hydroxybutyrate concentrations in rat brain regions and plasma. J Chromatogr B Analyt Technol Biomed Life Sci 807:287–291 [DOI] [PubMed] [Google Scholar]
- Fung HL, Tsou PS, Bulitta JB, Tran DC, Page NA, Soda D, Mi Fung S. (2008) Pharmacokinetics of 1,4-butanediol in rats: bioactivation to γ-hydroxybutyric acid, interaction with ethanol, and oral bioavailability. AAPS J 10:56–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galicia M, Nogue S, Miró O. (2011) Liquid ecstasy intoxication: clinical features of 505 consecutive emergency department patients. Emerg Med J 28:462–466 [DOI] [PubMed] [Google Scholar]
- Gobaille S, Hechler V, Andriamampandry C, Kemmel V, Maitre M. (1999) γ-Hydroxybutyrate modulates synthesis and extracellular concentration of gamma-aminobutyric acid in discrete rat brain regions in vivo. J Pharmacol Exp Ther 290:303–309 [PubMed] [Google Scholar]
- Gorman JM, Raymond RG, Uy J, Ross D, Martinez J, Fyer AJ, Liebowitz MR, Klein DF. (1988) Hyperventilation occurs during lactate-induced panic. J Anxiety Disord 2:193–202 [Google Scholar]
- Hechler V, Ratomponirina C, Maitre M. (1997) γ-Hydroxybutyrate conversion into GABA induces displacement of GABAB binding that is blocked by valproate and ethosuximide. J Pharmacol Exp Ther 281:753–760 [PubMed] [Google Scholar]
- Kaupmann K, Cryan JF, Wellendorph P, Mombereau C, Sansig G, Klebs K, Schmutz M, Froestl W, van der Putten H, Mosbacher J, et al. (2003) Specific γ-hydroxybutyrate-binding sites but loss of pharmacological effects of γ-hydroxybutyrate in GABAB1-deficient mice. Eur J Neurosci 18:2722–2730 [DOI] [PubMed] [Google Scholar]
- Laborit H. (1964) Sodium 4-hydroxybutyrate. Int J Neuropharmacol 3:433–451 [DOI] [PubMed] [Google Scholar]
- Lettieri J, Fung HL. (1976) Absorption and first-pass metabolism of 14C-γ-hydroxybutyric acid. Res Commun Chem Pathol Pharmacol 13:425–437 [PubMed] [Google Scholar]
- Lettieri J, Fung HL. (1978) Improved pharmacological activity via pro-drug modification: comparative pharmacokinetics of sodium γ-hydroxybutyrate and γ-butyrolactone. Res Commun Chem Pathol Pharmacol 22:107–118 [PubMed] [Google Scholar]
- Lettieri JT, Fung HL. (1979) Dose-dependent pharmacokinetics and hypnotic effects of sodium γ-hydroxybutyrate in the rat. J Pharmacol Exp Ther 208:7–11 [PubMed] [Google Scholar]
- Li J, Stokes SA, Woeckener A. (1998) A tale of novel intoxication: seven cases of γ-hydroxybutyric acid overdose. Ann Emerg Med 31:723–728 [DOI] [PubMed] [Google Scholar]
- Liechti ME, Kupferschmidt H. (2004) γ-Hydroxybutyrate (GHB) and γ-butyrolactone (GBL): analysis of overdose cases reported to the Swiss Toxicological Information Centre. Swiss Med Wkly 134:534–537 [DOI] [PubMed] [Google Scholar]
- Maitre M. (1997) The γ-hydroxybutyrate signalling system in brain: organization and functional implications. Prog Neurobiol 51:337–361 [DOI] [PubMed] [Google Scholar]
- Mason PE, Kerns WP., 2nd (2002) γ Hydroxybutyric acid (GHB) intoxication. Acad Emerg Med 9:730–739 [DOI] [PubMed] [Google Scholar]
- Metcalfe HK, Monson JP, Welch SG, Cohen RD. (1986) Inhibition of lactate removal by ketone bodies in rat liver. Evidence for a quantitatively important role of the plasma membrane lactate transporter in lactate metabolism. J Clin Invest 78:743–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris ME, Hu K, Wang Q. (2005) Renal clearance of γ-hydroxybutyric acid in rats: increasing renal elimination as a detoxification strategy. J Pharmacol Exp Ther 313:1194–1202 [DOI] [PubMed] [Google Scholar]
- Morris ME, Morse BL, Baciewicz GJ, Tessena MM, Acquisto NM, Hutchinson DJ, DiCenzo R. (2011) Monocarboxylate transporter inhibition with osmotic diuresis increases γ-hydroxybutyrate renal elimination in humans: a proof-of-concept study. J Clinic Toxicol 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson M, Ho HP, Annerbrink K, Thylefors J, Eriksson E. (2002) Respiratory responses to intravenous infusion of sodium lactate in male and female Wistar rats. Neuropsychopharmacology 27:85–91 [DOI] [PubMed] [Google Scholar]
- Palatini P, Tedeschi L, Frison G, Padrini R, Zordan R, Orlando R, Gallimberti L, Gessa GL, Ferrara SD. (1993) Dose-dependent absorption and elimination of γ-hydroxybutyric acid in healthy volunteers. Eur J Clin Pharmacol 45:353–356 [DOI] [PubMed] [Google Scholar]
- Sporer KA, Chin RL, Dyer JE, Lamb R. (2003) γ-Hydroxybutyrate serum levels and clinical syndrome after severe overdose. Ann Emerg Med 42:3–8 [DOI] [PubMed] [Google Scholar]
- Substance Abuse and Mental Health Services Administration (2011) Drug Abuse Warning Network, 2008: National Estimates of Drug-Related Emergency Department Visits. HHS Publ. No. SMA 11-4618 Substance Abuse and Mental Health Services Administration, Center for Behavioral Health Statistics and Quality, Rockville, MD [Google Scholar]
- Tappy L, Cayeux MC, Chioléro R. (1996) Effects of sodium lactate on ventilation and acid-base balance in healthy humans. Clin Physiol 16:393–401 [DOI] [PubMed] [Google Scholar]
- Wang Q, Darling IM, Morris ME. (2006) Transport of γ-hydroxybutyrate in rat kidney membrane vesicles: role of monocarboxylate transporters. J Pharmacol Exp Ther 318:751–761 [DOI] [PubMed] [Google Scholar]
- Wang Q, Morris ME. (2007) The role of monocarboxylate transporter 2 and 4 in the transport of γ-hydroxybutyric acid in mammalian cells. Drug Metab Dispos 35:1393–1399 [DOI] [PubMed] [Google Scholar]
- Wang Q, Wang X, Morris ME. (2008a) Effects of l-lactate and d-mannitol on γ-hydroxybutyrate toxicokinetics and toxicodynamics in rats. Drug Metab Dispos 36:2244–2251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang Q, Morris ME. (2008b) Pharmacokinetic interaction between the flavonoid luteolin and γ-hydroxybutyrate in rats: potential involvement of monocarboxylate transporters. AAPS J 10:47–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zvosec DL, Smith SW, Porrata T, Strobl AQ, Dyer JE. (2011) Case series of 226 γ-hydroxybutyrate-associated deaths: lethal toxicity and trauma. Am J Emerg Med 29:319–332 [DOI] [PubMed] [Google Scholar]