

Abstract
AIM: To investigate the role of polo-like kinase 1 (PLK1) as a therapeutic target for hepatocellular carcinoma (HCC).
METHODS: PLK1 gene expression was evaluated in HCC tissue and HCC cell lines. Gene knockdown with short-interfering RNA (siRNA) was used to study PLK1 gene and protein expression using real-time reverse transcription polymerase chain reaction (RT-PCR) and Western blotting, and cell proliferation using 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2(4-sulfophenyl)-2H-tetrazolium (MTS) and bromodeoxyuridine (BrdU) assays. Apoptosis was evaluated using the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay, and caspase-inhibition assay. Huh-7 cells were transplanted into nude mice and co-cultured with PLK1 siRNA or control siRNA, and tumor progression was compared with controls.
RESULTS: RT-PCR showed that PLK1 was overexpressed 12-fold in tumor samples compared with controls, and also was overexpressed in Huh-7 cells. siRNA against PLK1 showed a reduction in PLK1 gene and protein expression of up to 96% in Huh-7 cells, and a reduction in cell proliferation by 68% and 92% in MTS and BrdU cell proliferation assays, respectively. There was a 3-fold increase in apoptosis events, and TUNEL staining and caspase-3 assays suggested that this was caspase-independent. The pan-caspase inhibitor Z-VAD-FMK was unable to rescue the apoptotic cells. Immnofluorescence co-localized endonuclease-G to fragmented chromosomes, implicating it in apoptosis. Huh-7 cells transplanted subcutaneously into nude mice showed tumor regression in siPLK1-treated mice, but not in controls.
CONCLUSION: Knockdown of PLK1 overexpression in HCC was shown to be a potential therapeutic target, leading to apoptosis through the endonuclease-G pathway.
Keywords: RNA, Polo-like kinase 1, Apoptosis, Endonuclease G, Forkhead box transcription factors, Nude mice
INTRODUCTION
Hepatocellular carcinoma (HCC) is the fifth most common solid tumor cancer in the world, and is ranked third in terms of mortality according to GLOBALCAN 2002[1]. Treatments for HCC other than liver transplantation, can benefit patients but are often plagued by recurrence of the tumor[2,3]. Those with advanced HCC have a poor prognosis. Recently, vascular endothelial growth factor inhibitors showed a survival benefit[4], albeit of only 3 mo, in the treatment of HCC. Therefore, this finding and the paucity of effective treatment options is a strong indicator for the investigation of new genomic targets that can provide better treatment outcomes for HCC patients. One genomic target that has been reported in many other tumors but only recently in HCC is polo-like kinase 1 (PLK1)[5-8].
PLK1 is a cell cycle protein that plays multiple roles in promoting cell cycle progression[9]. However, the most prominent role of PLK1 probably lies in regulating the spindle checkpoint at the M-phase[10]. Mutated PLK1 alleles induce abrupt spindle formation resulting in polyploid cells[11], and in mammalian cells depleted of PLK1, mitotic arrest with dysfunctional spindle assembly occurs[10]. PLK1 is a highly conserved protein that has unique polo-box binding domains that can bind to phosphopeptide and render it to be regulated spatially and temporally during the cell cycle by proteins that carry the optimal phosphopeptide motif[12].
PLK1 is overexpressed in many cancers and serves as a significant prognostic factor in cancers such as small-cell lung cancer, colon cancer and ovarian cancer[13]. In addition, high expression levels of PLK1 in melanoma and breast cancer correlate well with the metastatic potential of these tumors[14,15]. PLK1 overexpression may contribute to the deregulation of cell proliferation during oncogenesis by overcoming mitotic checkpoints[16]. In this study, we sought to determine if PLK1 was a potential therapeutic target for HCC.
MATERIALS AND METHODS
Patients and samples
Tumor and surrounding non-tumor liver tissues were obtained from 56 primary HCC patients who underwent liver resection in the National University Hospital from 1998 to 2002. The study was approved by the National University of Singapore Institutional Review Board, and National Healthcare Group Domain Specific Review Board. Informed consent was obtained from every patient for the use of excised tissue. The human hepatoma cell line HepG2 was obtained from American Type Culture Collection, Huh-7 cells from the Japanese Collection of Research Bioresources Cell Bank and HepG2.2.15 was kindly provided by Dr. Acs (Mt. Sinai Medical College, New York, NY, United States). HepG2 and Huh-7 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 0.1 mmol/L sodium pyruvate, 0.1 mmol/L L-glutamine, 0.1 mmol/L non-essential amino acids, 10% fetal calf serum, and 1X antibiotic-antimycotic solution (Invitrogen, Carlsbad, CA, United States). HepG2.2.15 cells were cultured in similar media with the addition of 0.4 mg/mL G418 (Sigma-Aldrich, St Louis, MO, United States). All cell lines were maintained in an incubator with 5% CO2 at 37 °C.
RNA extraction and cDNA synthesis
Total RNA was extracted from cell lines or patient tissues using RNeasy Mini kits (Qiagen, Hilden, Germany) or Trizol (Invitrogen), respectively according to the manufacturer’s protocol. Reverse transcription was carried out in 26 μL aliquots of solution containing 5 μg of total RNA, 2.5 ng Oligo d(T)18 and RNase-free water, which was then incubated for 15 min at 72 °C in a thermal cycler. After 10 min of cooling at 4 °C, 24 μL of the RT-enzyme mixture containing 5 μL 10 mmol/L dNTPs, 5 μL 0.1 mol/L dithiothreitol, 10 μL 5 × first-strand buffer, 2 μL SuperScript II reverse transcriptase (Invitrogen) and 2 μL recombinant RNasin ribonuclease inhibitor (Promega, Madison, WI, United States) was added to obtain a final volume of 50 μL. The mixture was then incubated for 90 min at 42 °C followed by 15 min incubation at 72 °C, and was subsequently cooled to 10 °C.
Real-time quantitative reverse transcription polymerase chain reaction
Primers and probes for PLK1 and FOXM1 were from TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA, United States). In brief, 20 μL reaction was set up containing 4 μL cDNA, 10 μL 2 × TaqMan Universal PCR Master Mix (Applied Biosystems), 1 μL 20 × TaqMan Gene Expression Assay Mix of corresponding target gene, 1 μL 20 × human hypoxanthine-guanine phosphoribosyltransferase endogenous control (Applied Biosystems)[17] and 4 μL RNase-free water. The reaction was run on the ABI Prism 7000 Sequence Detection System (Applied Biosystems) with the following profile: 1 cycle at 50 °C for 2 min, 1 cycle at 95 °C for 10 min, then 40 cycles at 95 °C for 15 s and at 60 °C for 1 min. No-template control and TaqMan Control Total RNA (Human) (Applied Biosystems) as reference control were also included where applicable. Relative quantification was done using the 2-∆∆Ct method.
Short interfering RNA transfection
Short interfering RNA (siRNA) targeting human PLK1 (si-PLK1) and FOXM1 were from siGENOME On-TargetPlus Set of 4 duplex (Dharmacon, Chicago, IL, United States) and On-Target plus siCONTROL Non-targeting pool (si-NT) (Dharmacon) was used as negative control. The mixture containing 4 siRNA duplexes was transfected into cells using Lipofectamine RNAiMAX Transfection Reagent (Invitrogen) according to the manufacturer’s protocol. No-treatment and transfection-reagent-only controls were also included where suitable. The transfection period was 48 h for all assays or tests unless otherwise specified.
Cell proliferation assays
The 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2(4-sulfophenyl)-2H-tetrazolium (MTS) assay was performed using the CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay (Promega) according to the manufacturer’s protocol. The BrdU (5-bromo-2-deoxyuridine) assay was performed using Cell Proliferation enzyme-linked immunosorbent assay (ELISA), BrdU (chemiluminescent) (Roche Applied Science, Indianapolis, IN, United States) according to the manufacturer’s protocol.
Western blot analysis
Cells were lysed using Complete Lysis-M (Roche Applied Science) according to the manufacturer’s protocol. The protein concentration was determined using BCA Protein Assay Kit (Pierce, Rockford, IL, United States). Protein (30 μg) was mixed 1:1 with Laemmli sample buffer (Bio-Rad Laboratories, Hercules, CA, United States) supplemented with 5% β-mercaptoethanol and heated at 95 °C for 5 min followed by 5 min incubation on ice. The sample was then loaded onto a 10% Tris-HCl Ready Gel (Bio-Rad) and subsequently electrotransferred to a Hybond-P polyvinylidene difluoride membrane (Amersham Pharmacia Biotech, Buckinghamshire, United Kingdom). The membrane was blocked for an hour at 22 °C with 5% Blotting-Grade Blocker and non-fat dry milk (Bio-Rad). After blocking, the membrane was incubated with mouse anti-human Plk1 (F-8) antibody (Santa Cruz Biotechnology, Santa Cruz, CA, United States) at 1:500 for 1 h at 22 °C followed by incubation with Cruz Marker Compatible goat anti-mouse IgG antibody (Santa Cruz) at 1:50 000 for 1 h at 22 °C. The reaction was detected using the ECL plus system (Amersham) and developed using Hyperfilm ECL (Amersham). Mouse anti-human β-actin (C4) antibody (Santa Cruz) diluted at 1:10 000 was used as loading control. The Blots were analyzed with the Quantity One software (Bio-Rad).
Immunofluorescence imaging
Cells were cultured on Lab-Tek II 2-chambered glass slides (Nunc, Rochester, NY, United States). Twenty 4 h after siRNA transfection, the cells were fixed in 4% buffered paraformaldehyde for 30 min at 4 °C. The cells were then permeabilized with 0.5% Triton X-100 in phosphate-buffered solution for 10 min and subsequently blocked in 2% bovine serum albumin for 10 min. The cells were incubated with primary antibody diluted at 1:100 for 1 h at 22 °C followed by an hour’s incubation at 22 °C with fluorescein isothiocyanate (FITC)-conjugated secondary antibody diluted at 1:400. The cells were subsequently mounted using Vectashield with 4’,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA, United States) for nuclei staining. Primary antibodies used were FITC-conjugated mouse anti-α-tubulin antibody (clone DM1A) (Sigma-Aldrich), goat polyclonal anti-human endonuclease G (Santa Cruz) and goat polyclonal anti-human apoptosis-inducing factor (Santa Cruz). The secondary antibody used was donkey anti-goat IgG-FITC (Santa Cruz). Fluorescence images were captured using Olympus Fluoview FV500 or BX60F5 (Olympus, Center Valley, PA, United States).
Chromosome fragmentation detection assay, terminal deoxynucleotidyl transferase dUTP nick end labeling assay and caspase-3 activity assay
Chromosome fragmentation was detected using Cell Death Detection ELISAPLUS (Roche) according to the manufacturer’s protocol. Thapsigargin (Sigma-Aldrich) treatment was used as positive control. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was carried out after 24 h of transfection using the DeadEnd Fluorometric TUNEL system (Promega) according to the manufacturer’s protocol. Fluorescence images were captured using Olympus BX60F5 (Olympus). The caspase-3 activity assay (Roche) was performed according to the manufacturer’s protocol.
Caspase inhibition assay
Cells were transfected with siRNA in the presence of cell permeable pan-caspase inhibitor Z-VAD-FMK (Sigma-Aldrich) at 100 μmol/L for 48 h. Cell proliferation was subsequently determined with MTS assay. Camptothecin (Sigma-Aldrich) at 5 μmol/L was used to induce apoptosis in HepG2 cells. For camptothecin treatment, HepG2 cells were incubated with the caspase inhibitor for an hour before camptothecin was added and remained till the end of incubation. An equal amount of DMSO was used in the negative control where appropriate.
In vivo nude mice experiment
Nude mice were transplanted subcutaneously with 2 × 106 Huh-7 cells in 100 μL Matrigel (Sigma-Aldrich). The treatment was started 1 wk after the transplantation. Eighteen tumor-bearing nude mice were divided equally into a group (n = 6) that received si-PLK1 treatment, a group that received si-NT treatment (n = 6), and a control group that received no treatment (n = 6). Treatment groups received intratumoral injections of 1 nmol siRNA coupled with Lipofectamine RNAiMAX Transfection Reagent (Invitrogen) every alternate day. The control group was injected with saline instead. Tumor sizes recorded before treatment were calculated by the formula: volume (mm3) = (width)2 × length/2. Animal experiments were carried out in compliance with the guidelines of the Laboratory Animals Centre, National University of Singapore and were approved by the National University of Singapore Institutional Animal Care and Use Committee.
Statistical analysis
Statistical analysis was carried out using Microsoft Excel or SPSS. P values of less than 0.05 were deemed significant. All data was expressed as mean ± SE unless otherwise specified.
RESULTS
Baseline characteristics of the patients
The patients, 49 male, 7 female, age range 32-82 years (mean, 56 years), were hepatitis B-positive and were Asian (Table 1).
Table 1.
Correlation between polo-like kinase-1 gene expression and clinicopathological parameters in 56 patients with hepatocellular carcinoma
| Clinicopathological parameters | n | Relative PLK1 gene expression (mean ± SE) | P value |
| Age (yr) | |||
| < 65 | 42 | 18.35 ± 2.95 | 0.5701 |
| ≥ 65 | 14 | 16.15 ± 5.11 | |
| Sex | |||
| Male | 49 | 17.24 ± 2.74 | 0.4361 |
| Female | 7 | 21.72 ± 6.83 | |
| Histologic type of tumor | |||
| Well differentiated | 2 | 14.00 ± 13.10 | 0.8462 |
| Moderately differentiated | 41 | 17.39 ± 2.92 | |
| Poorly differentiated | 13 | 19.69 ± 5.87 | |
| Liver cirrhosis | |||
| Present | 44 | 16.05 ± 2.42 | 0.4971 |
| Absent | 12 | 24.22 ± 7.83 | |
| Pathological stage | |||
| I | 1 | 5.68 | 0.8492 |
| II | 5 | 14.11 ± 6.67 | |
| III | 32 | 17.68 ± 2.93 | |
| IV | 18 | 19.72 ± 5.74 |
P value was determined by the Mann-Whitney U test;
P value was determined by the Kruskal-Wallis test. PLK1 gene expression in every hepatocellular carcinoma tissues was quantified relative to the non-tumor tissues counterparts using 2-∆∆Ct method. PLK1: Polo-like kinase-1.
PLK1 gene expression in HCC patients and correlation with clinicopathological parameters
Gene expression of 10 candidate genes (PLK1, forkhead box M1, pituitary tumor-transforming gene 1, ubiquitin specific peptidase 21, reticulocalbin 2, dual specificity phosphatase 12, ubiquitin specific peptidase 1, S100 calcium binding protein P, ubiquitin specific peptidase 5, X-box binding protein 1) indentified from a literature survey of genes shown to be upregulated in tumors but not well documented in HCC, were chosen for real-time quantitative reverse transcription polymerase chain reaction (RT-PCR) analysis in 10 sets of paired HCC tumor/adjacent non-tumor tissue (data not shown). PLK1 was found to be consistently upregulated in HCC tumor tissue. Gene expression of PLK1 was then investigated in 46 other patient samples (a total of 56 HCC tumors) using real-time quantitative RT-PCR. The median PLK1 gene expression was about 12 times higher in 50% of the HCC tumors when compared with non-tumor tissues (Figure 1A).
Figure 1.
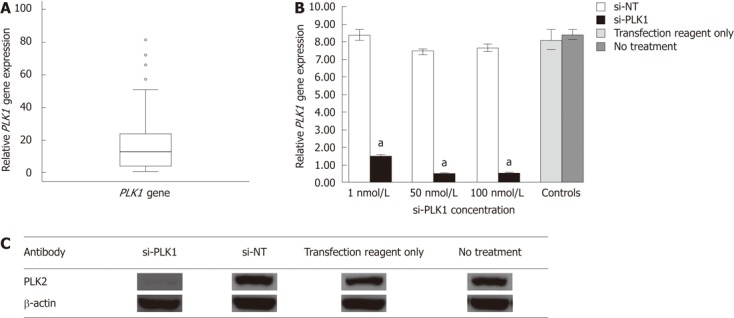
Upregulation of polo-like kinase 1 gene expression in 56 hepatocellular carcinoma tumors, efficiency of short-interfering RNA in silencing the polo-like kinase 1 gene, and protein expression in Huh-7 cells. A: Boxplot showing the minimum, 25th percentile, median, 75th percentile and maximum relative polo-like kinase 1 (PLK1) gene expression. Circles represent statistical outliers. PLK1 gene expression in all hepatocellular carcinoma (HCC) tissue was quantified relative to the non-tumor tissue counterpart using the 2-∆∆Ct method; B: Knockdown with short interfering PLK1 (si-PLK1) at 1 nmol/L, 50 nmol/L and 100 nmol/L successfully silenced PLK1 gene expression (using the 2-∆∆Ct method by quantitative real-time RT-PCR) by 83%, 95% and 96% respectively, compared with short interfering non-targeting (si-NT), or controls. Data shown as mean ± SE, using the Student t-test (aP < 0.05); C: Western blotting showing reduction of PLK1 protein expression in Huh-7 cells, at si-PLK1 and si-NT (non-targeting si-RNA) concentrations of 50 nmol/L compared with si-NT or controls.
PLK1 siRNA successfully silenced PLK1 gene expression in Huh-7 cells
PLK1 gene expression in Huh-7 cell-line was about eight times higher than other human hepatoma cell lines (HepG2 and HepG2.2.15) as determined by real-time quantitative RT-PCR (data not shown). Hence, it was selected as in vitro model to study the effect of silencing PLK1 gene expression. PLK1 knockdown with 1 nmol/L, 50 nmol/L and 100 nmol/L si-PLK1 was able to silence of PLK1 gene expression by 83%, 95% and 96%, respectively, compared with the Huh-7 cells transfected with an equal concentration of si-NT (Figure 1B). The reduction in PLK1 gene expression by si-PLK1 corresponded to the reduction observed in PLK1 protein expression level. Using 50 nmol/L si-PLK1, PLK1 protein expression was reduced by 95% when compared with the si-NT transfected Huh-7 cells (Figure 1C). Therefore, si-PLK1 was shown to be efficient and specific in silencing PLK1 gene and protein expression in Huh-7 cells.
Silencing of PLK1 reduced cell proliferation in Huh-7 cells
Transfection of si-PLK1 caused a significant reduction in Huh-7 cells proliferation as measured by the MTS cell proliferation assay (Figure 2A) and BrdU cell proliferation assay (Figure 2B), but with no apparent dose-response. On average, si-PLK1-treated Huh-7 cells showed 68% and 92% reductions in cell proliferation in MTS and BrdU cell proliferation assays, respectively. In addition, Huh-7 cells that were transfected with si-PLK1 appeared to be binucleated (Figure 2C, left panel) while Huh-7 cells transfected with si-NT completed mitosis with functional spindle assembly (Figure 2C, right panel), indicative of its role in establishing functional spindle assembly.
Figure 2.
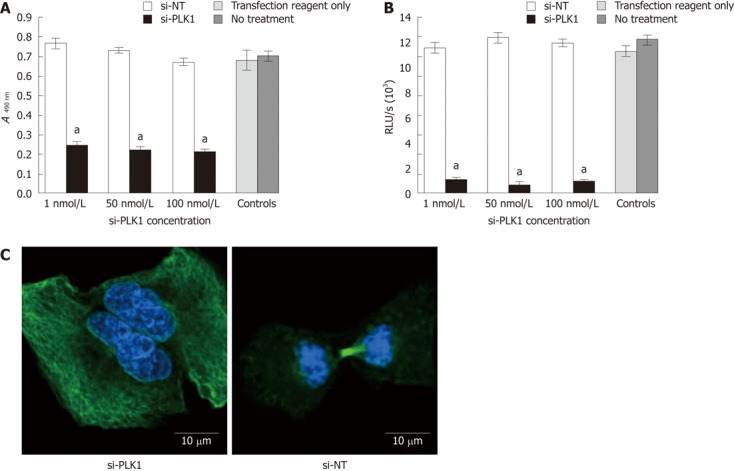
Reduction of cell proliferation by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2(4-sulfophenyl)-2H-tetrazolium assay and bromodeoxyuridine assay after silencing of polo-like kinase 1, and failure of mitosis after knockdown of polo-like kinase 1. A: Knockdown of polo-like kinase 1 (PLK1) reduced cell proliferation in Huh-7 cells in the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2(4-sulfophenyl)-2H-tetrazolium (MTS) cell proliferation assay by a mean of 65% compared with short interfering non-targeting (si-NT) or controls; B: Knockdown of PLK1 reduced cell proliferation in the bromodeoxyuridine cell proliferation assay in Huh7 cells by a mean of 93% with 50 nmol/L short-interfering PLK1 (si-PLK1) compared with si-NT or controls; C: Confocal fluorescence images show the si-PLK1 transfected Huh-7 cells (left panel) were binucleated, depicting failure in completing mitosis due to the lack of a functional spindle assembly. The right panel shows a functional spindle assembly in Huh-7 cells transfected with si-NT. Huh-7 cells were processed for confocal imaging after 24 h of transfection either with 50 nmol/L si-PLK1 or si-NT; α-tubulins were stained with fluorescein isothiocyanate-conjugated antibody and nuclei were counterstained with 4',6-diamidino-2-phenylindole. Data are shown as mean ± SE, using the Student t-test (aP < 0.05).
Silencing of PLK1-induced apoptosis in Huh-7 cells
Nuclear fragmentation expressed as the enrichment factor (sample absorbance/absorbance of the non-transfected control) > 1, indicates enrichment of mono- and oligo-nucleosomes in the cytoplasm of the apoptotic cells due to DNA breakdown. The enrichment factor in Huh-7 cells that were transfected with si-PLK1 was 3-fold higher than in the Huh-7 cells transfected with si-NT (Figure 3A). In addition, TUNEL staining of si-PLK1-transfected Huh-7 cells helped to identify and visualize apoptotic cells with fragmented chromosomes (Figure 3B). To examine the apoptosis pathway, caspase-3 activity was analyzed in si-PLK1 transfected Huh-7 cells at three different time points (Figure 3C). There was activation of caspase-3 to over 500 U (compared with 2350 U in positive controls), but this was only detected at 48 h after transfection (Figure 3C). This was an unexpected finding as the TUNEL assay had already showed prominent nuclear fragmentation in apoptotic cells 24 h after transfection, and suggests that the apoptosis pathway induced by PLK1 knockdown in Huh-7 cells may be caspase-independent.
Figure 3.
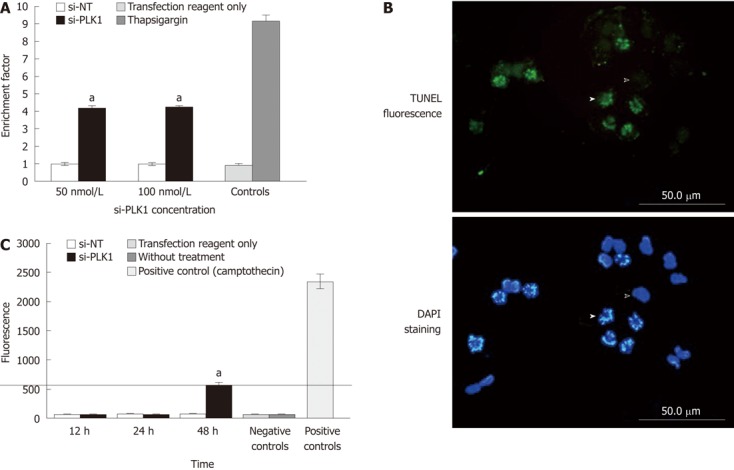
Increased apoptosis, apoptosis by terminal deoxynucleotidyl transferase dUTP nick end labeling staining and caspase 3 activity after knockdown of polo-like kinase 1. A: Increased apoptosis; apoptosis measured by enrichment factor [enrichment factor (sample absorbance/control absorbance) > 1 indicates nuclear fragmentation]. Huh-7 cells transfected with short-interfering polo-like kinase 1 (si-PLK1) showed a 4-fold increase in enrichment factor compared with short-interfering non-targeting (si-NT) or the negative control, no difference was seen between the 50 nmol/L and 100 nmol/L si-PLK1. Thapsigargin (5 μmol/L) treated Huh-7 cells were positive controls. Data are shown as mean ± SE, using the Student t-test (aP < 0.05); B: Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) fluorescence images showing fragmented DNA labeled with fluorescein-12-dUTP using recombinant terminal deoxynucleotidyl transferase (upper image). The lower image is the corresponding 4’,6-diamidino-2-phenylindole (DAPI) stain showing an apoptotic Huh-7 cell (solid arrowhead) and a non-apoptotic Huh-7 cell (hollow arrowhead). The TUNEL staining colocalized the fragmented chromosomes to apoptotic cells while is absent in non-apoptotic cells with intact chromosomes. Huh-7 cells were processed for fluorescence imaging after 24 h of transfection with 50 nmol/L si-PLK1; C: Caspase 3 activity was measured using a fluorometric immunosorbent enzyme assay from Huh-7 cells transfected with 50 nmol/L si-PLK1 showed no change after 12 and 24 h, with an increase to just over 500 U at 48 h compared with si-NT and negative controls. Positive controls showed values over 2350 U (cell lysates from U937 cells treated with camptothecin, supplied with the kit). Data are shown as mean ± SE, using the Student t-test (aP < 0.05).
Apoptosis induced by silencing PLK1 gene expression is caspase-independent
A cell permeable pan-caspase inhibitor Z-VAD-FMK was applied to validate the requirement of caspase in apoptosis induced by silencing PLK1 gene expression in Huh-7 cells. By using the MTS cell proliferation assay as the functional end-point indicator for apoptosis, the pan-caspase inhibitor was unable to rescue the reduction in cell proliferation of Huh-7 cells transfected with si-PLK1 (Figure 4A). The finding clearly demonstrates that apoptosis in si-PLK1-transfected Huh-7 cells was caspase-independent. Two exclusively mitochondrial pro-apoptotic proteins, endonuclease G (EndoG) and apoptosis-inducing factor (AIF), have been identified to be involved in the caspase-independent apoptosis pathway[18]. In Figure 4B, EndoG was released from the mitochondria and colocalized in the fragmented chromosomes in apoptotic Huh-7 cells, indicating that EndoG was likely to be the main caspase-independent apoptotic effector. AIF was found to be absent and therefore was probably not involved in this particular apoptosis event (Figure 4B).
Figure 4.
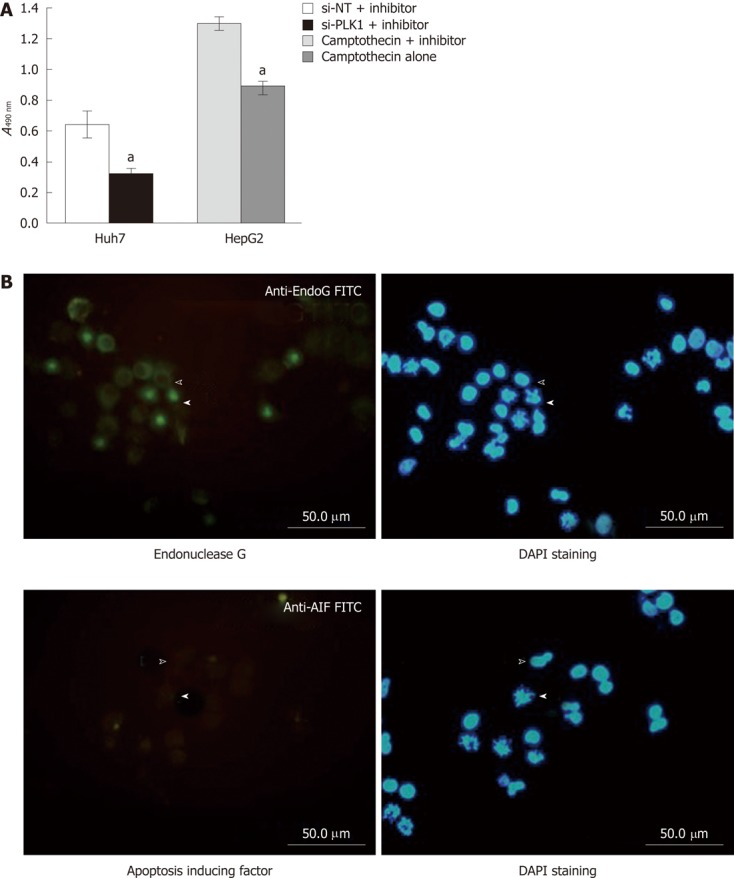
Lack of apoptosis protection and caspase-independent apoptosis. A: Lack of apoptosis protection by pan-caspase inhibitor Z-VAD-FML after knockdown of polo-like kinase 1 (PLK1). The MTS cell proliferation assay in Huh-7 cells transfected with either short interfering non-targeting (si-NT) or short interfering-PLK1 (si-PLK1) in the presence of the pan caspase inhibitor Z-VAD-FMK showed that the pan caspase inhibitor failed to protect si-PLK1 transfected Huh-7 cells from apoptosis. In contrast, HepG2 that were treated with camptothecin and Z-VAD-FMK were protected from apoptosis induced by camptothecin while those treated with camptothecin without Z-VAD-FMK were not protected from apoptosis. Data are shown as mean ± SE, using the Student t-test (aP < 0.05); B: Caspase-independent apoptosis likely due to endonuclease G. Huh-7 transfected with 50 nmol/L si-PLK1 for 24 h was probed with antibody against endonuclease G or apoptosis-inducing factor (AIF) and subsequently visualized with fluorescein isothiocyanate-conjugated secondary antibody (left panels). The corresponding 4’,6-diamidino-2-phenylindole (DAPI)-stained images are shown in the right panels. Block arrowheads and hollow arrowheads mark apoptotic and non-apoptotic Huh-7 cells respectively. Endonuclease G was positively stained in apoptotic cells and colocalized to the fragmented chromosomes (upper left panel), while AIF was found to be absent (lower left panel).
Targeting PLK1 in vivo impeded tumor growth
Huh-7 cells were subcutaneously transplanted into the flanks of nude mice and allowed to develop into HCC tumors. These tumors were later treated with repeated intratumoral injections of si-PLK1 every alternate day to evaluate the anti-tumor effect by targeting PLK1. The mean tumor volume of the si-PLK1 treatment group was significantly lower than the si-NT treatment group starting from Day 11 after five si-PLK1 treatments (Figure 5A and B). At the end of the treatment period, the mean tumor volume in the si-PLK1 treatment group was about 33% lower than in the si-NT treatment group.
Figure 5.
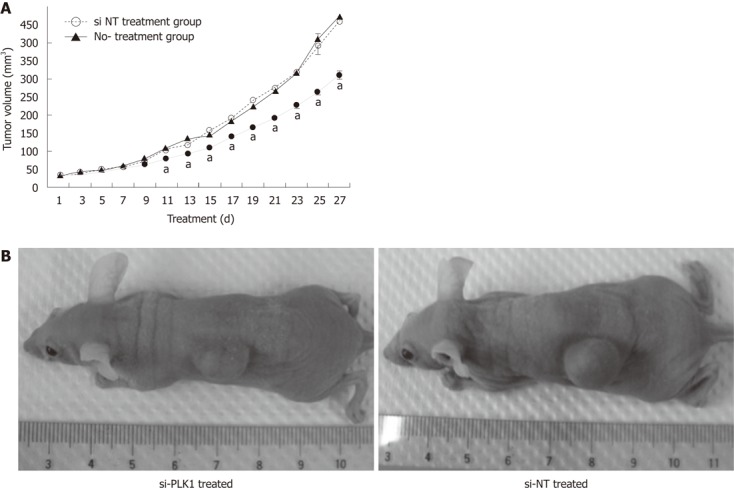
Reduction in hepatocellular carcinoma tumor size with knockdown of polo-like kinase 1. A: Treatment of short-interfering RNA (siRNA) against polo-like kinase 1 (PLK1) (si-PLK1, n = 6) in nude mice impeded the tumor growth compared with short interfering non-targeting (si-NT) (n = 6) or the non-treatment group (n = 6). Huh-7 tumor-bearing nude mice were treated with 1 nmol/L si-PLK1 intratumoral injections every alternate day for 27 d. Data are shown as mean ± SE, using the Student t-test (aP < 0.05); B: Images of nude mice with their tumors at day 27. The left panel shows a reduction in tumor size of subcutaneous tumors in nude mice treated with si-PLK1, while the right panel shows similar mice treated with si-NT.
DISCUSSION
PLK1 gene overexpression was seen in a majority of the HCC patients in this study, suggesting that PLK1 may have a role in the HCC tumorigenesis. Recently, PLK1 expression was significantly correlated with a higher recurrence rate in HCC patients[19]. Moreover, PLK1 overexpression has also been recently reported to be correlated with metastatic HCC disease[6] and poor prognosis[5] in HCC. More recently, Pelligrino et al[8] found that PLK1 silencing was associated with suppression of cell growth, while PLK2-4 silencing had the opposite effect. These studies indicate that PLK1 may have an important role in HCC.
We have shown that Huh-7 cell proliferation requires PLK1. Moreover, PLK1 knockdown in Huh-7 cells results in nuclear fragmentation, indicating apoptosis is present. Intriguingly, caspase activation is found to be unnecessary for PLK1-induced apoptosis. Consequently, the caspase-independent pathway is likely to be active, and the apoptotic effector appears to be endoG, which is a DNase that is exclusively mitochondrial. EndoG is released from mitochondria and translocates to the nucleus to cleave DNA independently of caspase upon apoptosis induction[20]. AIF, another potential effector candidate in the caspase-independent apoptosis pathway[21] was found not be involved in PLK1-induced apoptosis. It is not an essential requirement for the presence of both endoG and AIF to induce caspase-independent apoptosis. In galectin-1 induced T-cell apoptosis, which is independent of caspase, AIF and cytochrome c, only endoG was shown to be involved[22]. The induction of apoptosis that is independent of caspase by silencing PLK1 gene expression in Huh-7 cells has significant implications in the treatment of HCC. It is reported that caspase-1 and caspase-3 expressions are downregulated in HCC and this could contribute to the lack of efficacy of chemotherapy[23]. Consequently, a caspase-directed strategy is unlikely to be successful, and thus targeting PLK1 may be a more effective strategy in treatment of HCC patients.
Similar to other reports of PLK1 involvement in the proliferation of cell types such as breast cancer, lung cancer, prostate cancer and esophageal cancer[24-26], we have also shown that silencing of PLK1 significantly reduced cell proliferation and also impeded HCC growth in nude mice. PLK1 is therefore a promising gene candidate for targeted therapy in treating not only HCC but other solid tumors as well. The requirement of PLK1 in the tumorigenesis of HCC and other cancers implies it has a critical role in the final pathway for cancer cell proliferation. Overexpression of PLK1 may help to inactivate the spindle checkpoint persistently by targeting early-mitotic inhibitor 1, which is an inhibitor of anaphase promoting complex/cyclosome, that is degraded by ubiquitin ligase complexes[27]. However, the exact role of PLK1 overexpression in the tumorigenesis of HCC or other cancers is still unclear since the physiological role of PLK1 is not fully defined.
The results of our study suggest that targeting PLK1 in HCC treatment is a promising therapeutic option. In addition, normal diploid cells such as hTERT-RPE1 and MCF10A cells can survive similar PLK1 depletion that otherwise causes mitotic arrest and apoptosis in cancer cell lines[28]. Normal diploid cells still require PLK1 for survival, but at a much lower PLK1 level compared with cancer cells[29]. Consequently, controlling the extent of PLK1 depletion could minimize the deleterious effects on HCC patients. Currently, there are a number of PLK1 inhibitors in development such as ON 01910, DAP81, TAL, BTO-1/cyclopolin, and compound 1[30]. However, the most advanced agent is a novel small molecular inhibitor, BI2536, which inhibits the enzymatic activity of PLK1 protein with high selectivity (10 000-fold selectivity over other kinases) and high potency[31,32]. B12536 has now completed phase I clinical trials in patients with advanced solid tumors with reported anti-tumor activity, and neutropenia as a dose-limiting toxicity[33]. Phase II clinical trials of BI2536 are currently ongoing for various tumor indications[34]. Such inhibitors could probably be suitable in treating HCC or could be used in combination with other drugs, a strategy that is being explored in many cancers, for example, breast cancer[35].
In conclusion, PLK1 is overexpressed in HCC and the silencing experiments have shown significant antiproliferative effects in vitro and in vivo, through induction of a caspase-independent apoptosis pathway that could lead to increased sensitivity of chemotherapy in HCC patients. Thus, PLK1 is a promising therapeutic target for HCC.
COMMENTS
Background
The pathogenesis of hepatocellular carcinoma (HCC) is likely to be a multi-gene process. One gene that has been described in many cancers but only recently in HCC is polo-like kinase 1 (PLK1), a kinase that is critical to spindle formation during the metaphase.
Research frontiers
PLK1 is emerging as an important therapeutic target as a number of new agents are currently undergoing clinical trials.
Innovations and breakthroughs
The authors show that PLK1 knockdown results in a reduction in PLK1 gene and protein expression with resultant reduction in cell proliferation and increase in apoptosis. The authors also show that the increase in apoptosis is caspase-independent and involves EndoG and apoptosis-inducing factor. Knockdown of PLK1 in subcutaneous tumors transplanted into nude mice showed impairment of tumor growth.
Applications
The study indicates that PLK1 inhibitors should be evaluated in clinical trials of HCC, either as monotherapy or combination therapy.
Terminology
PLK1: Polo-like kinase, an important kinase required for spindle formation in metaphase; caspase-induced apoptosis is a key pathway for apoptosis; cell proliferation is a classic method to evaluate tumor cell growth - blocking proliferation implies that tumor growth can be blocked; si-RNA knockdown: Short interfering RNAs are short complementary RNAs that bind specifically to the mRNA produced by the gene of interest, hence blocking the signal for protein expression.
Peer review
This is a good descriptive study in which authors investigate the role of PLK1 as a therapeutic target for HCC.The results are interesting and suggest knockdown of PLK1 overexpression in HCC was shown to be a potential therapeutic target, leading to apoptosis through the endonuclease-G pathway.
Footnotes
Supported by The National University of Singapore Grants, No. R-172-000-001-731 and No. R-172-000-024-731
Peer reviewers: Jiping Wang, MD, PhD, Division of Surgical Oncology, Brigham and Women’s Hospital, Harvard Medical School, 75 Francis Street, Boston, MA 02115, United States; Fausto Catena, MD, PhD, Department of General, Emergency and Transplant Surgery, St Orsola- Malpighi University Hospital, Via Massarenti 9, 40139 Bologna, Italy
S- Editor Cheng JX L- Editor Cant MR E- Editor Zheng XM
References
- 1.Ferlay J, Bray F, Pisani P, Parkin DM. Globocan 2002: Cancer incidence, mortality and prevalence worldwide. Lyon: IARC press; 2004. [Google Scholar]
- 2.Bruix J, Llovet JM. Prognostic prediction and treatment strategy in hepatocellular carcinoma. Hepatology. 2002;35:519–524. doi: 10.1053/jhep.2002.32089. [DOI] [PubMed] [Google Scholar]
- 3.Poon RT, Fan ST, Tsang FH, Wong J. Locoregional therapies for hepatocellular carcinoma: a critical review from the surgeon’s perspective. Ann Surg. 2002;235:466–486. doi: 10.1097/00000658-200204000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 5.He ZL, Zheng H, Lin H, Miao XY, Zhong DW. Overexpression of polo-like kinase1 predicts a poor prognosis in hepatocellular carcinoma patients. World J Gastroenterol. 2009;15:4177–4182. doi: 10.3748/wjg.15.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang XQ, Zhu YQ, Lui KS, Cai Q, Lu P, Poon RT. Aberrant Polo-like kinase 1-Cdc25A pathway in metastatic hepatocellular carcinoma. Clin Cancer Res. 2008;14:6813–6820. doi: 10.1158/1078-0432.CCR-08-0626. [DOI] [PubMed] [Google Scholar]
- 7.Studach LL, Rakotomalala L, Wang WH, Hullinger RL, Cairo S, Buendia MA, Andrisani OM. Polo-like kinase 1 inhibition suppresses hepatitis B virus X protein-induced transformation in an in vitro model of liver cancer progression. Hepatology. 2009;50:414–423. doi: 10.1002/hep.22996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pellegrino R, Calvisi DF, Ladu S, Ehemann V, Staniscia T, Evert M, Dombrowski F, Schirmacher P, Longerich T. Oncogenic and tumor suppressive roles of polo-like kinases in human hepatocellular carcinoma. Hepatology. 2010;51:857–868. doi: 10.1002/hep.23467. [DOI] [PubMed] [Google Scholar]
- 9.Barr FA, Silljé HH, Nigg EA. Polo-like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol. 2004;5:429–440. doi: 10.1038/nrm1401. [DOI] [PubMed] [Google Scholar]
- 10.Xie S, Xie B, Lee MY, Dai W. Regulation of cell cycle checkpoints by polo-like kinases. Oncogene. 2005;24:277–286. doi: 10.1038/sj.onc.1208218. [DOI] [PubMed] [Google Scholar]
- 11.Sunkel CE, Glover DM. polo, a mitotic mutant of Drosophila displaying abnormal spindle poles. J Cell Sci. 1988;89(Pt 1):25–38. doi: 10.1242/jcs.89.1.25. [DOI] [PubMed] [Google Scholar]
- 12.Lowery DM, Lim D, Yaffe MB. Structure and function of Polo-like kinases. Oncogene. 2005;24:248–259. doi: 10.1038/sj.onc.1208280. [DOI] [PubMed] [Google Scholar]
- 13.Takai N, Hamanaka R, Yoshimatsu J, Miyakawa I. Polo-like kinases (Plks) and cancer. Oncogene. 2005;24:287–291. doi: 10.1038/sj.onc.1208272. [DOI] [PubMed] [Google Scholar]
- 14.Ahr A, Karn T, Solbach C, Seiter T, Strebhardt K, Holtrich U, Kaufmann M. Identification of high risk breast-cancer patients by gene expression profiling. Lancet. 2002;359:131–132. doi: 10.1016/S0140-6736(02)07337-3. [DOI] [PubMed] [Google Scholar]
- 15.Kneisel L, Strebhardt K, Bernd A, Wolter M, Binder A, Kaufmann R. Expression of polo-like kinase (PLK1) in thin melanomas: a novel marker of metastatic disease. J Cutan Pathol. 2002;29:354–358. doi: 10.1034/j.1600-0560.2002.290605.x. [DOI] [PubMed] [Google Scholar]
- 16.Eckerdt F, Yuan J, Strebhardt K. Polo-like kinases and oncogenesis. Oncogene. 2005;24:267–276. doi: 10.1038/sj.onc.1208273. [DOI] [PubMed] [Google Scholar]
- 17.de Kok JB, Roelofs RW, Giesendorf BA, Pennings JL, Waas ET, Feuth T, Swinkels DW, Span PN. Normalization of gene expression measurements in tumor tissues: comparison of 13 endogenous control genes. Lab Invest. 2005;85:154–159. doi: 10.1038/labinvest.3700208. [DOI] [PubMed] [Google Scholar]
- 18.Niikura Y, Dixit A, Scott R, Perkins G, Kitagawa K. BUB1 mediation of caspase-independent mitotic death determines cell fate. J Cell Biol. 2007;178:283–296. doi: 10.1083/jcb.200702134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen XJ, Wu LM, Xu XB, Feng XW, Xie HY, Zhang M, Shen Y, Wang WL, Liang TB, Zheng SS. [Expression and prognostic value of Polo-like kinase 1, E-cadherin in the patients with hepatocellular carcinoma] Zhonghua Waike Zazhi. 2007;45:1354–1358. [PubMed] [Google Scholar]
- 20.Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412:95–99. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- 21.Cregan SP, Dawson VL, Slack RS. Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene. 2004;23:2785–2796. doi: 10.1038/sj.onc.1207517. [DOI] [PubMed] [Google Scholar]
- 22.Hahn HP, Pang M, He J, Hernandez JD, Yang RY, Li LY, Wang X, Liu FT, Baum LG. Galectin-1 induces nuclear translocation of endonuclease G in caspase- and cytochrome c-independent T cell death. Cell Death Differ. 2004;11:1277–1286. doi: 10.1038/sj.cdd.4401485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fujikawa K, Shiraki K, Sugimoto K, Ito T, Yamanaka T, Takase K, Nakano T. Reduced expression of ICE/caspase1 and CPP32/caspase3 in human hepatocellular carcinoma. Anticancer Res. 2000;20:1927–1932. [PubMed] [Google Scholar]
- 24.Spänkuch-Schmitt B, Bereiter-Hahn J, Kaufmann M, Strebhardt K. Effect of RNA silencing of polo-like kinase-1 (PLK1) on apoptosis and spindle formation in human cancer cells. J Natl Cancer Inst. 2002;94:1863–1877. doi: 10.1093/jnci/94.24.1863. [DOI] [PubMed] [Google Scholar]
- 25.Reagan-Shaw S, Ahmad N. Silencing of polo-like kinase (Plk) 1 via siRNA causes induction of apoptosis and impairment of mitosis machinery in human prostate cancer cells: implications for the treatment of prostate cancer. FASEB J. 2005;19:611–613. doi: 10.1096/fj.04-2910fje. [DOI] [PubMed] [Google Scholar]
- 26.Bu Y, Yang Z, Li Q, Song F. Silencing of polo-like kinase (Plk) 1 via siRNA causes inhibition of growth and induction of apoptosis in human esophageal cancer cells. Oncology. 2008;74:198–206. doi: 10.1159/000151367. [DOI] [PubMed] [Google Scholar]
- 27.Moshe Y, Boulaire J, Pagano M, Hershko A. Role of Polo-like kinase in the degradation of early mitotic inhibitor 1, a regulator of the anaphase promoting complex/cyclosome. Proc Natl Acad Sci USA. 2004;101:7937–7942. doi: 10.1073/pnas.0402442101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu X, Lei M, Erikson RL. Normal cells, but not cancer cells, survive severe Plk1 depletion. Mol Cell Biol. 2006;26:2093–2108. doi: 10.1128/MCB.26.6.2093-2108.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lei M, Erikson RL. Plk1 depletion in nontransformed diploid cells activates the DNA-damage checkpoint. Oncogene. 2008;27:3935–3943. doi: 10.1038/onc.2008.36. [DOI] [PubMed] [Google Scholar]
- 30.Taylor S, Peters JM. Polo and Aurora kinases: lessons derived from chemical biology. Curr Opin Cell Biol. 2008;20:77–84. doi: 10.1016/j.ceb.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 31.Steegmaier M, Hoffmann M, Baum A, Lénárt P, Petronczki M, Krssák M, Gürtler U, Garin-Chesa P, Lieb S, Quant J, et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol. 2007;17:316–322. doi: 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- 32.Lénárt P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, Rettig WJ, Kraut N, Peters JM. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr Biol. 2007;17:304–315. doi: 10.1016/j.cub.2006.12.046. [DOI] [PubMed] [Google Scholar]
- 33.Mross K, Frost A, Steinbild S, Hedbom S, Rentschler J, Kaiser R, Rouyrre N, Trommeshauser D, Hoesl CE, Munzert G. Phase I dose escalation and pharmacokinetic study of BI 2536, a novel Polo-like kinase 1 inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2008;26:5511–5517. doi: 10.1200/JCO.2008.16.1547. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt M, Bastians H. Mitotic drug targets and the development of novel anti-mitotic anticancer drugs. Drug Resist Updat. 2007;10:162–181. doi: 10.1016/j.drup.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 35.Spänkuch B, Kurunci-Csacsko E, Kaufmann M, Strebhardt K. Rational combinations of siRNAs targeting Plk1 with breast cancer drugs. Oncogene. 2007;26:5793–5807. doi: 10.1038/sj.onc.1210355. [DOI] [PubMed] [Google Scholar]