

Abstract
AIM: To investigate the effect of zinc finger protein A20 on chronic liver allograft dysfunction in rats.
METHODS: Allogeneic liver transplantation from DA rats to Lewis rats was performed. Chronic liver allograft dysfunction was induced in the rats by administering low-dose tacrolimus at postoperative day (POD) 5. Hepatic overexpression of A20 was achieved by recombinant adenovirus (rAd.)-mediated gene transfer administered intravenously every 10 d starting from POD 10. The recipient rats were injected with physiological saline, rAdEasy-A20 (1 × 109 pfu/30 g weight) or rAdEasy (1 × 109 pfu/30 g weight) every 10 d through the tail vein for 3 mo starting from POD 10. Liver tissue samples were harvested on POD 30 and POD 60.
RESULTS: Liver-transplanted rats treated with only tacrolimus showed chronic allograft dysfunction with severe hepatic fibrosis. A20 overexpression ameliorated the effects on liver function, attenuated liver allograft fibrosis and prolonged the survival of the recipient rats. Treatment with A20 suppressed hepatic protein production of tumor growth factor (TGF)-β1, interleukin-1β, caspase-8, CD40, CD40L, intercellular adhesion molecule-1, vascular cell adhesion molecule-1 and E-selectin. A20 treatment suppressed liver cell apoptosis and inhibited nuclear factor-κB activation of Kupffer cells (KCs), liver sinusoidal endothelial cells (LSECs) and hepatic stellate cells (HSCs), and it subsequently decreased cytokine mRNA expression in KCs and LSECs and reduced the production of TGF-β1 in HSCs.
CONCLUSION: A20 might prevent chronic liver allograft dysfunction by re-establishing functional homeostasis of KCs, LSECs and HSCs.
Keywords: Chronic allograft dysfunction, Liver transplantation, Zinc finger protein A20, Rat
INTRODUCTION
Although the incidence of chronic rejection at 5 years after transplantation has decreased from 15%-20% in the 1980s to an expected incidence of 3%-5% in current recipients, probably because of the introduction of the novel drug tacrolimus[1,2], the incidence of chronic liver allograft dysfunction is still high in long-surviving recipients. Chronic liver allograft dysfunction, which results in the loss of approximately 2000 liver grafts every year, has a significant impact on liver graft function and long-term survival. It has been reported that a significant proportion of long-term liver allografts presented features of a hepatitis-like reaction that was not attributable to known viral agents or other agents[3,4]. Chronic liver allograft dysfunction is now considered to be a result of various causes of hepatic injury, including immune and non-immune factors. Currently, research is focused on non-immune factors that may lead to chronic liver allograft dysfunction, including the use of donor organs of marginal quality, the use of organs from brain injury/brain-dead donors, the presence of ischemia-reperfusion injury, Kupffer cell (KC) activation, interleukin and growth factor production, damage caused by immunosuppressive drugs (chronic toxicity of damage) and cytomegalovirus infection. Because of the numerous causative factors, the prevention and treatment of chronic liver graft dysfunction represents a considerable challenge.
Pathological changes observed in cases of chronic liver allograft dysfunction include arterial proliferative occlusive disease and/or bile duct disappearance, liver cell death and eventually liver fibrosis. Bile duct disappearance is considered to be a result of arterial proliferative occlusive disease. Molecular changes include increased hepatic expression of tumor growth factor (TGF)-β, interleukin (IL)-1β, caspase-1 and caspase-8[5]. IL-1 has been shown to contribute to chronic rejection[6]. IL-1 is produced by activated macrophages and many other cell types, including injured endothelial cells (ECs), and it stimulates smooth-muscle proliferation in vitro and increases the adhesive properties of the vascular endothelium. Overproduction of TGF-β is a chief cause of tissue fibrosis in various organs[7]. TGF-β induces the phenotypic transition of hepatic stellate cells (HSC) into proliferating myofibroblast-like cells, thus enhancing production of extracellular components[8]. The cellular and molecular mechanisms of chronic liver allograft dysfunction are still not completely clear, and the currently available drug treatments are ineffective.
The process of liver fibrosis is well-understood, and the basic steps can be summarised as follows: (1) various sources of liver damage induce KC activation; (2) activated KCs express and produce a variety of cytokines and co-stimulating molecules, such as TNF-α, IL-6, TGF-β, IL-1β and CD40L[9-11]; and (3) cytokines and co-stimulating molecules stimulate HSC activation and stimulation of myofibroblasts, which synthesize a large amount of extracellular matrix, resulting in liver fibrosis. In the process of hepatic fibrosis, nuclear factor (NF)-κB may play an important central regulatory role by regulating functional changes of hepatocytes, KCs and HSCs[9,11,12].
NF-κB is a key nuclear factor involved in the regulation of KC activation. In addition to the production of pro-inflammatory cytokines, such as TNF-α, IL-1β, TGF-β, IL-6 and IL-8, activated KC also express the co-stimulatory molecule CD40L, which is an important characteristic of chronic liver allograft dysfunction[13]. Expression of inflammatory mediators can stimulate the nuclear translocation of NF-κB in KCs via autocrine or paracrine pathways and induce the production of additional inflammatory mediators, leading to an “inflammatory cascade”, which results not only in liver damage, but also leads to the rapid stimulation of HSC activation and proliferation. Thus, inhibition of NF-κB activation in KCs may down-regulate the expression of inflammatory mediators, such as TNF-α, TGF-β, IL-1 and CD40L, and thereby suppress the liver inflammatory response.
Although the role of NF-κB in liver graft arterial lesions is not completely clear, NF-κB plays a key regulatory role in non-organ transplant atherosclerosis. In 1996, using a new type of mouse antibody (mAb α-p65 mAb), Brand demonstrated the presence of activated NF-κB in human atherosclerotic tissue for the first time[14]. Activation of NF-κB was identified in smooth muscle cells, macrophages and ECs in their study. Previous studies had shown that atherosclerosis involves activation of vascular ECs and proliferation of vascular smooth muscle cells which are subject to the regulation of NF-κB activation[15-19].
A20 is a zinc finger protein that was originally identified as a TNF-responsive gene in ECs[20]. A20 is expressed in multiple cell types, including fibroblasts, B cells, T cells and β cells, in response to a variety of stimuli that activate NF-κB, including IL-1, LPS, phorbol 12-myristate 13-acetate, H2O2 and CD40 ligand. In ECs and hepatocytes, A20 has a dual cytoprotective function[21-26]. A20 is anti-inflammatory, due to inhibition of NF-κB through a negative feedback loop, and it is antiapoptotic, due to inhibition of the caspase cascade at the level of initiator caspase-8[21-23]. A20 can also inhibit NF-κB activation induced by LPS, IL-1 and CD40 cross-linking through the negative feedback loop[24-26]. A20 curtails inflammation by inhibiting NF-κB activation, either through its association with IκB kinase-γ/NF-κB essential modifier within the signalosome or through its ubiquitin-editing functions[21,22,27,28].
A previous study indicated that reduced expression of A20 might be an important pathogenic contributor to an increased susceptibility to liver allograft ischemia/reperfusion (I/R) injury[29]. Ramsey et al[30] recently reported that A20 could protect mice from lethal liver I/R injury by increasing peroxisome proliferator-activated receptor-alpha expression. In addition, it has been shown that A20 expression is up-regulated in human renal allografts in response to immune injury inferred by acute rejection, and the result suggests that A20 could limit graft injury[31]. Our previous studies indicated that A20 expression was up-regulated in immature dendritic cells derived from rat liver allografts undergoing acute rejection[32]. Furthermore, A20 overexpression could inhibit NF-κB activation of liver sinusoidal endothelial cells (LSECs) in rat liver allografts and suppress acute rejection[32]. These results suggest that A20 may protect liver allografts from I/R injury and acute rejection.
Although the effects of A20 on lipopolysaccharide-induced acute toxic lethal hepatitis, liver regeneration, hepatic I/R injury and liver allograft rejection have been investigated, little is known about the effect of A20 on chronic liver allograft dysfunction. In this work, the effect of A20 on liver allograft chronic dysfunction induced by postoperative low-dose tacrolimus administration was investigated.
MATERIALS AND METHODS
Recombinant adenoviruses
The rAdEasy-A20 and the empty control rAdEasy containing green fluorescent protein were generated in our laboratory[33]. The NcoI→ SalI fragment (2332 bp) of the A20 gene, which was obtained from the plasmid pCAGGS-FLAGmA20 (a kind gift from Dr. Rudi Beyaert from Department of Molecular Biology, Flanders Interuniversity Institute of Biotechnology, University of Ghent, Belgium) and carries the entire mouse A20 cDNA sequence, was cloned into the shuttle plasmid pAdTrack-CMV. Homologous recombination took place between the resultant plasmid and the backbone plasmid pAdEasy-1 in Escherichia coli BJ5183, and the recombinant adenoviral plasmid was generated. The adenovirus was packaged in 293 cells, and the recombinant adenovirus rAdEasy-A20 was generated. The empty Ad vector (rAdEasy) was generated following the same principle.
Animal model
Inbred male DA (RT1a) and Lewis (RT1l) rats weighing 260-320 g were used as liver donors and recipients, respectively. The animals were maintained under standard conditions and treated according to the Guidelines for the Care and Use of Laboratory Animals of Sichuan University. Orthotopic liver transplantations (OLT) were performed with the two-cuff technique. All operations were performed under ether anesthesia under sterile conditions. Cefazolin (40 mg/kg, injected intramuscularly) was given after the implantation operation for 5 d to prevent infection. More than 90% of the rats survived this operative procedure. To induce chronic liver allograft dysfunction, a low dose of tacrolimus (0.1 mg/kg per day) was administered intramuscularly for 5 d after the implantation operation[5]. To examine chronic liver allograft dysfunction, recipient rats were given physiological saline (PS), rAdEasy-A20 (1 × 109 pfu/30 g weight) or rAdEasy (1 × 109 pfu/30 g weight) through the tail vein once every 10 d from postoperative day (POD) 10 for 3 mo. Five recipient rats per group were allowed to survive until they died. Ten recipient rats per group were killed on POD 30 and POD 60 before rAdEasy-A20 or rAdEasy injection. Blood samples were harvested from the inferior vena cava. The left lateral lobes and caudate lobes from 5 liver allografts per group were harvested for Western blotting, Masson staining and immunohistochemistry, and the other liver lobes were harvested for KC and LSEC isolation. Another 5 liver grafts per group were harvested for HSC isolation.
Histological analysis
Liver fibrosis was analyzed on POD 30 and POD 60. The liver tissue lobes were fixed in 10% neutral-buffered formalin embedded in paraffin. For histological analysis, the sections were stained with hematoxylin-eosin. For fibrosis analysis, the sections were stained with Masson stain.
Hepatic A20 expression
Liver graft specimens were harvested on POD 30 and POD 60. Immunohistochemistry was performed for hepatic A20 protein expression in five sections from per graft after the samples were fixed in 10% neutral-buffered formalin embedded in paraffin. The liver sections were incubated with a 1:100 dilution of anti-rabbit polyclonal A20 antibody (Abcam, ab45366) for 45 min and Envision™ for 45 min. The sections were counterstained with hematoxylin. Positive cells were counted at 400 × magnification. Ten random fields were observed in each of the liver portal tract areas.
Liver function assay
Serum samples from POD 30 and POD 60 were also analysed for alanine aminotransferase (ALT) and total bilirubin (TBIL) levels as indices of hepatocellular injury. The levels of ALT and TBIL were measured with an automatic biochemical analyser using diagnostic kits from Sigma Chemical Co.
Hepatic protein production study
TGF-β1, IL-1β, caspase-1, caspase-8, CD40, CD40L, Intercellular adhesion molecule (ICAM)-1, vascular cell adhesion molecule (VCAM)-1 and E-selectin protein levels in the liver allografts were analysed by Western blotting. Liver tissue lysates were electrophoresed on sodium dodecylsulfate polyacrylamide gel electrophoresis gels and transferred to polyvinylidene chloride (PVDC) membranes for Western blotting analysis. Briefly, the PVDC membranes were incubated in a blocking buffer for 1 h at room temperature followed by incubation for 2 h with Abs raised against TGF-β1, IL-1β, caspase-1, caspase-8, CD40, CD40L, ICAM-1, VCAM-1 and E-selectin. The membranes were washed and incubated for 1 h with horseradish peroxidase-labelled immunoglobulin G. Immunoreactive bands were visualised using enhanced chemiluminescence detection reagent. The bands were quantified using a scanning densitometer from Bio-Image Analysis System. The results were expressed as the relative optical density.
Hepatic cells apoptosis
The paraffin sections of liver grafts on POD 30 and 60 were analysed for apoptotic cells using the terminal transferase dUTP nick end-labelling (TUNEL) method (in situ Cell Death Detection Kit, Roche Biochemicals, Mannheim, Germany). For all staining procedures, positive and negative cells were counted in 3 randomly selected fields under a light microscope. Quantitative analysis was performed using the Coulter EPICS Elite ESP cell sorting system, United States.
Isolation of KC and LSECs
KCs and LSECs were isolated using a modified method of Braet and colleagues[34]. In brief, the liver graft was perfused with Ca2+-Mg2+-free Hanks’ balanced salt solution followed by 0.6% collagenase A (Sigma type 1) via a polyethylene catheter inserted into the portal vein trunk. After incubation of the fragmented tissue in the same solution, the resulting cell suspension was centrifuged at 100 r/min for 10 min to remove the parenchymal cells. The supernatant containing a mixture of the hepatic nonparenchymal cell fraction was subsequently layered on top of a two- step Percoll gradient (25% to 50%) and centrifuged for 10 min at 900 r/min. The intermediate zone located between the two density layers, which was enriched with LSECs and KCs, was cultured for 20 min in plastic flasks, and the LSECs and KCs subsequently were further isolated based on the selective adherence of KCs to plastic flasks and the spreading of the LSECs on collagen.
Isolation of HSCs
Hepatic stellate cells were isolated from the liver allografts by a modified method that has been described previously[35]. Briefly, HSCs were isolated from the liver grafts by sequential in situ perfusion with collagenase and digestion with pronase. Suspensions of liberated HSCs were prepared by centrifugation on a double-layered (17%/11.5%) metrizamide solution (Sigma). After centrifugation at 1700 g for 15 min, the HSCs were harvested from the top of the upper layer. More than 90% pure and viable HSCs were routinely obtained using this procedure, as determined by ultraviolet-excited fluorescence microscopy and Trypan blue dye exclusion, respectively. Isolated HSCs were used for nuclear protein or RNA extraction.
NF-κB activation of LSECs, KCs and HSCs
The NF-κB activity of the LSCEs, KCs and HSCs was analysed with the electrophoretic mobility shift assay (EMSA) as previously described[33]. Nuclear proteins were extracted from LSCEs, KCs and HSCs. The protein concentration of the nuclear extracts was determined by Bradford assay[36]. Nuclear extracts were frozen on dry ice and stored at -80 °C until they were assessed in the EMSA. The double-stranded NF-κB consensus oligonucleotides (5’-AGTTGAGGGGACTTTCCCAGGC-3’, and 3’-TCAACTCCCCTGAAAGGGTCCG-5’)[37] used in EMSA were end-labelled with γ-32P adenosine triphosphate (3.7 × 105 Bq/L at 5 μL) using T4 polynucleotide kinase. The reaction products were separated in 6 % nondenaturing polyacrylamide gels subjected to gamma autoradiography at -70 °C for 48 h and were analysed with a gel imaging system.
mRNA expression of cytokines in LSECs and KCs
Analysis of mRNA expression of ICAM-1, VCAM-1, E-selectin, IL-1β and CD40 in LSECs and mRNA expression of TGF-β1, IL-1 and CD40 L in KCs was performed by semiquantitative reverse transcription polymerase chain reaction (RT-PCR) amplification and compared with the expression of the house-keeping gene β-actin using the one-step PCR Kit. Total RNA from LSECs and KCs was extracted using the TripureTM reagent. PCR was performed in a 25 μL reaction system. The PCR reaction produced a 513-bp product for ICAM-1, a 257-bp product for VCAM-1, a 239-bp product for E-selectin, a 388-bp product for CD40, a 395-bp product for CD40L, a 378-bp product for IL-1β, a 383 bp product for TGF-β1, and a 813-bp product for β-actin. The PCR products from each sample were subjected to electrophoresis in a 15 g/L agarose gel containing 0.5 mg/L ethidium bromide. Densitometrical analysis using NIH imaging software was performed for semiquantification of the PCR products. The mRNA expression of each target was evaluated by determining the ratio of the band intensity to β-actin and was presented as the percent of β-actin (%).
TGF-β1 protein level in HSCs
Supernatant samples from the HSCs were analysed for TGF-β1 using enzyme-linked immunosorbent assays (ELISA) according to the manufacturer’s instructions.
Statistical analysis
SPSS 13.0 statistical software (SPSS Inc., Chicago, IL) was used to analyse the relevant data. The results are expressed as the means ± SD. Significant differences between two groups or more were identified by the paired Student t test. P values less than 0.05 were considered statistically significant.
RESULTS
A20 protein over-expression in liver grafts by successful venous adenoviral gene transfer
Immunohistochemical staining confirmed significant hepatic A20 protein expression on POD 30 (data not shown) and POD 60 in the group of rats that received venous A20 adenovirus, whereas only some hepatic A20 protein expression was shown in the rats treated with rAdEasy and PS POD 30 (data not shown) and POD 60 (Figure 1A-C).
Figure 1.
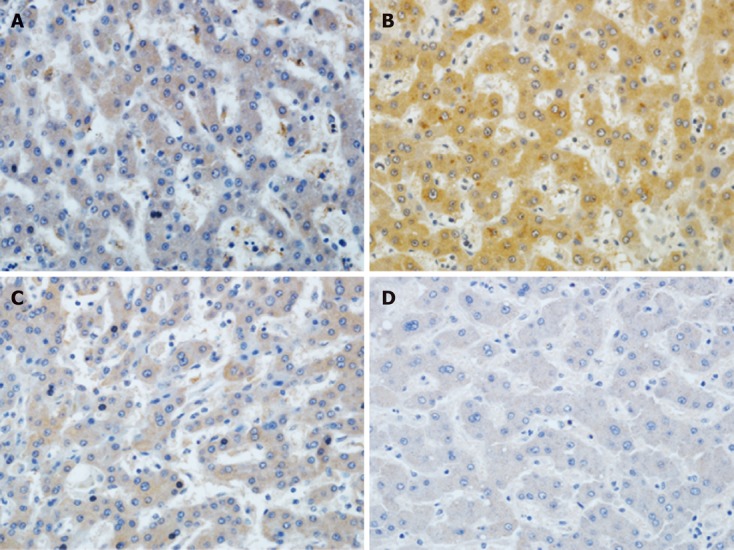
Immunohistochemical analysis of A20 expression in liver grafts on postoperative day 30. Representative immunohistochemical sections from FK506 + physiological saline (A) treated livers demonstrate lower A20 expression compared to the substantial number of A20-positive liver cells in the FK506 + A20 treated liver grafts (B). Furthermore, the expression of FK506 + rAdEasy (C) is also low. D: The results of the normal control group (brown staining) (original magnification, 400 ×).
Survival study
The survival days of the liver-grafted rats are shown in Table 1. The results suggested that the rats in the A20 treatment group survived longer than the rats in the PS and rAdEasy groups.
Table 1.
Effect of A20 on the survival of Lewis rats transplanted with Dark Agouti livers
| Treatment1 | Survival (d) | mean ± SD |
| FK506 + PS | 52, 57, 60, 72, 78 | 63.80 ± 10.83 |
| FK506 + rAdEasy A20 | 117, 121, 123, 128, 136 | 125.00 ± 7.31a |
| FK506 + rAdEasy | 44, 50, 57, 58, 67 | 55.20 ± 8.70 |
Lewis rats transplanted with Dark Agouti livers were given 0.1 mg/kg FK506 for 5 d after transplantation. Lewis rats receiving transplants were given rAdEasy-A20 (1 × 109 pfu/30 g weight) or rAdEasy or physiological saline (PS) once every 10 d from postoperative day (POD) 30 to POD 90.
P < 0.01 vs FK506 + PS and FK506 + rAdEasy treatment.
Liver fibrosis
Fibrosis of the liver allografts was detected on POD 30 and POD 60 with Masson staining. The histological findings showed that postoperative administration of low-dose tacrolimus without A20 treatment resulted in marked liver fibrosis on POD 60. However, tacrolimus combined with A20 treatment resulted in reduced hepatic fibrosis (Figure 2).
Figure 2.
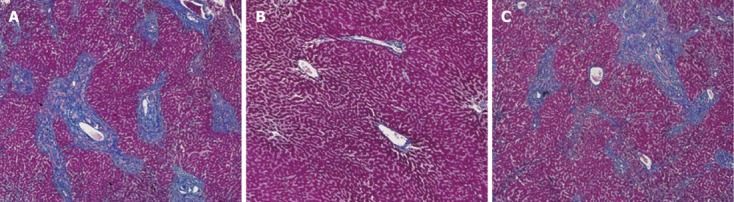
Representative liver fibrosis samples from FK506 + physiological saline treated group (A), FK506 + A20 treated group (B) and FK506 + rAdEasy -treated group (C) on postoperative day 60 (100 ×).
Serum ALT and TBIL levels
As shown in Table 2, the results showed that postoperative administration of low-dose tacrolimus led to a significant increase of TBIL and ALT levels on POD 30 and POD 60. A20 treatment markedly decreased serum TBIL and ALT levels on POD 30 and POD 60.
Table 2.
Serum total bilirubin and alanine aminotransferase levels after liver transplantation (n = 5)
| Group |
ALT (IU/L) |
TBIL(μmol/L) |
||
| POD 30 | POD 60 | POD 30 | POD 60 | |
| FK506 + PS | 163.41 ± 35.28 | 257.35 ± 42.78 | 85.72 ± 16.47 | 165.43 ± 24.63 |
| FK506 + A20 | 66.79 ± 17.56b | 90.28 ± 22.37bd | 37.61 ± 8.06b | 63.71 ± 11.38bd |
| FK506 + rAdEasy | 187.66 ± 43.54 | 282.75 ± 53.64d | 106.37 ± 21.35 | 182.93 ± 28.75d |
P < 0.01 vs FK506 + physiological saline (PS) or FK506 + rAdEasy;
P < 0.01 vs postoperative day (POD) 30. ALT: Alanine aminotransferase; TBIL: Total bilirubin.
A20 decreases hepatic protein production of TGF-β1, IL-1β, caspase-8, CD40, CD40L, ICAM-1, VCAM-1 and E-selectin
High levels of TGF-β1, IL-1β, caspase-1, caspase-8, CD40, CD40L, ICAM-1, VCAM-1 and E-selectin protein were detected in liver grafts from rats that did not receive A20 treatment liver graft on POD 30 (data not shown) and POD 60. However, A20 treatment markedly down-regulated the protein levels of TGF-β1, IL-1β, caspase-8, CD40, CD40L, ICAM-1, VCAM-1 and E-selectin in liver allografts (Figure 3).
Figure 3.
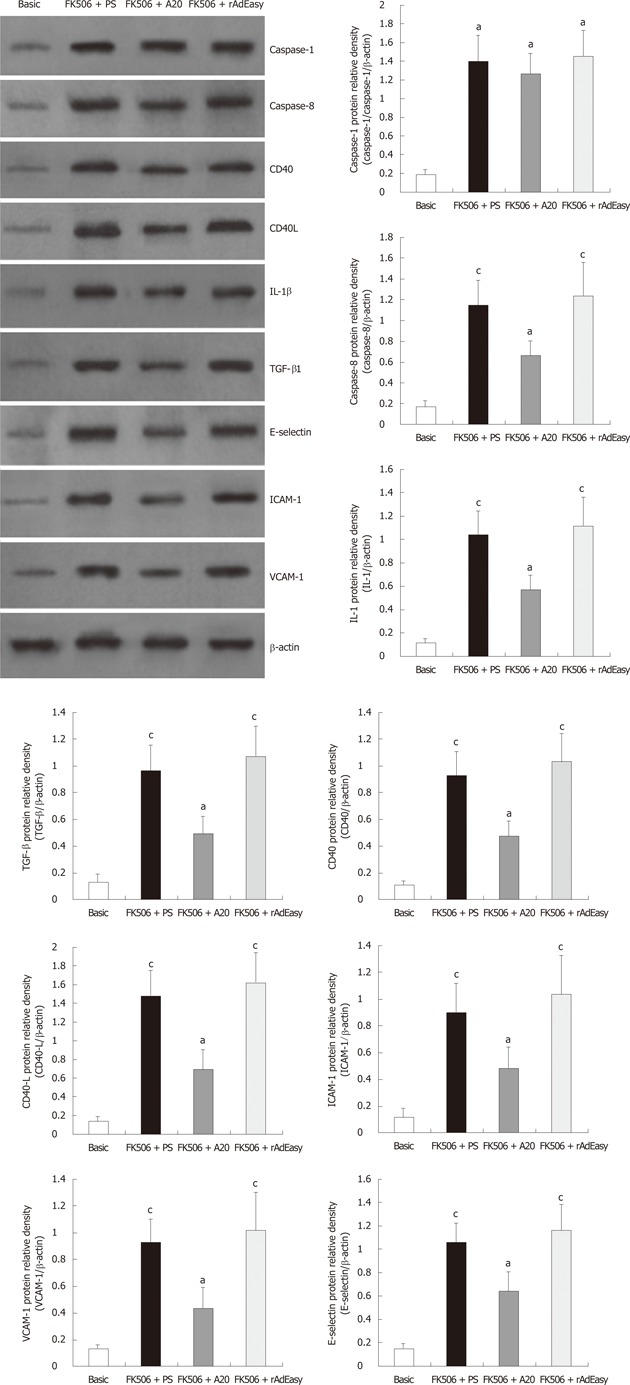
Western blotting analysis and densitometric analysis of associated cytokines in liver graft tissue. Data were expressed as the relative intensity vs β-actin. aP < 0.05 vs basic control; cP < 0.05 vs FK506 + A20. IL: Interleukin; TGF: Tumor growth factor; ICAM: Intercellular adhesion molecule; VCAM: Vascular cell adhesion molecule; PS: Physiological saline.
A20 treatment suppresses liver cell apoptosis
Liver cells apoptosis on POD 30 and POD60 were measured with the TUNEL assay. TUNEL staining revealed a decreased apoptosis index among the liver cells in the A20 group (11.83% ± 3.52% on POD 30, 14.66% ± 3.84% on POD 60) compared with that of the PS group (20.62% ± 5.36% on POD30, 32.78% ± 6.74% on POD 60) and rAdEasy group (21.58% ± 6.17% on POD 30, 35.27% ± 7.38% on POD 60) (P < 0.01) (Figure 4).
Figure 4.
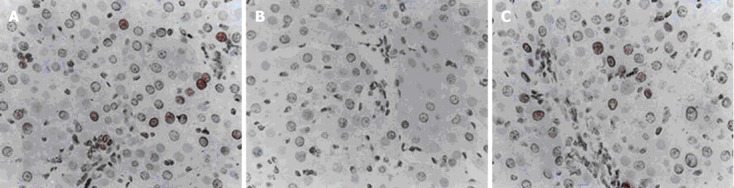
Terminal transferase dUTP nick end-labelling analysis of liver cells apoptosis (red stained) (original magnification, × 400). Overexpressed A20 in liver graft resulted in a significantly decreased number of transferase dUTP nick end-labelling-positive liver cells (B) compared with physiological salinegroup rats (A) and rAdeasy group rats (C) (P < 0.01).
A20 suppresses NF-κB activation of LSECs, KCs and HSCs
The EMSA showed that postoperative low-dose tacrolimus treatment led to a significant activation of NF-κB in LSECs, KCs and HSCs on POD 30 (data not shown) and POD 60, and A20 treatment markedly inhibited NF-κB activation in these cells (Figure 5).
Figure 5.
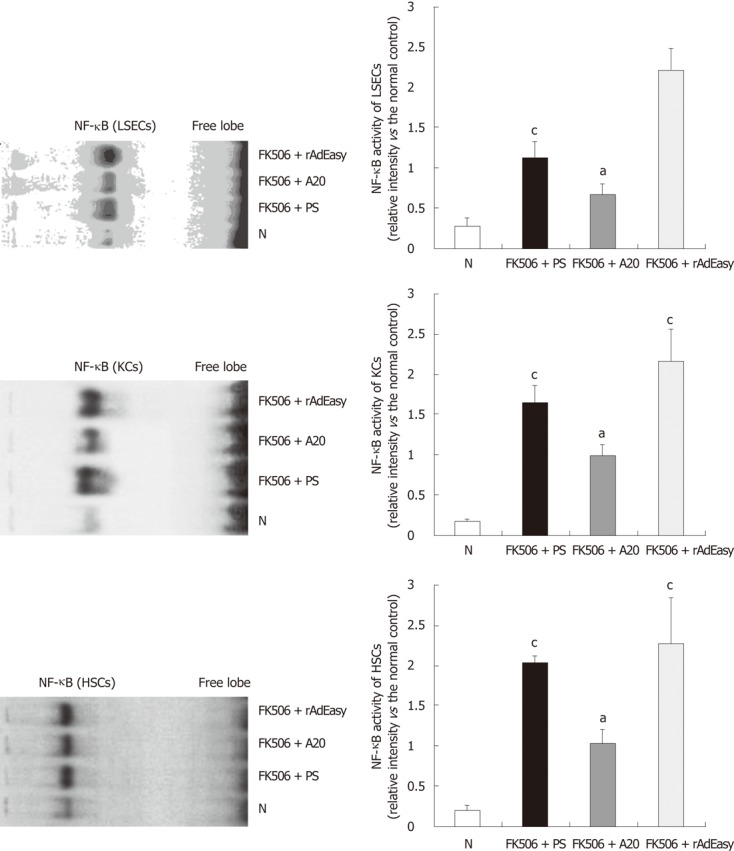
Effects of A20 on nuclear factor-κB activation in liver sinusoidal endothelial cells, Kupffer cells and hepatic stellate cells. Nuclear factor-κB (NF-κB) activation on postoperative day (POD) 60 was determined by electrophoretic mobility shift assay (EMSA). Data were expressed as the relative intensity vs the normal basic control (N). aP < 0.05 vs basic control; cP < 0.05 vs FK506 + A20. LSECs: Liver sinusoidal endothelial cells; KCs: Kupffer cells; HSCs: Hepatic stellate cells.
A20 suppresses cytokine mRNA expression in LSECs and KCs
High levels of ICAM-1, VCAM-1, E-selectin, IL-1β and CD 40 mRNA in LSECs, as well as high levels of TGF-β1, IL-1β and CD40L, in KCs on POD 30 and POD 60 were detected by RT-PCR. The increased mRNA expression levels of these cytokines were significantly reduced by A20 treatment (Figure 6).
Figure 6.
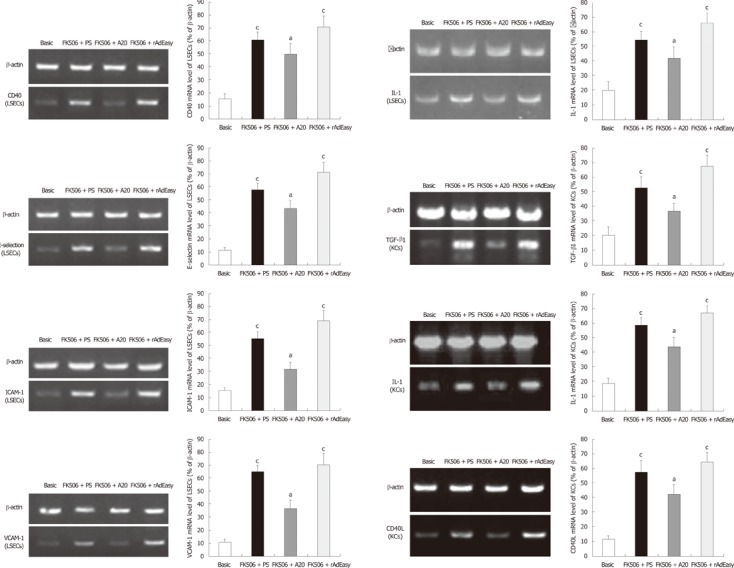
Cytokine mRNA expression in hepatic nonparenchymal cells on postoperative day 60 detected by reverse transcription polymerase chain reaction analysis. Data were expressed as the relative intensity vs β-actin. aP < 0.05 vs basic control; cP < 0.05 vs FK506 + A20. LSECs: Liver sinusoidal endothelial cells; ICAM: Intercellular adhesion molecule; KCs: Kupffer cells; VCAM: Vascular cell adhesion molecule; TGF: Tumor growth factor; IL: Interleukin.
A20 suppresses TGF-β1 protein production in HSCs
The ELISA showed that A20 overexpression significantly reduced TGF-β1 protein production in HSCs from liver allografts (Table 3).
Table 3.
Tumor growth factor-β1 levels in the conditioned media of cultured hepatic stellate cells (n = 5)
| Group |
TGF-β1 (pg/mL, mean ± SD) |
|
| POD 30 | POD 60 | |
| FK506 + PS | 1385.80 ± 186.84b | 2368.62 ± 211.58bd |
| FK506 + A20 | 595.76 ± 85.71 | 718.28 ± 97.12 |
| FK506 + rAdEasy | 1525.32 ± 202.66b | 2475.33 ± 194.93bd |
P < 0.01 vs FK506 + A20;
P < 0.01 vs postoperative day (POD) 30. TGF-β1: Tumor growth factor-β1.
DISCUSSION
To improve the survival of OLT patients, it is particularly important to protect liver grafts from chronic dysfunction. In the present study, we demonstrated that the zinc finger protein A20, a potent negative feedback inhibitor of NF-κB activation and a hepatoprotective gene, could suppress chronic liver allograft dysfunction in rats.
The identification of NF-κB as a key factor for the pathogenesis of allograft rejection suggests that NF-κB-targeted therapeutics might be useful in transplant patients. Although many drugs, such as corticosteroids and cyclosporin, can inhibit NF-κB activation[38-40], these immunosuppressants have few effects on chronic liver allograft dysfunction. Therefore, novel effective agents for chronic liver allograft dysfunction should be investigated.
Previous studies have identified A20 as a critical component of the physiologic hepatoprotective role of hepatocytes. The effects of A20 on lipopolysaccharide-induced acute toxic lethal hepatitis[22,23], liver regeneration[22,23,33], hepatic I/R injury[30] and liver allograft rejection[33] have been investigated. Furthermore, as a role for NF-κB is inferred in the pathological changes involved in chronic liver allograft dysfunction, such as liver cell death, arterial proliferative occlusive disease and/or bile duct disappearance, and eventually liver fibrosis, we reasoned that A20 would probably attenuate chronic liver allograft dysfunction. In the present study, we found that A20 is also an effective agent for chronic liver allograft dysfunction by showing that fibrosis was markedly attenuated in A20-overexpressing liver allografts compared with the controls. The suppressed NF-κB activation in LSECs, KCs and HSCs, the decreased production of TGF-β1, IL-1β, caspase-8, ICAM-1, VCAM-1, E-selectin, CD40 and CD40L, as well as the suppressed level of liver cell apoptosis, are possible mechanisms for these effects.
Overproduction of TGF-β1 is a chief cause of liver fibrosis. TGF-β1 is mainly produced by HSCs and KCs. HSC has been affirmed to be the main effector cell of liver fibrosis. As the main macrophage and proinflammatory cell, KCs not only perform phagocytosis, but they also release many proinflammatory cytokines, including TNF-α, IL-1, IL-6 and TGF-β1, meaning that the role of KCs in liver grafts may change during different phases, including the early phase of induction of hepatic I/R injury[41], the acute rejection phase in human liver allografts[42] and during the establishment of tolerance in the OLT model of transplantation from one Sprague-Dawley (SD) rat to another SD rat[43]. However, the role of KCs in chronic liver graft dysfunction has not been investigated. IL-1 has been shown to contribute to chronic rejection[6]. IL-1 produced by activated macrophages and many other cell types, including injured ECs, increases smooth muscle proliferation in vitro and the adhesive properties of the vascular endothelium. LSECs provide a barrier against the infiltration of the liver graft infiltrating mononuclear cells (LIMCs), and blocking of adhesion molecules, such as ICAM-1, on ECs interferes with recruitment of sinusoidal NK-like cells into the rat liver[44]. NF-κB activation is the key regulatory factor of nuclear transcription of TGF-β and IL-1 in these cells; therefore, inhibition of NF-κB activation could inhibit transcription of TGF-β and IL-1. In our previous study, we showed that A20 could inhibit NF-κB activation and apoptosis in LSECs, which subsequently suppresses ICAM-1 mRNA expression and recruitment of LIMCs, including T cells and NK/NKT cells[33]. Thus, we reasoned that LSECs and KCs could play important effect on the development of chronic liver allograft dysfunction. Therefore, functional changes of KCs and LSECs in liver allografts, including NF-κB activation and transcription of TGF-β and IL-1, were investigated in the present study.
Low-dose tacrolimus was administrated post-OLT for 5 d to induce liver allograft dysfunction in the present study. Short-term postoperative administration of low-dose tacrolimus resulted in significant liver fibrosis on POD 60 and reduced liver function on POD 30 and POD 60. Excessive NF-κB activation and a high level of mRNA expression of ICAM-1,VCAM-1, E-selectin, IL-1β and CD40 in LSECs were found on POD 30 and POD 60. Similar changes were detected in KCs and HSCs, including marked NF-κB activation and high levels of TGF-β1, IL-1β and CD40L mRNA expression in KCs, as well as elevated TGF-β1 protein levels in HSCs. Increased hepatic protein production of TGF-β1, IL-1β, ICAM-1, VCAM-1, E-selectin, CD40 and CD40L were also detected on POD30 and POD 60. These results suggested that excessive NF-κB activation and the associated production and secretion of cytokines by LSECs, KCs and HSCs may be the important steps inducing chronic liver allograft dysfunction. This hypothesis was confirmed by the results obtained with the combined A20 treatment. Liver fibrosis and liver function were significantly improved by the combined A20 treatment. Furthermore, the NF-κB activity in LSECs, KCs and HSCs was significantly down-regulated by A20 treatment, and consequently the elevated cytokines mRNA expression and protein production were more suppressed. Suppression of ICAM-1, VCAM-1 and E-selectin production in LSECs could inhibit the recruitment of LIMCs into the liver graft[33], reducing the hepatic injury caused by LIMCs. The down-regulated IL-1β secretion in LSECs and KCs might theoretically suppress chronic rejection[6]. The CD40/CD40 L signalling pathway is a potent activator of ECs and a promoter of atherosclerosis. A20 works at multiple levels to protect ECs from CD40/CD40L-mediated activation and apoptosis. A20-based therapy could be beneficial for the treatment of vascular diseases, such as atherosclerosis and transplant-associated vasculopathy[15]. A20 can also inhibit NF-κB activation induced by LPS, IL-1 and CD40 cross-linking through a negative feedback loop[24-26]. Previous data suggested that FasL expression on APCs and phagocytosis of apoptotic T cells by FasL+ KCs were indicators of acute and chronic rejection activity in human liver allografts[41]. In the present study, the A20-induced decrease in the expression of IL-1 and CD40 in LSECs, as well as IL-1 and CD40L in KCs, might inhibit NF-κB activation in LSECs through a negative feedback loop and protect LSECs from apoptosis, subsequently inhibiting recruitment of LIMCs into the liver graft. Suppression of NF-κB activation in KCs could inhibit hepatic ischemia/reperfusion injury, which represents an important cause of chronic liver allograft dysfunction. The decreased expression of TGF-β1 in KCs and the suppressed NF-κB activation in HSCs by A20 might inhibit the transition from HSCs to myofibroblast-like cells and consequently suppress TGF-β1 protein production in HSCs. The present study also revealed suppressed TGF-β1 production and reduced fibrosis in combined A20-treated liver grafts.
A20 has a dual cytoprotective function in ECs and hepatocytes. In addition to its anti-inflammatory function, A20 is also antiapoptotic through inhibition of the caspase cascade at the level of initiator caspase 8[21-23]. A20 can also protect hepatocytes from TNF-mediated apoptosis[21,22]. Furthermore, it has been well-established that hepatocytes undergo apoptotic cell death in the course of rejection of a liver graft, and apoptosis is a mechanism of cell death in liver allograft rejection[45,46]. FasL expression on activated NK cells was augmented[47,48], and FasL ligation to Fas expressed on hepatocytes could mediate hepatocyte apoptosis[49,50]. In the present study, high levels of caspase-8 and caspase-1 protein were demonstrated in the liver grafts with chronic dysfunction, and caspase-8 but not caspase-1 production was markedly decreased by A20 treatment. This result was the opposite of the findings presented in the previous study[5] in which hepatocyte growth factor significantly suppressed the production of caspase-1 but not caspase-8 in liver allografts with chronic dysfunction. However, the A20-induced decrease in hepatic caspase-8 production observed in the present study was consistent with the report by Daniel et al[23], who demonstrated that A20 could protect ECs from TNF-, Fas-, and NK-mediated cell death by inhibiting caspase-8 activation. Our previous study[33] also showed a marked down-regulation of the number of LIMCs by A20, including a more prominent decrease in the subproportion of NK and NKT cells in the liver allograft. Thus, we could hypothesise that A20 could inhibit infiltration of LIMC (including NK cell) by suppression of LSEC caspase-8 activation in the liver allograft and consequently attenuate hepatic injury, including acute rejection and chronic dysfunction. In the present study, liver cell (including hepatocyte) apoptosis increased chronic dysfunction progressed, and the apoptosis indices in the PS group and the rAdEasy group on POD 60 were markedly higher than on POD 30 in the same groups. However, A20 treatment significantly inhibited liver cell apoptosis, and the results showed that the apoptosis index on POD 30 and POD 60 in A20 group were similar.
In summary, A20 could protect the liver allograft from chronic dysfunction, which might be caused by the re-established functional homeostasis of KCs, LSECs and HSCs, as well as the suppressed liver cell apoptosis.
COMMENTS
Background
In spite of the introduction of the use of tacrolimus in liver transplantation recipients, the incidence of chronic liver allograft dysfunction is still high in long-surviving recipients. Chronic liver allograft dysfunction, which results in the loss of approximately 2000 liver grafts every year, has a great impact on liver graft function and long-term survival. The mechanisms of chronic liver allograft dysfunction are still unclear, and there is no effective treatment. The possible factors that induce chronic liver allograft dysfunction include immune and non-immune events.
Research frontiers
Many important events, such as activation of liver graft sinusoidal endothelial cells (LSECs), activation of Kupffer cells and hepatic satellite cells, atherosclerosis, hepatic fibrosis and liver cells’ apoptosis through nuclear factor-κB (NF-κB) activation, were reproduced in the rat model of chronic liver allograft dysfunction, and the effects of the zinc finger protein A20 on chronic liver allograft dysfunction were investigated, including the effects of A20 on recipient survival, hepatic fibrosis and liver function, NF-κB activation in LSECs, Kupffer cells and hepatic stellate cells, liver cell apoptosis, and the expression of the related cytokines.
Innovations and breakthroughs
This study sought to elucidate the cellular mechanisms of chronic liver allograft dysfunction. This study also intended to provide a theoretical basis for the use of A20 as a treatment of liver allograft chronic dysfunction.
Applications
The study results suggest that the zinc finger protein A20 may be an effective tool for the prevention and treatment for chronic liver dysfunction after liver transplantation.
Terminology
A20 is a zinc finger protein originally identified as a tumor necrosis factor-responsive gene in endothelial cells (ECs). A20 is expressed in multiple cell types, including fibroblasts, B cells, T cells, and β cells, in response to a variety of stimuli that activate NF-κB, including interleukin-1, lipopolysaccharides, phorbol 12-myristate 13-acetate, H2O2 and CD40 ligand. In ECs and hepatocytes, A20 has a dual cytoprotective function. It has been shown that A20 is a protective gene for the liver.
Peer review
This is an original study in which the authors provide strong evidence demonstrating the important finding that the zinc finger protein A20 might protect liver allografts from chronic dysfunction. These results are easily comprehensible and convincing. Additionally, the study explores the cellular and molecular mechanisms of chronic liver allograft dysfunction.
Footnotes
Supported by The National Natural Science Foundation of China, No. 30872529; the PhD Program Fund of the Ministry of Education of China, No. 20030610078; and the Chinese Postdoctoral Science Foundation, No. 2003033531
Peer reviewer: Dr. Yasuhiko Sugawara, Artificial Organ and Transplantation Division, Department of Surgery, Graduate School of Medicine, University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan
S- Editor Wu X L- Editor Ma JY E- Editor Xiong L
References
- 1.Blakolmer K, Jain A, Ruppert K, Gray E, Duquesnoy R, Murase N, Starzl TE, Fung JJ, Demetris AJ. Chronic liver allograft rejection in a population treated primarily with tacrolimus as baseline immunosuppression: long-term follow-up and evaluation of features for histopathological staging. Transplantation. 2000;69:2330–2336. doi: 10.1097/00007890-200006150-00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.European FK506 Multicentre Liver Study Group. Randomised trial comparing tacrolimus (FK506) and cyclosporin in prevention of liver allograft rejection. Lancet. 1994;344:423–428. [PubMed] [Google Scholar]
- 3.Wong T, Nouri-Aria KT, Devlin J, Portmann B, Williams R. Tolerance and latent cellular rejection in long-term liver transplant recipients. Hepatology. 1998;28:443–449. doi: 10.1002/hep.510280223. [DOI] [PubMed] [Google Scholar]
- 4.Slapak GI, Saxena R, Portmann B, Gane E, Devlin J, Calne R, Williams R. Graft and systemic disease in long-term survivors of liver transplantation. Hepatology. 1997;25:195–202. doi: 10.1002/hep.510250136. [DOI] [PubMed] [Google Scholar]
- 5.Tashiro H, Fudaba Y, Itoh H, Mizunuma K, Ohdan H, Itamoto T, Asahara T. Hepatocyte growth factor prevents chronic allograft dysfunction in liver-transplanted rats. Transplantation. 2003;76:761–765. doi: 10.1097/01.TP.0000083040.50727.CE. [DOI] [PubMed] [Google Scholar]
- 6.Tilney NL, Whitley WD, Diamond JR, Kupiec-Weglinski JW, Adams DH. Chronic rejection--an undefined conundrum. Transplantation. 1991;52:389–398. doi: 10.1097/00007890-199109000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Friedman SL. Seminars in medicine of the Beth Israel Hospital, Boston. The cellular basis of hepatic fibrosis. Mechanisms and treatment strategies. N Engl J Med. 1993;328:1828–1835. doi: 10.1056/NEJM199306243282508. [DOI] [PubMed] [Google Scholar]
- 8.Border WA, Noble NA. Transforming growth factor beta in tissue fibrosis. N Engl J Med. 1994;331:1286–1292. doi: 10.1056/NEJM199411103311907. [DOI] [PubMed] [Google Scholar]
- 9.Hellerbrand C, Jobin C, Iimuro Y, Licato L, Sartor RB, Brenner DA. Inhibition of NFkappaB in activated rat hepatic stellate cells by proteasome inhibitors and an IkappaB super-repressor. Hepatology. 1998;27:1285–1295. doi: 10.1002/hep.510270514. [DOI] [PubMed] [Google Scholar]
- 10.Elsharkawy AM, Wright MC, Hay RT, Arthur MJ, Hughes T, Bahr MJ, Degitz K, Mann DA. Persistent activation of nuclear factor-kappaB in cultured rat hepatic stellate cells involves the induction of potentially novel Rel-like factors and prolonged changes in the expression of IkappaB family proteins. Hepatology. 1999;30:761–769. doi: 10.1002/hep.510300327. [DOI] [PubMed] [Google Scholar]
- 11.Schwabe RF, Schnabl B, Kweon YO, Brenner DA. CD40 activates NF-kappa B and c-Jun N-terminal kinase and enhances chemokine secretion on activated human hepatic stellate cells. J Immunol. 2001;166:6812–6819. doi: 10.4049/jimmunol.166.11.6812. [DOI] [PubMed] [Google Scholar]
- 12.Hellerbrand C, Jobin C, Licato LL, Sartor RB, Brenner DA. Cytokines induce NF-kappaB in activated but not in quiescent rat hepatic stellate cells. Am J Physiol. 1998;275:G269–G278. doi: 10.1152/ajpgi.1998.275.2.G269. [DOI] [PubMed] [Google Scholar]
- 13.Gaweco AS, Wiesner RH, Yong S, Krom R, Porayko M, Chejfec G, McClatchey KD, Van Thiel DH. CD40L (CD154) expression in human liver allografts during chronic ductopenic rejection. Liver Transpl Surg. 1999;5:1–7. doi: 10.1002/lt.500050108. [DOI] [PubMed] [Google Scholar]
- 14.Brand K, Page S, Rogler G, Bartsch A, Brandl R, Knuechel R, Page M, Kaltschmidt C, Baeuerle PA, Neumeier D. Activated transcription factor nuclear factor-kappa B is present in the atherosclerotic lesion. J Clin Invest. 1996;97:1715–1722. doi: 10.1172/JCI118598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Longo CR, Arvelo MB, Patel VI, Daniel S, Mahiou J, Grey ST, Ferran C. A20 protects from CD40-CD40 ligand-mediated endothelial cell activation and apoptosis. Circulation. 2003;108:1113–1118. doi: 10.1161/01.CIR.0000083718.76889.D0. [DOI] [PubMed] [Google Scholar]
- 16.Arvelo MB, Badrichani AZ, Stroka DM, Grey ST, Bach FH, Ferran C. A novel function for A20 in smooth muscle cells: inhibition of activation and proliferation. Transplant Proc. 1999;31:858–859. doi: 10.1016/s0041-1345(98)01804-1. [DOI] [PubMed] [Google Scholar]
- 17.Patel VI, Daniel S, Longo CR, Shrikhande GV, Scali ST, Czismadia E, Groft CM, Shukri T, Motley-Dore C, Ramsey HE, et al. A20, a modulator of smooth muscle cell proliferation and apoptosis, prevents and induces regression of neointimal hyperplasia. FASEB J. 2006;20:1418–1430. doi: 10.1096/fj.05-4981com. [DOI] [PubMed] [Google Scholar]
- 18.Daniel S, Patel VI, Shrikhande GV, Scali ST, Ramsey HE, Csizmadia E, Benhaga N, Fisher MD, Arvelo MB, Ferran C. The universal NF-kappaB inhibitor a20 protects from transplant vasculopathy by differentially affecting apoptosis in endothelial and smooth muscle cells. Transplant Proc. 2006;38:3225–3227. doi: 10.1016/j.transproceed.2006.10.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brand K, Page S, Walli AK, Neumeier D, Baeuerle PA. Role of nuclear factor-kappa B in atherogenesis. Exp Physiol. 1997;82:297–304. doi: 10.1113/expphysiol.1997.sp004025. [DOI] [PubMed] [Google Scholar]
- 20.Opipari AW, Boguski MS, Dixit VM. The A20 cDNA induced by tumor necrosis factor alpha encodes a novel type of zinc finger protein. J Biol Chem. 1990;265:14705–14708. [PubMed] [Google Scholar]
- 21.Arvelo MB, Cooper JT, Longo C, Daniel S, Grey ST, Mahiou J, Czismadia E, Abu-Jawdeh G, Ferran C. A20 protects mice from D-galactosamine/lipopolysaccharide acute toxic lethal hepatitis. Hepatology. 2002;35:535–543. doi: 10.1053/jhep.2002.31309. [DOI] [PubMed] [Google Scholar]
- 22.Longo CR, Patel VI, Shrikhande GV, Scali ST, Csizmadia E, Daniel S, Sun DW, Grey ST, Arvelo MB, Ferran C. A20 protects mice from lethal radical hepatectomy by promoting hepatocyte proliferation via a p21waf1-dependent mechanism. Hepatology. 2005;42:156–164. doi: 10.1002/hep.20741. [DOI] [PubMed] [Google Scholar]
- 23.Daniel S, Arvelo MB, Patel VI, Longo CR, Shrikhande G, Shukri T, Mahiou J, Sun DW, Mottley C, Grey ST, et al. A20 protects endothelial cells from TNF-, Fas-, and NK-mediated cell death by inhibiting caspase 8 activation. Blood. 2004;104:2376–2384. doi: 10.1182/blood-2003-02-0635. [DOI] [PubMed] [Google Scholar]
- 24.Cooper JT, Stroka DM, Brostjan C, Palmetshofer A, Bach FH, Ferran C. A20 blocks endothelial cell activation through a NF-kappaB-dependent mechanism. J Biol Chem. 1996;271:18068–18073. doi: 10.1074/jbc.271.30.18068. [DOI] [PubMed] [Google Scholar]
- 25.Beyaert R, Heyninck K, Van Huffel S. A20 and A20-binding proteins as cellular inhibitors of nuclear factor-kappa B-dependent gene expression and apoptosis. Biochem Pharmacol. 2000;60:1143–1151. doi: 10.1016/s0006-2952(00)00404-4. [DOI] [PubMed] [Google Scholar]
- 26.Ferran C, Stroka DM, Badrichani AZ, Cooper JT, Wrighton CJ, Soares M, Grey ST, Bach FH. A20 inhibits NF-kappaB activation in endothelial cells without sensitizing to tumor necrosis factor-mediated apoptosis. Blood. 1998;91:2249–2258. [PubMed] [Google Scholar]
- 27.Zhang SQ, Kovalenko A, Cantarella G, Wallach D. Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKgamma) upon receptor stimulation. Immunity. 2000;12:301–311. doi: 10.1016/s1074-7613(00)80183-1. [DOI] [PubMed] [Google Scholar]
- 28.Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 29.Liang TB, Man K, Kin-Wah Lee T, Hong-Teng Tsui S, Lo CM, Xu X, Zheng SS, Fan ST, Wong J. Distinct intragraft response pattern in relation to graft size in liver transplantation. Transplantation. 2003;75:673–678. doi: 10.1097/01.TP.0000048490.24429.89. [DOI] [PubMed] [Google Scholar]
- 30.Ramsey HE, Da Silva CG, Longo CR, Csizmadia E, Studer P, Patel VI, Damrauer SM, Siracuse JJ, Daniel S, Ferran C. A20 protects mice from lethal liver ischemia/reperfusion injury by increasing peroxisome proliferator-activated receptor-alpha expression. Liver Transpl. 2009;15:1613–1621. doi: 10.1002/lt.21879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Avihingsanon Y, Ma N, Csizmadia E, Wang C, Pavlakis M, Giraldo M, Strom TB, Soares MP, Ferran C. Expression of protective genes in human renal allografts: a regulatory response to injury associated with graft rejection. Transplantation. 2002;73:1079–1085. doi: 10.1097/00007890-200204150-00011. [DOI] [PubMed] [Google Scholar]
- 32.Xu MQ, Wang W, Xue L, Yan LN. NF-kappaB activation and zinc finger protein A20 expression in mature dendritic cells derived from liver allografts undergoing acute rejection. World J Gastroenterol. 2003;9:1296–1301. doi: 10.3748/wjg.v9.i6.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu MQ, Yan LN, Gou XH, Li DH, Huang YC, Hu HY, Wang LY, Han L. Zinc finger protein A20 promotes regeneration of small-for-size liver allograft and suppresses rejection and results in a longer survival in recipient rats. J Surg Res. 2009;152:35–45. doi: 10.1016/j.jss.2008.04.029. [DOI] [PubMed] [Google Scholar]
- 34.Braet F, De Zanger R, Sasaoki T, Baekeland M, Janssens P, Smedsrød B, Wisse E. Assessment of a method of isolation, purification, and cultivation of rat liver sinusoidal endothelial cells. Lab Invest. 1994;70:944–952. [PubMed] [Google Scholar]
- 35.Iwamoto H, Nakamuta M, Tada S, Sugimoto R, Enjoji M, Nawata H. A p160ROCK-specific inhibitor, Y-27632, attenuates rat hepatic stellate cell growth. J Hepatol. 2000;32:762–770. doi: 10.1016/s0168-8278(00)80245-7. [DOI] [PubMed] [Google Scholar]
- 36.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 37.Xu J, Xie J, Bao M, Li Z, Yang Z. NF-kappaB/I-kappaB pathway during ischemia reperfusion injury of rat liver. Chin Med J (Engl) 2003;116:1146–1149. [PubMed] [Google Scholar]
- 38.Fujihara SM, Cleaveland JS, Grosmaire LS, Berry KK, Kennedy KA, Blake JJ, Loy J, Rankin BM, Ledbetter JA, Nadler SG. A D-amino acid peptide inhibitor of NF-kappa B nuclear localization is efficacious in models of inflammatory disease. J Immunol. 2000;165:1004–1012. doi: 10.4049/jimmunol.165.2.1004. [DOI] [PubMed] [Google Scholar]
- 39.Okada M, Okamoto T, Yamada S, Yamada S, Itoh T, Mori A, Saheki K, Takatsuka H, Wada H, Tamura A Y, et al. Successful treatment of chronic graft-versus-host disease with sulfasalazine in allogeneic bone marrow transplantation. Acta Haematol. 1999;102:107–109. doi: 10.1159/000040981. [DOI] [PubMed] [Google Scholar]
- 40.May MJ, Ghosh S. Signal transduction through NF-kappa B. Immunol Today. 1998;19:80–88. doi: 10.1016/s0167-5699(97)01197-3. [DOI] [PubMed] [Google Scholar]
- 41.Oikawa K, Ohkohchi N, Sato M, Masamume A, Satomi S. Kupffer cells play an important role in the cytokine production and activation of nuclear factors of liver grafts from non-heart-beating donors. Transpl Int. 2002;15:397–405. doi: 10.1007/s00147-002-0435-8. [DOI] [PubMed] [Google Scholar]
- 42.Miyagawa-Hayashino A, Tsuruyama T, Egawa H, Haga H, Sakashita H, Okuno T, Toyokuni S, Tamaki K, Yamabe H, Manabe T, et al. FasL expression in hepatic antigen-presenting cells and phagocytosis of apoptotic T cells by FasL+ Kupffer cells are indicators of rejection activity in human liver allografts. Am J Pathol. 2007;171:1499–1508. doi: 10.2353/ajpath.2007.070027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen Y, Liu Z, Liang S, Luan X, Long F, Chen J, Peng Y, Yan L, Gong J. Role of Kupffer cells in the induction of tolerance of orthotopic liver transplantation in rats. Liver Transpl. 2008;14:823–836. doi: 10.1002/lt.21450. [DOI] [PubMed] [Google Scholar]
- 44.Luo D, Vanderkerken K, Bouwens L, Kuppen PJ, Baekeland M, Seynaeve C, Wisse E. The role of adhesion molecules in the recruitment of hepatic natural killer cells (pit cells) in rat liver. Hepatology. 1996;24:1475–1480. doi: 10.1002/hep.510240629. [DOI] [PubMed] [Google Scholar]
- 45.Yamamoto H, Ohdan H, Shintaku S, Asahara T, Ito H, Dohi K. Role of the bcl-2/bax pathway in hepatocyte apoptosis during acute rejection after rat liver transplantation. Transpl Int. 1998;11 Suppl 1:S179–S184. doi: 10.1007/s001470050456. [DOI] [PubMed] [Google Scholar]
- 46.Krams SM, Egawa H, Quinn MB, Villanueva JC, Garcia-Kennedy R, Martinez OM. Apoptosis as a mechanism of cell death in liver allograft rejection. Transplantation. 1995;59:621–625. [PubMed] [Google Scholar]
- 47.Hsieh CL, Obara H, Ogura Y, Martinez OM, Krams SM. NK cells and transplantation. Transpl Immunol. 2002;9:111–114. doi: 10.1016/s0966-3274(02)00033-3. [DOI] [PubMed] [Google Scholar]
- 48.Kojima Y, Kawasaki-Koyanagi A, Sueyoshi N, Kanai A, Yagita H, Okumura K. Localization of Fas ligand in cytoplasmic granules of CD8+ cytotoxic T lymphocytes and natural killer cells: participation of Fas ligand in granule exocytosis model of cytotoxicity. Biochem Biophys Res Commun. 2002;296:328–336. doi: 10.1016/s0006-291x(02)00841-0. [DOI] [PubMed] [Google Scholar]
- 49.Ariki N, Morimoto Y, Yagi T, Oyama T, Cyouda Y, Sadamori H, Inagaki M, Urushihara N, Iwagaki H, Tanaka N. Activated T cells and soluble molecules in the portal venous blood of patients with cholestatic and hepatitis C virus-positive liver cirrhosis. Possible promotion of Fas/FasL-mediated apoptosis in the bile-duct cells and hepatocyte injury. J Int Med Res. 2003;31:170–180. doi: 10.1177/147323000303100302. [DOI] [PubMed] [Google Scholar]
- 50.Wang J, Li W, Min J, Ou Q, Chen J. Fas siRNA reduces apoptotic cell death of allogeneic-transplanted hepatocytes in mouse spleen. Transplant Proc. 2003;35:1594–1595. doi: 10.1016/s0041-1345(03)00438-x. [DOI] [PubMed] [Google Scholar]