

Abstract
Activity of voltage-gated Na channels (Nav) is modified by alternative splicing. However, whether altered splicing of human Navs contributes to epilepsy remains to be conclusively shown. We show here that altered splicing of the Drosophila Nav (paralytic, DmNav) contributes to seizure-like behavior in identified seizure mutants. We focus attention on a pair of mutually exclusive alternate exons (termed K and L), which form part of the voltage sensor (S4) in domain III of the expressed channel. The presence of exon L results in a large, non-inactivating, persistent INap. Many forms of human epilepsy are associated with an increase in this current. In wild-type (WT) Drosophila larvae, ∼70–80% of DmNav transcripts contain exon L, and the remainder contain exon K. Splicing of DmNav to include exon L is increased to ∼100% in both the slamdance and easily-shocked seizure mutants. This change to splicing is prevented by reducing synaptic activity levels through exposure to the antiepileptic phenytoin or the inhibitory transmitter GABA. Conversely, enhancing synaptic activity in WT, by feeding of picrotoxin is sufficient to increase INap and promote seizure through increased inclusion of exon L to 100%. We also show that the underlying activity-dependent mechanism requires the presence of Pasilla, an RNA-binding protein. Finally, we use computational modeling to show that increasing INap is sufficient to potentiate membrane excitability consistent with a seizure phenotype. Thus, increased synaptic excitation favors inclusion of exon L, which, in turn, further increases neuronal excitability. Thus, at least in Drosophila, this self-reinforcing cycle may promote the incidence of seizure.
Introduction
Alternative splicing involves the substitution, removal, and/or inclusion of exonic sequences within a pre-mRNA to produce transcripts encoding related protein isoforms (Li et al., 2007). Estimates indicate that ∼95% of human genes are alternatively spliced (Pan et al., 2008; Wang et al., 2008). How splicing influences function of voltage-gated Na channel (Nav) transcripts, and whether such changes promote seizure, is complicated by the genetic redundancy present in the mammalian genome. Recent reports suggest, however, that Navs show altered splicing in mesial temporal lobe epilepsy and that a single nucleotide polymorphism is sufficient to influence splicing of exon 5N in Nav1.1, an effect that is associated with altered sensitivity to established antiepileptic drugs and possibly increased risk of febrile seizures (Heinzen et al., 2007; Schlachter et al., 2009; Thompson et al., 2011).
In contrast to mammals, the genome of the fruitfly Drosophila melanogaster contains only one Nav channel homolog: encoded by paralytic (Feng et al., 1995; Mee et al., 2004). This, coupled with the high degree of structural and functional homology, makes DmNav an ideal model with which to study the role of alternative splicing of this ion channel family (Lin et al., 2009). Our previous work described the complete pattern of alternative splicing of DmNav isolated from Drosophila embryonic CNS (Lin et al., 2009). In particular, we identified a pair of mutually exclusive, membrane-spanning exons (termed K and L) that markedly affect the magnitude of the persistent current (INap) that arises from incomplete inactivation of the channel (Kiss, 2008). The magnitude of INap ranges from 4.1 to 9.5% of peak transient current (INat) in transcripts containing exon L. In contrast, inclusion of exon K reduces this to 1.5–2.4%. Although relatively small compared with INat, the effect INap has on membrane excitability can be substantial (Darbon et al., 2004; Li et al., 2004; Vervaeke et al., 2006). Indeed, a number of mutations in Nav channels, seemingly causative of human epilepsy, specifically increase INap (Stafstrom, 2007; Ragsdale, 2008). Intriguingly, we recently reported that the seizure phenotype characteristic of the larval Drosophila slamdance (sda) mutant is also associated with an increased INap in central motoneurons. In contrast, the magnitude of INat was not affected (Marley and Baines, 2011). How loss of the sda gene, which encodes the fly homolog of mammalian aminopeptidase N (APN), results in heightened seizures remains unknown (Zhang et al., 2002). In mammals, APN is widely expressed and catalyzes the removal of basic and neutral amino acids from the N terminals of peptides (Inagaki et al., 2010). Intriguingly, the related insulin-regulated amino peptidase has been implicated to contribute to seizure, primarily through an as yet undefined interaction with angiotensin IV (Stragier et al., 2008). However, the precise mechanistic details also remain unknown.
We show here that the choice to splice either exons K or L is perturbed in the sda mutant to favor exclusive inclusion of L. This change is rescued by pretreatment of sda larvae with either the antiepileptic phenytoin or the inhibitory transmitter GABA and recapitulated in wild type (WT) by exposure to picrotoxin (PTx), a known proconvulsive. These observations are indicative that the underlying splicing mechanism is activity dependent. Increased inclusion of exon L requires the presence of Pasilla (Ps), a known RNA-binding protein that we and others have previously shown regulates splicing of exons K and L (Park et al., 2004; Lin et al., 2009). Finally, we use a computational approach to show that increasing INap is sufficient to increase membrane excitability consistent with the sda epileptic phenotype. Together, our results indicate that increased synaptic activity influences the decision to splice in exon L, which, in turn, may promote seizure.
Materials and Methods
Fly stocks.
Flies (of either sex) were maintained on standard cornmeal medium at 25°C. Gal4RRa (J. Jaynes, Thomas Jefferson University, Philadelphia, PA) was used to drive UAS–GFPCD8 in the aCC motoneuron in third-instar larvae. In earlier larval stages, this line also expresses in the RP2 motoneuron and, to a lesser extent, in the pCC interneuron. However, by late wall climbing, third-instar-only aCC expression remains robust (Fig. 1). WT was Canton-S. A UAS–ps RNAi was obtained from Bloomington Drosophila Stock Center (stock #33426).
Figure 1.

Splicing of alternate exons K and L is altered in the sda mutant. A, Schematic of the DmNav transcript highlighting common spliced exons. Exons J, I, A, B, E, F, and H are subject to cassette-based splicing (i.e., can be present or absent), whereas exons C/D and K/L are mutually exclusive (i.e., one or other are present but not both). Inset, Exons K and L differ by 16 aa. B, Whole-cell voltage-clamp recordings from a third-instar aCC neuron reveals an increased INap in sda compared with WT. In contrast, INat is not different (for a full description, see Marley and Baines, 2011). C, GAL4RRa is sufficient to express GFP (UAS–GFPCD8 shown) in only aCC neurons by late-stage, wall-climbing third-instar larvae. A wide-field deconvolved fluorescent image shows three segments of the ventral nerve cord each containing two aCC motoneurons. Anterior is to the top. Scale bar, 10 μm. D, Analysis of splicing of DmNav, in aCC neurons isolated by FACS, shows that inclusion of exon L is greatly increased in the sda mutant (70.2 vs 96%, p ≤ 0.01). Preexposure of sda larvae to the antiepileptic drug phenytoin (phy) is sufficient to partially rescue this change (76.5%, p ≤ 0.05). Only functional DmNav splice variants are included in this analysis. Thus, DmNav69, in which K and L are coexpressed in the sda mutant, is not included because it is nonfunctional (see Materials and Methods).
Fluorescence-activated cell sorting.
CNSs (50–200) were dissected from late-wall-climbing third-instar larvae in saline (Marley and Baines, 2011) and then dissociated by incubating in 1× PBS containing 1 mg/ml Collagenase/Dispase II (Roche) for 2 h at room temperature. After a wash in 1× PBS, cells were triturated in Schneider's Drosophila medium (Invitrogen) and strained through a 35 μm nylon mesh filter (BD Biosciences) to remove larger chunks of material. Green fluorescence-positive cells were sorted and collected using a BD FACS-Aria cell sorter (BD Biosciences). Sorted cells were tested for purity by PCR. The dorsal motoneuron aCC expresses the transcription factor even-skipped (eve) but not the transcription factors islet or lim3 (Landgraf and Thor, 2006) and as a glutamatergic cell also expresses the vesicular glutamate transporter (vGlut) and not dopamine decarboxylase (Ddc; required for monoamine transmitter synthesis). PCR in isolated cells shows considerable enrichment for eve and vGlut but compared with CNS only trace levels (<10%) for islet, lim3, or Ddc. Thus, we estimate purity to be ≥90%.
Cloning of DmNav splice variants.
Total RNA was extracted from fluorescence-activated cell sorted cells using the RNeasy micro kit (QIAGEN). cDNA synthesis was performed in a total volume of 20 μl. A primer specific to DmNav (5′-GTGTGAAAAAGGATCCAAATATGA-3′) (0.2 μg), located at exon 28, and random hexamer (0.2 μg) were mixed with RNA and made up to 12 μl with RNase-free water. The mix was incubated at 65°C for 5 min to denature RNA, followed by incubation on ice for 2 min. To this was added 4 μl of reaction buffer (in mm: 250 Tris-HCl, 250 KCl, 20 MgCl2, and 50 DTT), 2 μl of 10 mm dNTPs, 1 μl of RNase inhibitor, and 1 μl of RevertAid M-MuLV (monkey murine leukemia virus) reverse transcriptase (RevertAid First Strand cDNA Synthesis kit; Fermentas). The reaction was incubated at 25°C for 10 min, 42°C for 60 min, followed by 70°C for 10 min. To amplify DmNav open-reading frames (ORFs), PCR primers were designed to flank the region between exon I and exon L: forward primer, 5′-GAACTCTAGATGGCCAAGAGTCCGACGTATT-3′ (introduces XbaI site); reverse primer, 5′-GCCTGCGGCCGCATTGCGATTTGGTATGATCTCGTG-3′ (introduces NotI site). The PCR mixture consisted of 2 μl of cDNA, 2 μl of Elongase Enzyme Mix (Invitrogen), dNTPs at a final concentration of 0.2 mm each, 7.6 pmol of each primer, and 1× PCR buffer with a final Mg2+ concentration of 1.3 mm. Cycling conditions were as follows: initial denaturation at 94°C for 30 s; 40 cycles of 94°C for 30 s, 58°C for 30 s, and 68°C for 4 min; a final extension step at 68°C for 10 min. This amplifies an ORF that includes all known spliced exons with the exception of exon J (Lin et al., 2009, their Fig. 1). PCR shows that exon J is not expressed in the third-instar CNS.
The PCR product was double digested by XbaI and NotI (Fermentas), excised from an agarose gel (0.7%) after electrophoresis, and purified using the QIAquick Gel Extraction kit (QIAGEN). The purified PCR product was then ligated into a pGH19 vector. The ligation mix was used to transform MAX Efficiency DH5α Competent Cells (Invitrogen) according to the protocol of the manufacturer but incubating transformants at 30°C instead of 37°C. DNA was prepared using the QIAprep Spin Miniprep kit (QIAGEN), and the concentration was determined using a NanoVue spectrophotometer (GE Healthcare).
Determination of exon inclusion.
Plasmids were denatured by heating at 95°C for 5 min and then 1 μl spotted (2 ng/μl) on to nylon membranes (Roche). Prehybridization was performed at 45°C in DIG Easy Hyb buffer (Roche), followed by hybridization with 10 pmol digoxigenin (DIG)-labeled oligonucleotides, which were labeled following the protocol in the DIG Oligonucleotide 3′-End Labeling kit (Roche). The membranes were washed in 1× SSC with 0.1% SDS for 10 min at various temperatures depending on the melting temperature of oligonucleotides. The primers and washing conditions are described in Table 1. A subsequent DIG nucleic acid color detection kit (Roche) was used to visualize hybrid molecules. Frequency of inclusion was determined by dividing the number of sense variants (i.e., those that express functional channels) containing a particular spliced exon (e.g., exon L) by the total number of sense variants identified. Splice variants that do not express functional channels (i.e., either lacking both exons K and L or containing both) are shown for completeness but excluded from analysis of exon inclusion.
Table 1.
Primers and washing conditions used to identify spliced exons in DmNav clones
| Probe | Sequence | Temperature |
|---|---|---|
| Exon I | GCGTTATACAAAGACAACCAGCACCT | 60°C |
| Exon A | ATCCTTACCTGGTTCACCGTTTAACA | 45°C |
| Exon B | GTCTCCGTTTACTATTTCCCAACA | 45°C |
| Exon C | CATGCAGCTGTTTGGCAAGAACTACACA | 60°C |
| Exon D | AATGCAACTGTTCGGAAAGAATTATCAT | 55°C |
| Exon E | AGAGGACCAACCAGATCAGTTGGATTT | 55°C |
| Exon F | CAAAGGCGTTTGTCGTTGTATATCTGC | 55°C |
| Exon H | ACCAAGACAATAGACTGGAACACGAGC | 60°C |
| Exon K | AATTAATTTGGCCGCGGTCTG | 55°C |
| Exon L | GCTTATCAACTTCGTTGCTTCACTTGT | 45°C |
Real-time PCR.
A Taqman probe method (Lightcycler Taqman Master; Roche) was used for quantification of exons K and L. Cycle threshold (Ct) values, as defined by the default setting, were measured using a Lightcycler 1.5 real-time PCR machine (Roche). The thermal profile used was 10 min at 95°C, followed by 40 cycles of 10 s at 95°C, followed by 20 s at 65°C, and finally 20 s at 72°C. Absolute molecule number for each transcript was calculated by comparing with a standard curve established by measuring the Ct values of known amounts of cDNA. Samples were measured in duplicate from five independent isolations of RNA. The real-time primer pairs and TaqMan probe were designed with the aid of Primer Express 2.0 (Invitrogen) and purchased from Eurofins MWG Operon. Primer sequences (5′ to 3′) were as follows: L exon, TGGCTCGATTTCGTGATTGTC and CCACCAGCTCCAACAAGTGAA; K exon, TGATTGTCATGCTGTCGCTAATTAAT and GAAAGGCGGGCACATCATC; Taqman probe for L exon, TAMRA-TGGTATCGCTTATCAACTTCGTTG-BHQ2; and Taqman probe for K exon, FAM-TGGCCGCGGTCTGGTCCG-BHQ1.
Seizure induction and electrophysiology.
Wall-climbing third-instar larvae were subjected to electric shock to test seizure severity, with or without previous feeding of drugs, as described previously (Marley and Baines, 2011). For drug feeding studies, larvae were raised on food containing drug for ≥24 h before analysis. PTx and phenytoin were acquired from Sigma-Aldrich. Mean recovery times (MRTs) shown represent the average time for larvae to resume normal crawling behavior. Synaptic currents were measured from third-instar aCC motoneurons using whole-cell voltage clamp (Multiclamp 700B amplifier; Molecular Devices) also as described previously (Marley and Baines, 2011). Voltage-gated sodium currents were recorded from Xenopus oocytes using standard two-electrode voltage clamp (Axoclamp 2B amplifier; Molecular Devices) as described previously (Lin et al., 2009). To eliminate the contribution of endogenous currents, currents were recorded first in the absence and then in the presence of TTX. Subtraction of the latter from the former isolated the Na+ conductance. Three mixtures of cRNAs were injected to mimic the splicing observed in WT, sda, and sda-fed phenytoin aCC neurons. For each mixture, only those variants present in three or more copies were included in this mixture (for details, see Results). The choice to include only those variants present in three or more copies was based on number of variants identified in some genotypes, many of which were only present in two copies and as such made little contribution (<10%) to overall variant frequency.
Voltage dependence of activation was determined by applying 100 ms voltage steps in 10 mV increments (−80 to 0 mV) from a holding potential of −90 mV. Currents evoked were normalized by dividing by the peak current to derive I/Imax. These values were then plotted against membrane potential (Origin 8; Microcal) and the half-activation (V½) calculated from that plot. Voltage-dependent inactivation was determined by applying 100 ms prepulses (−80 to 0 mV in 10 mV increments) before stepping to a −10 mV test potential (50 ms). Current amplitudes at the test potential were again normalized to the peak current obtained. V½ inactivation was determined by plots of I/Imax versus membrane potential. To calculate the magnitude of INap, the voltage steps eliciting the largest transient current and largest persistent current (recorded at 100 ms after onset of voltage jump) were used. These were not necessarily from the same test potential.
Statistics.
Significance of changes in splicing frequencies observed was tested using either a χ2 test (Fig. 1D) or a Student's t test (all other figures). All data shown are means ± SE.
Computational model of the aCC motoneuron.
We developed a simple ball-and-stick model of the third-instar larval aCC motoneuron. Construction of this model will be explained in detail elsewhere. This model has two compartments of 4 pF capacitance each, one being a passive compartment representing the soma, connected with an axial conductance of 1 nS to an axonal compartment with active channels INat, INap, and fast, slow, and leak IK. Following the Hodgkin–Huxley formalism, currents were modeled as I = ḡmph(V − E), where m and h are gating variables calculated with differential equations of the form dm/dt = (m∞ − m)τm. The steady state of the gate was defined as m∞ =1/(1 + exp((V − V½)/k)). The parameters in these equations for each of the currents are given in Table 2. The model was simulated using the XPP-Aut software (Ermentrout, 2002) using the Euler numerical integration method with a step size parameter of 0.001 ms. There are more complex mammalian sodium channel models with multiple inactivation mechanisms and that model microscopic channel properties as Markov states (Kahlig et al., 2006). However, we have chosen to use a Hodgkin–Huxley model formalism because the current study focuses on the effects of the macroscopic persistent current remaining after fast inactivation. Because of this, it should be noted that our model cannot replicate activity resulting from slow inactivation (e.g., slow recovery from inactivation) and assumes independence of activation and inactivation gates.
Table 2.
Parameters of the currents included in the active axonal compartment of the motoneuron model
| Channel | Rev. E (mV) | ḡ | p | Active V½ (mV) | Active k (mV) | Inactive V½ (mV) | Inactive k (mV) | τm (ms) | τh (ms) |
|---|---|---|---|---|---|---|---|---|---|
| Nat | 45 | 100 | 3 | −29.13 | −8.92 | −40 | 6.04 | ||
| Nap | 45 | 0.82 | 1 | −48.77 | −3.68 | 1 | |||
| Kslow | −80 | 50 | 4 | −12.85 | −19.91 | ||||
| Kfast | −80 | 20.1 | 4 | −17.55 | −7.27 | −45 | 6 |
The transient sodium (Nat) current parameters were calculated from published embryonic Drosophila motoneuron observations (O'Dowd and Aldrich 1988). The persistent sodium (Nap) current model was fit to data from oocyte recordings of DmNav10. Both fast inactivating (Kfast) and slow (Kslow) K currents were modeled using data recorded from third-instar motoneurons. Kfast was modeled with a second inactivation gate, with constant 116 ms decay time constant, that only contributed to 5% of its response. The leak K current had a conductance of 0.3 nS in the soma and 6.8 nS in the axon. A holding current of −12 pA was applied to the model motoneuron to keep its membrane potential at the physiological rest value of −60 mV.
Results
Splicing of exons K and L in DmNav is altered in the sda mutant
The sda mutant is one of a collection of mutants collectively termed the “bang-sensitives.” These mutants are recognized models of epilepsy (Muraro et al., 2008). In a previous study, we showed that motoneurons in the sda mutant exhibit increased persistent (INap), but no change in transient (INat), voltage-gated Na+ current (Marley and Baines, 2011). A change to only one of the two current components, carried by the same channel protein, is inconsistent with a change in gene expression. Instead, we hypothesized that this change may be attributable to altered alternative splicing of the DmNav pre-mRNA transcript to favor inclusion of exon L rather than its mutually exclusive alternative exon K (Fig. 1A). This is because expressed channels containing exon L exhibit increased INap, with no change to INat, compared with expressed channels containing exon K (Lin et al., 2009).
To determine experimentally whether a change of splicing underlies our reported increase in magnitude of INap in sda motoneurons (Fig. 1B; Marley and Baines, 2011), we cloned and analyzed DmNav transcripts isolated from third-instar aCC motoneurons (the same neuron used for electrophysiology). Cells were isolated by fluorescence-activated cell sorting (FACS) based on expression of GFP in aCC using the GAL4RRa driver (Fig. 1C). ORFs of DmNav were amplified by PCR and cloned into E. coli, and splicing was determined using exon-specific probes. For this analysis, we included only those transcripts present in two or more copies. We considered that transcripts present as single copies may result from random splicing errors. However, even when all functional clones (i.e., those that produce functional channels when expressed in Xenopus oocytes) are included, regardless of copy number, there were no qualitative differences.
Analysis of exon composition of individual DmNav clones, derived from both WT and sda aCC motoneurons, showed a significant increase in inclusion of exon L in the latter. Thus, in those transcripts that will result in functional channels (i.e., contain only one of exon K or L), the inclusion of exon L was increased from 70.2% in WT to 96% in sda (p ≤ 0.01; Fig. 1D, Table 3). We also analyzed splicing of K/L in another bang-sensitive mutation, easily-shocked (eas), that also exhibits an increased INap relative to WT (Marley and Baines, 2011). We observed the same striking shift in inclusion of exon L in DmNav transcripts isolated from aCC (95.7%, p ≤ 0.01; data not shown).
Table 3.
Splicing of DmNav in the aCC motoneuron in WT, sda, and phenytoin-fed sda backgrounds
| Genotype/drug | DmNav | J | I | A | B | C | D | E | F | H | K | L | # Clones | % |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | 33 | I | A | B | D | L | 19 | 40.4 | ||||||
| 58 | I | A | B | D | K | 9 | 19.1 | |||||||
| 30 | I | A | B | D | F | L | 7 | 14.9 | ||||||
| 54 | I | A | B | D | F | K | 5 | 10.6 | ||||||
| 55 | I | A | B | D | H | L | 4 | 8.5 | ||||||
| 32 | I | A | B | D | F | H | L | 3 | 6.4 | |||||
| sda | 31 | I | B | D | F | L | 13 | 22.8 | ||||||
| 43 | I | B | D | L | 11 | 19.3 | ||||||||
| 55 | I | A | B | D | H | L | 9 | 15.8 | ||||||
| 33 | I | A | B | D | L | 7 | 12.3 | |||||||
| 69 | I | A | B | D | K | L | 7 | 12.3 | ||||||
| 30 | I | A | B | D | F | L | 5 | 8.8 | ||||||
| 59 | I | A | D | L | 3 | 5.3 | ||||||||
| 58 | I | A | B | D | K | 2 | 3.5 | |||||||
| sda + phenytoin | 30 | I | A | B | D | F | L | 12 | 23.5 | |||||
| 31 | I | B | D | F | L | 7 | 13.7 | |||||||
| 33 | I | A | B | D | L | 7 | 13.7 | |||||||
| 58 | I | A | B | D | K | 7 | 13.7 | |||||||
| 32 | I | A | B | D | F | H | L | 4 | 7.8 | |||||
| 43 | I | B | D | L | 4 | 7.8 | ||||||||
| 54 | I | A | B | D | F | K | 3 | 5.9 | ||||||
| 55 | I | A | B | D | H | L | 3 | 5.9 | ||||||
| 34 | I | B | D | F | H | L | 2 | 3.9 | ||||||
| 60 | I | B | D | K | 2 | 3.9 | ||||||||
The inclusion of exon L increases from 70.2% in WT to 96% in sda (p ≤ 0.01). Exposure of sda larvae to phenytoin rescues the inclusion of L to 76.5% (p ≤ 0.05). Note that DmNav69 isolated from sda contains both exons K and L. Nomenclature of DmNav splice variants has been described previously (Lin et al., 2009). Only those splice variants present in two or more copies are shown isolated from 50, 59, and 56 clones, respectively. DmNavs shown in bold (i.e., 69) are nonfunctional in that they do not give rise to functional channels. These clones are shown for reference only and have not been used for determination of frequency of inclusion for either exons K and L. The letters J–L denote alternative exons in DmNav, and the presence of the letter denotes inclusion in the transcript (for details, see Lin et al., 2009).
To determine how global this change to splicing is, we also analyzed splicing in whole CNS. Our results showed an identical trend to aCC (87.8 vs 100% inclusion of exon L, WT vs sda, respectively, p ≤ 0.01; Table 4). Thus, the change to favor inclusion of exon L is shared by the majority, if not all, central neurons in the sda mutant. It is also interesting to note that DmNav transcripts isolated from WT CNS show increased inclusion of exon L compared with aCC (87.8 vs 70.2%, respectively).
Table 4.
Splicing of DmNav in all central neurons in WT and sda backgrounds
| Genotype/drug | DmNav | J | I | A | B | C | D | E | F | H | K | L | # Clones | % |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | 31 | I | B | D | F | L | 64 | 30.6 | ||||||
| 30 | I | A | B | D | F | L | 49 | 23.4 | ||||||
| 33 | I | A | B | D | L | 22 | 10.5 | |||||||
| 32 | I | A | B | D | F | H | L | 19 | 9.1 | |||||
| 43 | I | B | D | L | 12 | 5.7 | ||||||||
| 26 | I | B | D | F | K | 10 | 4.8 | |||||||
| 58 | I | A | B | D | K | 7 | 3.3 | |||||||
| 34 | I | B | D | F | H | L | 6 | 2.9 | ||||||
| 54 | I | A | B | D | F | K | 4 | 1.9 | ||||||
| 62 | I | A | B | D | E | F | L | 4 | 1.9 | |||||
| 42 | I | B | D | E | F | 3 | 1.4 | |||||||
| 55 | I | A | B | D | H | L | 3 | 1.4 | ||||||
| 41 | I | A | B | D | E | L | 2 | 1.0 | ||||||
| 47 | I | A | B | D | F | H | K | 2 | 1.0 | |||||
| 48 | I | B | D | F | H | K | 2 | 1.0 | ||||||
| sda | 30 | I | A | B | D | F | L | 8 | 21.1 | |||||
| 33 | I | A | B | D | L | 7 | 18.4 | |||||||
| 31 | I | B | D | F | L | 4 | 10.5 | |||||||
| 43 | I | B | D | L | 4 | 10.5 | ||||||||
| 73 | I | B | D | E | 3 | 7.9 | ||||||||
| 32 | I | A | B | D | F | H | L | 2 | 5.3 | |||||
| 41 | I | A | B | D | E | L | 2 | 5.3 | ||||||
| 29 | I | B | D | E | L | 2 | 5.3 | |||||||
| 52 | I | A | B | D | 2 | 5.3 | ||||||||
| 67 | I | B | D | 2 | 5.3 | |||||||||
| 84 | I | A | B | D | E | 2 | 5.3 | |||||||
| WT + PTx | 31 | I | B | D | F | L | 12 | 32.4 | ||||||
| 30 | I | A | B | D | F | L | 12 | 32.4 | ||||||
| 33 | I | A | B | D | L | 4 | 10.8 | |||||||
| 52 | I | A | B | D | 3 | 8.1 | ||||||||
| 43 | I | B | D | L | 2 | 5.4 | ||||||||
| 32 | I | A | B | D | F | H | L | 2 | 5.4 | |||||
| 73 | I | B | D | E | 2 | 5.4 | ||||||||
| sda + GABA | 30 | I | A | B | D | F | L | 68 | 45.0 | |||||
| 31 | I | B | D | F | L | 35 | 23.2 | |||||||
| 33 | I | A | B | D | L | 19 | 12.6 | |||||||
| 32 | I | A | B | D | F | H | L | 8 | 5.3 | |||||
| 34 | I | B | D | F | H | L | 6 | 4.0 | ||||||
| 26 | I | B | D | F | K | 5 | 3.3 | |||||||
| 54 | I | A | B | D | F | K | 4 | 2.6 | ||||||
| 29 | I | B | D | E | L | 2 | 1.3 | |||||||
| 43 | I | B | D | L | 2 | 1.3 | ||||||||
| 47 | I | A | B | D | F | H | K | 2 | 1.3 | |||||
The inclusion of exon L increases from 87.8% in WT to 100% in sda (p ≤ 0.01). Feeding PTx to WT increases the inclusion of exon L to 100% (compared with 87.8% in WT, p ≤ 0.01). Exposure of sda larvae to GABA rescues the inclusion of L to 92.6% (compared with 100% in sda, p ≤ 0.01). Nomenclature of DmNav splice variants has been described previously (Lin et al., 2009). Only those splice variants present in two or more copies are shown isolated from 233 (WT, comprising 3 replicates of 66, 85, and 82), 50 (sda), 50 (WT + PTx), and 171 (sda + GABA, comprising 3 replicates of 77, 27, and 67) clones, respectively. DmNavs shown in bold (i.e., 52, 67, 73, and 84) do not give rise to functional channels. These clones are shown for reference only and have not been used for determination of frequency of inclusion for exons K and L. The letters J–L denote alternative exons in DmNav, and the presence of the letter denotes inclusion in the transcript (for details, see Lin et al., 2009).
There are two potential caveats to this analysis: first, our cloning methodology may have introduced a bias for particular transcripts, and second, the number of clones probed (∼50) may be insufficient to sample the complete array of splice variants present. To address the first, we determined the relative abundance of both exons K and L by qRT-PCR, which is not subject to bias (see Materials and Methods). Our analysis of WT CNS showed that exon L represented 86 ± 0.8% of total DmNav transcript, with the remaining 14 ± 0.8% being exon K. In contrast, L increased to 94 ± 1.3% in sda and K reduced to 6 ± 1.3% (p ≤ 0.01, n = 5). We conclude from this that the cloning method we use is free from bias. To address the second caveat, we cloned and analyzed between 50 and 80 DmNav splice variants from WT CNS three separate times. Our analysis shows an essentially identical hierarchy of DmNav splice variants (data not shown). The reproducibility between these three determinations is indicative that our sampling is sufficient to report the full range of endogenous DmNav splice variants present.
We noted the existence of seemingly nonfunctional splice variants (either containing both exons K and L or lacking both exons) in sda. Seven (12.3%) identical clones (DmNav69) were isolated from aCC in sda in which exons K and L coexist (Table 3). Transcripts were isolated from sda whole CNS that lacked both exons K and L (23.8%: variants 73, 52, 67, and 84; Table 4). Expression of either variant type in Xenopus oocytes failed to produce a functional channel, consistent with a failure of splicing in these instances (data not shown). Nonfunctional splice variants were not observed in WT. Thus, although the nature of the splicing error seems to differ, splicing in both aCC and whole CNS in the sda mutant shows a consistent increase in both inclusion of exon L and in the frequency of nonfunctional splicing. The increase in exon L is consistent with, and indeed likely underlies, the increase in INap observed in aCC in the sda (and eas) mutant (Marley and Baines, 2011).
The change to splicing of K/L in sda is rescued by the antiepileptic phenytoin
Analysis of action-potential-dependent synaptic currents in sda aCC motoneurons reveals a hyperexcitability phenotype with significantly increased amplitude and duration of individual currents (Marley and Baines, 2011). This is consistent with an overall increase in network excitability, possibly because of the increased INap in this mutant. The antiepileptic drug phenytoin targets the mammalian Nav channel and specifically reduces INap (Segal and Douglas, 1997; Lampl et al., 1998). Chronic feeding of phenytoin (0.4 mg/ml) to sda larvae is sufficient to rescue both synaptic current amplitudes and INap and, presumably by doing so, is also able to fully suppress seizures (Marley and Baines, 2011). In contrast, INat was not affected by this amount of phenytoin, although at higher doses this current component was also significantly reduced (R.M. and R.A.B., unpublished data).
Thus, we asked whether the mechanism of action of this antiepileptic drug, in Drosophila, includes a change to splicing of DmNav. Analysis of DmNav splicing in aCC isolated from third-instar sda larvae that had been raised on food containing phenytoin (0.4 mg/ml) showed a significant rescue in inclusion of exon L toward WT levels (76.5%, p ≤ 0.05; Fig. 1D, Table 3). This rescue suggests that the increased inclusion of exon L observed in sda is not a direct consequence of the loss of Sda protein but is most likely attributable to the hyperexcitability phenotype that occurs in its absence.
Splicing of DmNav is not limited to a choice of either exon K or L. As shown in Figure 1A, there are many additional splicing events, the majority of which are cassette events in which an exon can be either present or absent (exons J, I, A, B, E, F, and H) in addition to one other mutually exclusive splice at exon C or D (for a full description of splicing, see Lin et al., 2009). Comparison of splicing of these additional exons in aCC, between WT and sda, shows additional phenytoin-reversible changes in inclusion of exons A and B (Table 3). However, only the change in exon A is mirrored in analysis of splicing in whole CNS (data not shown). Thus, although we cannot rule out activity-dependent changes to splicing of other exons, the consistent effect we observe in the sda mutant across neuron subtypes is seemingly limited to the choice to splice either exons A and K/L. The physiological consequence of the decision to splice K or L is expression of DmNav channels that exhibit differing magnitudes of INap (Lin et al., 2009). In contrast, the contribution of exon A to channel function is not so well understood. Our previous analysis of structure function for a range of splice variants did not identify any obvious contribution of this exon to channel kinetics (Lin et al., 2009).
The decision to include exon L is activity dependent
The restoration of the WT ratio of splicing for exons K/L in sda larvae fed phenytoin is consistent with the underlying regulatory mechanism being activity dependent. To test this further, we manipulated neural activity by raising larvae on food containing either the proconvulsant and GABAA-receptor antagonist PTx to increase activity or the anticonvulsant and inhibitory neurotransmitter GABA to decrease activity. Feeding PTx (0.3 mg/ml) to WT larvae significantly increased the frequency of excitatory synaptic currents in aCC (shown in Fig. 2A), consistent with a hyperexcitable phenotype (25.9 ± 5.8 vs 14.4 ± 2.2 per min, p ≤ 0.05). Amplitudes of currents were not significantly different (data not shown). Ingestion of this toxin is also sufficient to induce seizure-like activity as evidenced by a significantly increased MRT to electric shock (27.9 ± 6.0 vs 154 ± 41 s, p ≤ 0.01; Fig. 2C). We analyzed splicing of DmNav from whole CNS isolated from these PTx-fed larvae. Feeding of PTx is sufficient to mirror the effect of the sda mutation in that inclusion of exon L, in functional variants, becomes saturated (87.8 vs 100% WT vs PTx, p ≤ 0.01; Fig. 2D, Table 4). The frequency of nonfunctional splicing also increased in WT fed PTx: two splice variants (13.5% of the total) were present that lacked both exons K and L (DmNavs 52 and 73). Thus, increasing neural activity mimics the sda phenotype in that it is sufficient to alter splicing of DmNav to favor a complete inclusion of exon L.
Figure 2.
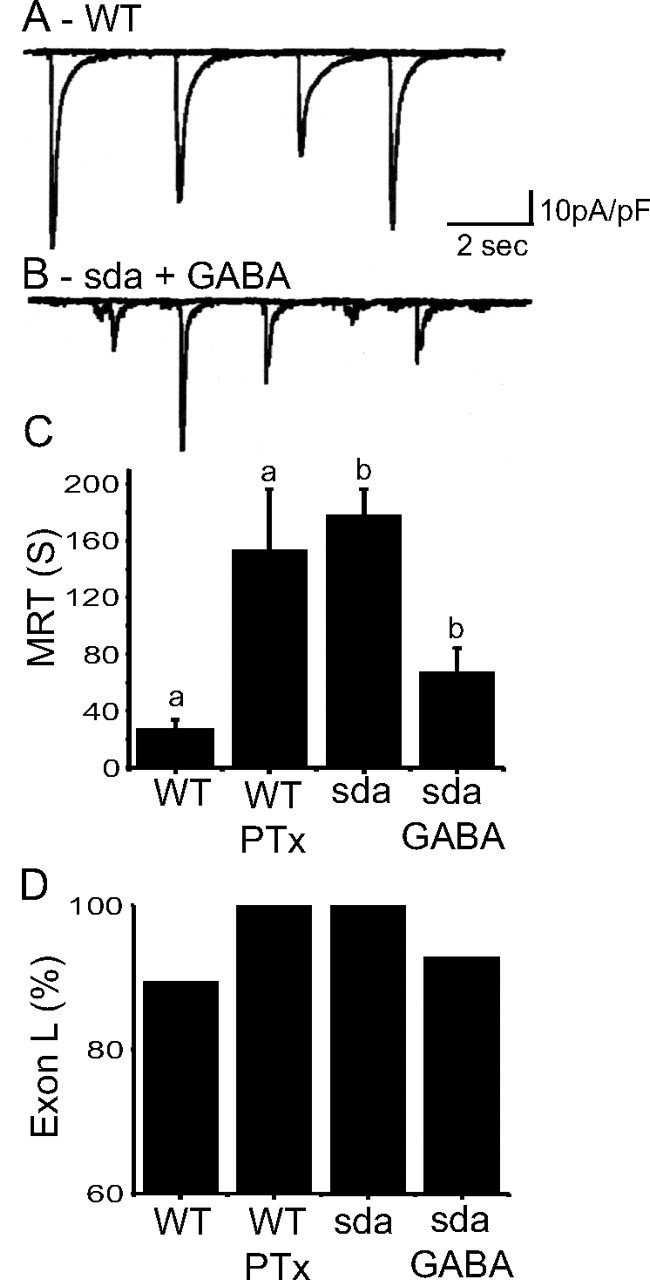
Splicing of K/L is influenced by activity. A, Overlaid traces to show examples of typical whole-cell voltage-clamp recordings (Vh = −60 mV) of action-potential-dependent synaptic currents from a WT aCC motoneuron in a wall-climbing third-instar larva. This activity represents the output of the locomotor central pattern generator (Baines, 2003). B, Feeding of GABA (1 mg/ml) to sda larvae is sufficient to dramatically reduce amplitudes of synaptic currents recorded in aCC (average values, 68.9 ± 5.4 vs 28.9 ± 3.5 pA/pF, WT vs GABA, p ≤ 0.01, n ≥ 8). C, Feeding WT larvae PTx is also sufficient to greatly increase their MRT to electroshock (27.9 ± 6.0 vs 154 ± 41 s). This is a measure of susceptibility to seizure (see Materials and Methods). In contrast, feeding GABA (1 mg/ml) to sda larvae is sufficient to rescue MRT (169.5 ± 16.9 vs 64.2 ± 15.5 s). Identical letters denote p ≤ 0.01, n ≥ 20. D, Analysis of splicing of DmNav in whole CNS shows that exposure of WT larvae to PTx is sufficient to saturate inclusion of exon L to 100% (p ≤ 0.01). In contrast, feeding of GABA to sda larvae is sufficient to rescue inclusion of exon K (i.e., a reduction in exon L to 92.6%, p ≤ 0.01).
To reduce activity levels in sda, we fed GABA (1 mg/ml) to third-instar larvae. This was sufficient to significantly reduce the amplitude of excitatory synaptic currents recorded in aCC in this genotype (68.9 ± 5.4 vs 28.9 ± 3.5 pA/pF, p ≤ 0.01; Fig. 2B). Frequency was not significantly different (data not shown). Feeding of GABA is also sufficient to significantly reduce the sda MRT for seizure activity induced by electric shock (169.5 ± 16.9 vs 64.2 ± 15.5 s, sda vs sda/GABA, p ≤ 0.01; Fig. 2C). Analysis of DmNav splicing in whole CNS isolated from sda shows that ingestion of GABA results in a modest, but significant, reduction of inclusion of exon L (100 ± 0 vs 92.6 ± 0.4%, sda vs sda/GABA, analysis repeated three times, p ≤ 0.01; Fig. 2D, Table 4). Thus, although the mechanism of action of PTx and GABA are clearly different, the consequence of these actions to aCC is complimentary in that PTx increases synaptic excitation, whereas GABA decreases it. Together, these data are indicative of a positive correlation between the frequency of inclusion of exon L in DmNav and the level of network (synaptic) activity in the CNS, consistent with this splicing decision being regulated by activity.
Activity regulates splicing through Ps
Our previous work identified the RNA-binding protein Ps to influence the choice to splice either exon K or L (Lin et al., 2009). Specifically, we showed that inclusion of exon K is significantly increased in a ps loss-of-function mutant. This observation suggested that the presence of Ps is required for inclusion of exon L. Because inclusion of exon L is significantly increased in both the sda mutant and WT fed PTx, we questioned whether the underlying mechanism required the presence of this protein. To test this, we first combined the sda and ps mutants to produce larvae heterozygous for both nulls (i.e., sda+/−, ps+/−). It should be noted that sda heterozygotes (sda+/−), similar to homozygous nulls, exhibit 100% saturation of exon L in DmNav (Fig. 3) and increased seizure-like behavior in response to electric shock (191.8 ± 20.6 s, p ≤ 0.01; data not shown). In this regard, sda is similar to many of the bang-sensitive Drosophila mutants that exhibit seizure-like activity when heterozygous for the mutation (Zhang et al., 2002). This effect is consistent with threshold levels of WT protein being required for a normal, non-seizure, phenotype.
Figure 3.

Ps is required for increased inclusion of exon L. The increased inclusion of exon L observed in sda is rescued by loss of one copy of ps (sda+/−, ps+/−). Similarly, the increase in exon L inclusion that results after feeding PTx to WT larvae is also rescued when one copy of ps is removed. Figure reports splicing of L in whole CNS. Identical letters denote p ≤ 0.05.
Analysis of splicing, in whole CNS, in the sda+/−, ps+/− double-heterozygote shows a clear reduction in inclusion of exon L compared with sda+/− alone (88.9 ± 2.4 vs 100 ± 0%, p ≤ 0.05; Fig. 3, Table 5). Analysis of seizure severity also shows significant rescue as a result of loss of one copy of ps (MRT's 191.8 ± 20.6 vs 78.4 ± 50 s, sda+/−, ps+/+ vs sda+/−, ps+/−, p ≤ 0.01; data not shown). A similar rescue in both inclusion of exon L (90.6%) and MRT to electric shock (63.7 ± 16.5 s) was also observed after pan-neuronal expression (GAL4elaV) of a ps RNAi transgene in the sda background (data not shown).
Table 5.
Ps is required for increased inclusion of exon L
| Genotype/drug | DmNav | J | I | A | B | C | D | E | F | H | K | L | # Clones | % |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sda+/−, ps+/− | 31 | I | B | D | F | L | 20 | 25.6 | ||||||
| 30 | I | A | B | D | F | L | 13 | 16.7 | ||||||
| 33 | I | A | B | D | L | 11 | 14.1 | |||||||
| 29 | I | B | D | E | L | 6 | 7.7 | |||||||
| 43 | I | B | D | L | 6 | 7.7 | ||||||||
| 26 | I | B | D | F | K | 5 | 6.4 | |||||||
| 34 | I | B | D | F | H | L | 3 | 3.8 | ||||||
| 58 | I | A | B | D | K | 3 | 3.8 | |||||||
| 84 | I | A | B | D | E | 3 | 3.8 | |||||||
| 41 | I | A | B | D | E | L | 2 | 2.6 | ||||||
| 50 | I | A | B | D | F | 2 | 2.6 | |||||||
| 63 | I | D | F | L | 2 | 2.6 | ||||||||
| 65 | I | B | D | F | 2 | 2.6 | ||||||||
| ps+/− + PTx | 30 | I | A | B | D | F | L | 27 | 29.0 | |||||
| 31 | I | B | D | F | L | 26 | 28.0 | |||||||
| 33 | I | A | B | D | L | 15 | 16.1 | |||||||
| 32 | I | A | B | D | F | H | L | 8 | 8.6 | |||||
| 43 | I | B | D | L | 5 | 5.4 | ||||||||
| 26 | I | B | D | F | K | 4 | 4.3 | |||||||
| 47 | I | A | B | D | F | H | K | 2 | 2.2 | |||||
| 54 | I | A | B | D | F | K | 2 | 2.2 | ||||||
| 55 | I | A | B | D | H | L | 2 | 2.2 | ||||||
| 60 | I | B | D | K | 2 | 2.2 | ||||||||
Loss of one copy of ps rescues the increased inclusion of exon L normally seen in whole CNS in the sda mutation (100 vs 88.9%; compared with 87.8% in WT controls, p ≤ 0.05; see Table 4). Similarly, the effect of feeding PTx to WT larvae is also rescued by loss of one copy of ps (100 vs 89.0%, p ≤ 0.05). Nomenclature of DmNav splice variants has been described previously (Lin et al., 2009). Only those splice variants present in two or more copies are shown isolated from 100 (comprising duplicates of 50 and 50) and 113 (comprising duplicates of 52 and 61) clones, respectively. DmNavs shown in bold (i.e., 50, 65, and 84) are nonfunctional. These clones are shown for reference only and have not been used for determination of frequency of inclusion for exons K and L. The letters J–L denote alternative exons in DmNav, and the presence of the letter denotes inclusion in the transcript (for details, see Lin et al., 2009).
We have also shown that feeding PTx to WT larvae is sufficient to recapitulate the sda phenotype (Fig. 2). Again, loss of one copy of ps (ps+/−) is sufficient to reduce inclusion of exon L in WT larvae fed PTx (100 to 89.0%, respectively, p ≤ 0.05; Fig. 3, Table 5). Loss of one copy of ps also significantly rescued PTx-induced seizure severity (31.2 ± 5.5 vs 161.1 ± 42 s, ps+/− vs ps+/+, p ≤ 0.01; data not shown). Thus, loss of one copy of ps diminishes both the change to splicing of DmNav and seizure severity attributable to either the sda mutation or the proconvulsive PTx. We conclude that the underlying and likely activity-dependent splicing mechanism requires Ps.
Altered kinetics of INa attributable to different splicing in the sda mutation
To determine how the altered splicing of DmNav, which we observe in sda aCC (Table 3), influences the kinetics of the expressed INa, we expressed cRNA in Xenopus oocytes. For each condition tested (WT, sda, and sda + phenytoin), a mixture of cRNAs that recapitulates the relative ratios of each individual clone was injected (Fig. 4A). To reduce the number of expression constructs that had to be made, we included only those variants present in three or more copies. In the case of the sda mixture, this included DmNav69, which contains both K and L and does not produce a functional channel when expressed in isolation. Restriction of mixtures to just those clones present in three or more copies did not significantly change the overall percentage of exon L inclusion for each mixture. We cannot, therefore, rule out the unlikely possibility that minor transcripts have a disproportionate effect on membrane excitability. There is also no a priori reason to expect that the currents observed in Xenopus oocytes will exactly recapitulate those in neurons, in which other mechanisms (e.g., protein phosphorylation and/or binding of interacting proteins) may alter channel activity.
Figure 4.

Altered splicing of DmNav in sda increases INap. A, Traces of INa recorded from heterologous expression of DmNav cRNA mixtures in Xenopus oocytes. Traces show currents evoked by Δ10 mV depolarizing voltage steps from a holding potential of −90 mV (full details are provided by Lin et al., 2009). Inset, For each trace, a section of INap is shown with a magnified amplitude. Expression of the sda DmNav mixture increases INap compared with WT. Expression of the DmNav mixture expressed in aCC isolated from larvae fed phenytoin (sda + phy) shows a reduced INap compared with that from sda alone. B, Average values for INap for the three DmNav cRNA mixtures expressed (for composition, see Table 3). Values shown are 4.0 ± 0.2, 5.3 ± 0.2, and 4.6 ± 0.2%, respectively (identical letters denote p ≤ 0.05, n ≥ 8). P/T ratio, Persistent/transient ratio. C, Voltage dependence of activation is significantly hyperpolarized in the sda and sda + phenytoin DmNav mixtures (p ≤ 0.05, n ≥ 8). D, Voltage dependence of inactivation is not significantly different between the three conditions (p > 0.05, n ≥ 8). Protocols for activation and inactivation are shown by Lin et al. (2009).
To analyze INa that results from splicing in WT aCC, we injected a mixture of cRNA comprising DmNavs 33, 58, 30, 54, 55, and 32 at a ratio of 40:19:15:11:8.5:6.4 (70.2% exon L inclusion). Analysis of the expressed current showed an INap that was 4.0% of the maximal INat (Fig. 4A,B). This is somewhat less than the relative proportion of INap (∼25%) observed in recordings from aCC (Fig. 1B). Because Na+ channels are localized outside of the cell body, from where we record, in this neuron (Baines and Bate, 1998) the high axial resistance that connects the soma to the dendritic regions of the cell likely influences recordings. Moreover, this resistance will disproportionately reduce the fast INat rather than the slower INap and as such will yield an underestimate of the former.
Identical expression of the sda DmNav mixture in oocytes (100% exon L: DmNavs 31, 43, 55, 33, 69, 30, and 59) increased INap to 5.3% (p ≤ 0.01; Fig. 4A,B). In contrast, expression of the DmNav mixture observed in sda aCC treated with phenytoin (78.7% exon L: DmNavs 30, 31, 33, 58, 32, 43, 54, and 55) showed a significant rescue in INap (4.6%, p ≤ 0.05; Fig. 4A,B). We also analyzed voltage-dependent activation and inactivation of INat for each of the three mixtures tested. Voltage dependence of activation was significantly hyperpolarized for both the sda and sda + phenytoin DmNav mixtures compared with WT (half-activation voltages were −30 ± 0.8, −33.5 ± 1.2, and −34.5 ± 1.5 mV for WT, sda, sda + phenytoin, respectively, p ≤ 0.05; Fig. 4C). In contrast, no significant differences were observed for voltage dependence of inactivation (half-inactivation voltages were −43.7 ± 0.6, −45.3 ± 0.9, and −45.0 ± 0.6 mV for WT, sda, sda + phenytoin, respectively, p > 0.05; Fig. 4D). We conclude that the changes to splicing observed in the sda mutation are sufficient to significantly increase both INap and V½ activation, which is consistent with a seizure phenotype. However, only the effect on INap is reversible on exposure to the antiepileptic phenytoin.
The persistent Na+ current promotes membrane excitability
The sda mutation exhibits increased seizure-like activity that is associated with an increased INap, and both are suppressed by phenytoin (Marley and Baines, 2011). Although the precise mechanism of how an increased INap can lead to neural instability remains to be determined, many studies have linked INap to increased ability to fire action potentials in a variety of neurons from insects to mammals (Kiss, 2008). However, how this current affects excitability is complicated by the existence of neuronal homeostatic compensatory mechanisms that act to regulate action potential firing to maintain physiologically relevant activity (Baines et al., 2001; Baines, 2003; Turrigiano and Nelson, 2004). Thus, to determine the uncompensated contribution of INap to membrane excitability requires removal of homeostatic regulation. To achieve this, we modeled the basic biophysical properties of the third-instar larval aCC motoneuron to allow us to manipulate individual conductances in silico to obviate the activity of homeostatic regulation.
The model we developed (see Materials and Methods) incorporates INat, INap, and fast, slow, and leak IK. The model also localizes INa and fast and slow IK to the axonal compartment to mirror the localization observed in vivo. The model does not include ICa and Ca2+-dependent IK(Ca). The aCC neuron continues to fire action potentials in Ca2+-free saline, indicative that these latter currents are not essential for spiking (Fig. 5A). Comparison of spiking between the aCC neuron (in an external saline containing Cd2+ to block ICa/IK(Ca)) and the model show excellent agreement over a physiologically relevant range (Fig. 5A–C). Modeling the percentage increase that we observe in the ratio between INap/INat in the sda DmNav mixture expressed in Xenopus oocytes relative to the WT mixture (a 32.5% increase over WT, 5.3 vs 4%, sda vs WT) elicits a significant increase in firing frequency (49 vs 102 Hz, WT vs sda). Modeling the INap/INat observed for the sda + phenytoin DmNav mixture (a 15% increase over WT, 4.6 vs 4%, sda/phenytoin vs WT) partially rescues firing frequency to 82 Hz (Fig. 5D). Thus, we conclude that increasing INap in aCC is likely to increase excitability, which is entirely consistent with, and likely responsible for, a seizure phenotype. However, in vivo such changes in membrane excitability may be partially masked as a result of the activity of homeostatic mechanisms that strive to maintain action potential firing within predetermined limits (Marley and Baines, 2011).
Figure 5.

INap enhances membrane excitability. A, Current-clamp recording from a third-instar aCC motoneuron showing response to current injections of 5, 25, and 45 pA for 500 ms. Extracellular saline contained Cd2+ to block ICa. B, Ball-and-stick model of aCC motoneuron showing simulated responses to the same current injections. C, Model approximates aCC motoneuron firing response to current injection. Firing rate is calculated as inverse of mean interspike interval. D, Increasing the ratio of persistent (INap) to transient (INat) components in the model to mimic the changes observed in DmNav mixtures, expressed in Xenopus oocytes, shows a clear relationship between magnitude of INap and firing rate. WT (4% INap/INat) is set to zero, and the increases in ratio observed in sda (5.3% INap/INat representing a 32.5% increase over WT) and sda + phenytoin (phy; 4.6% INap/INat representing a 15% increase over WT) are shown. Simulated action potentials are from a 10 pA current injection for 500 ms.
To further support our conclusion that INap increases firing frequency in the aCC motoneuron, we adopted two approaches. First, we used acute application of phenytoin at a dose (30 μm) that we have shown previously inhibits just this current component (by ∼45%) but does not markedly inhibit INat (Marley and Baines, 2011). This approach has the added benefit that it eliminates complications attributable to homeostasis. In the presence of phenytoin, both WT and sda aCC neurons fire fewer action potentials (reducing firing by ∼16 and 25%, respectively; Fig. 6A,B). Second, we expressed ps RNAi in just aCC (using Gal4RRa), which, by reducing inclusion of exon L, also specifically reduces INap (INap, 7.2 ± 1.8 vs 14.2 ± 2.1 pA/pF, p < 0.05; INat, 31 ± 3.4 vs 29.4 ± 2.4 pA/pF, p > 0.05, sda + RNAi vs sda, n ≥ 7; data not shown). Expression of ps RNAi in aCC also significantly reduces action potential firing (∼45%; Fig. 6C). That action potential firing is reduced when INap is partially blocked is entirely consistent with, and indeed supportive of, our conclusion that this current component regulates neuronal membrane excitability.
Figure 6.

Blocking INap reduces membrane excitability. A, Action potentials (APs) evoked by injection of constant depolarizing current (500 ms) in WT aCC are fewer in number in the presence of phenytoin (phy; 30 μm). B, This same dose of phenytoin also reduces action potential firing in sda aCC neurons. Note that sda neurons fire fewer action potentials than WT aCC even in the absence of phenytoin, an effect that is most likely attributable to synaptic homeostasis (Marley and Baines, 2011). C, Expression of ps RNAi in WT aCC neurons also results in a reduction in action potential firing compared with controls (GAL4RRa > UAS–GFP). Reduction of ps increases splicing of exon K and a smaller INap as a consequence (Lin et al., 2009). Values shown are averages from n ≥ 5.
Discussion
We show that a key splicing decision in DmNav is influenced by the level of synaptic excitation present in the CNS. Thus, increasing synaptic excitation, through either genetic (e.g., sda) or pharmacological (e.g., PTx) means, is sufficient to favor inclusion of exon L at the expense of the mutually exclusive exon K. Splicing of these exons dramatically influences the magnitude of INap carried by the expressed channel (Lin et al., 2009). Increased inclusion of L results in a larger INap, which, in turn, we predict further increases action potential firing. These observations provide experimental support for the premise that self-reinforcing cycles of activity contribute to the emergence of epilepsy in susceptible individuals (Blumenfeld et al., 2008). Moreover, early intervention to break these cycles may offer the exciting prospect of preventing certain types of epilepsy from developing (Marley and Baines, 2011).
Although a linkage between synaptic activity and splicing of ion channels has been reported previously (Ule et al., 2006; Li et al., 2007), whether this mode of regulation represents the norm remains to be determined. For example, splicing of exon 20 of the NMDA receptor 1 (NR1) is activity dependent. Increased activity promotes splicing to favor the C2 variant, whereas activity blockade results in a predominance of the C2′ variant. The C2′ variant accelerates NR1 trafficking from the ER to the synapse (Mu et al., 2003). Activity, or more specifically the lack of it, decreases the inclusion of the stress axis-regulated exon (STREX) in the mammalian BK K+ channel (also known as Slowpoke). Based on homologous expression studies, this change is predicted to decrease action potential firing in neurons expressing this variant (Xie and McCobb, 1998). Our demonstration that activity influences a splice decision in an Nav channel is, to our knowledge, the first such report for this channel type. Importantly, it also extends the number of known examples of such regulation. We have recently conducted a structure–function analysis for the more common splice variants of DmNav identified in embryonic CNS (Lin et al., 2009). Of particular relevance for this study are the mutually exclusive exons K and L. These exons, which differ in 16 of 41 aa, are membrane spanning and are located in homology domain III/S4, which forms part of the voltage sensor (Catterall, 2010). These exons influence the magnitude of INap in expressed channels. When L is present, INap ranges from 4.1 to 9.5% of the peak transient current, and this value drops to 1.5–2.4% when K is included (Lin et al., 2009).
The molecular origin of INap is still unclear but is believed to result from incomplete inactivation of the channel (Stafstrom, 2007; Kiss, 2008). Significantly, many channelopathies identified from human epilepsy sufferers show amino acid substitutions in Nav channels that, when expressed, produce channels with larger than normal INap (Rhodes et al., 2004; Kahlig et al., 2006; Estacion et al., 2010; Chen et al., 2011). This current component is also increased in motoneurons of seizure-sensitive Drosophila mutants, including the sda mutant used here (Marley and Baines, 2011). Most human Nav channels exhibit >50% identity to exon K/L, including a region of 11 residues that are identical (Lin et al., 2009). Although this region (termed exon 18 in mammalian Navs) is also subject to splicing, the outcome differs: a full-length channel containing exon 18 (termed 18A), a truncated channel containing an alternate exon 18 that encodes a stop codon (18N), or a channel that lacks either exon 18A/N (Δ18) (Oh and Waxman, 1998; Plummer et al., 1998; Diss et al., 2001). Intriguingly, splicing of this region in Nav is developmentally regulated, keeping open the possibility that aberrant splicing during embryogenesis could lead to altered patterns of activity that may predispose susceptible individuals to epilepsy.
An increase in INap has been shown to be associated with, and even causative of, epilepsy in a number of studies. One such example is provided by pilocarpine induction of status epilepticus in rat. Such treatment promotes the appearance of spontaneous recurrent seizures after 1–4 weeks, yet recordings from CA1 pyramidal cells show an associated 1.5-fold increase in INap and a switch to burst firing within 1 week (Chen et al., 2011). Similar observations have been reported in other neuron types, for example, entorhinal cortex layer 5 (Agrawal et al., 2003), indicative that acute increases in synaptic excitation may be sufficient to increase this current component in all temporal lobe structures. The underlying mechanism(s) is unknown but has been speculated to include transcriptional and/or posttranslational modifications of Navs (Chen et al., 2011). Our results highlight that changes to splicing may also contribute to this phenomenon. We show that the known RNA-binding protein Ps is required for the activity-dependent increase in inclusion of exon L in DmNav. Ps and its mammalian homologs Nova-1 and Nova-2 are predicted to bind to [T/C]CA[T/C] motifs in pre-mRNAs; multiple copies of these motifs are located in the downstream introns of both exons K and L in DmNav (Park et al., 2004; W.-H.L. and R.A.B., unpublished observations). A comparative analysis has recently concluded that the RNA regulatory map between Ps and Nova-1/2 is highly conserved between fly and mammals and that putative pre-mRNA targets of Nova-1/2 include Nav1.1 and Nav1.5 (Licatalosi et al., 2008; Brooks et al., 2011). Thus, it is probable that splicing of mammalian Navs are regulated by the Nova proteins. Significantly, transcription of at least Nova-2 is activity dependent and is downregulated after treatment with pilocarpine (Jelen et al., 2010). Whether activity also regulates expression of ps is unknown.
Epilepsy as a consequence of excitation/inhibition imbalance
The human brain has been estimated to contain ∼100 billion neurons, each of which receives an average of 10,000 synapses (Spitzer and Borodinsky, 2008). Because stable circuit function requires matching of presynaptic and postsynaptic activity (Turrigiano and Nelson, 2004), it is perhaps surprising, given this level of complexity, that epilepsy is not more prevalent. That it is not is attributable to regulatory mechanisms that continually monitor and, when required, modify both synaptic connectivity and levels of presynaptic and postsynaptic activity. These mechanisms are likely to be particularly important during neurogenesis when both neurons and neural circuits first form. Thus, from the outset, neurons are required not only to make appropriate connections but also to express suitable mixtures of ion channels to enable them to become functional members of individual networks. Once in a network, these same neurons must then continually monitor the level of excitatory and inhibitory synaptic drive to which they are exposed and adapt accordingly (Spitzer et al., 2000, 2002; Demarque and Spitzer, 2012). It has been hypothesized that the establishment of an excitation/inhibition balance in adult cortical neurons, critical for circuit stability, may arise from developmental coregulation of developing glutamatergic and GABAergic synapses (Zhang et al., 2011).
Significantly, perturbation of electrical activity in the early developing CNS is sufficient to evoke homeostatic changes in numbers of glutamatergic and GABAergic neurons (Borodinsky et al., 2004). Thus, it is possible that any alterations to activity patterns early in the development of the CNS may manifest as changes to the excitation/inhibition balance in mature neural networks. Such altered networks may be prone to seizure-like activity. The early CNS of sda larvae shows increased synaptic activity (Marley and Baines, 2011), which may be a consequence of the altered splicing of DmNav that we show here. Increased inclusion of exon L is likely to promote self-reinforcing cycles of activity that may disturb the excitation/inhibition balance of the developing CNS, possibly resulting in a seizure-like phenotype in more mature larvae. Consistent with this hypothesis is the observation that feeding phenytoin to WT larvae is sufficient to promote a seizure phenotype that is associated with a significant increase in INap in the aCC (and RP2) motoneuron (Marley and Baines, 2011). It is notable that this drug is also proconvulsive in WT rats (Callaghan and Schwark, 1980; Rundfeldt et al., 1990). Analysis of splicing of DmNav isolated from whole CNS from WT fed phenytoin also shows complete saturation of exon L (100%, p ≤ 0.05; data not shown). Thus, it seems likely that perturbation of neural activity in WT, mediated by phenytoin, is also sufficient to induce a change in DmNav splicing.
Our results also show that both splicing of K/L and seizure-like activity can be manipulated in the postembryonic larval CNS. Thus, feeding of PTx to WT, or GABA to sda, is sufficient to either promote or reduce inclusion of exon L and the associated increase in seizure-like activity. This observation is important because it suggests that the consequence of embryonic patterns of neural activity can be overwritten at a later stage. However, whether the effects of these manipulations persist long after exposure to the causative agent has ceased has yet to be determined. Persistence of effect was, however, observed in our previous study, which showed that exposure to a subthreshold amount of phenytoin during embryogenesis is sufficient to prevent the appearance of seizure-like activity in subsequent sda larvae (Marley and Baines, 2011). Analysis of DmNav splicing in aCC neurons isolated from such “treated” sda larvae shows an expected reduction in inclusion of exon L (W.-H.L. and R.A.B., unpublished data). The most parsimonious conclusion is that the presence of phenytoin, during embryogenesis, effectively capped hyperexcitability and, by doing so, uncoupled the positive feedback that we predict leads to circuit instability. Clearly, this possibility demands additional investigation.
In summary, we show that increased synaptic activity is sufficient to alter splicing of DmNav to promote inclusion of alternate exon L. This splicing increases the magnitude of INap of the expressed channel protein isoforms, which, in turn, promotes membrane excitability. This cycle of events may offer a possible mechanistic explanation of the long appreciated phenomenon that “seizures beget seizures.” For example, in the kindling model, multiple successive small electrical stimuli that are initially without obvious effect eventually lead to full-blown seizure-like behavior (Blumenfeld et al., 2008).
Footnotes
This work was supported by the Biotechnology and Biological Sciences Research Council UK (R.A.B.) and by a Burroughs Wellcome Career Award at the Scientific Interface (A.A.P.). We are grateful to Verena Wolfram, David Sattelle, and members of the Baines and Prinz groups for help and advice during the course of this work. Logesh Dharmar and Fred Sieling contributed to constructing the computational model.
References
- Agrawal N, Alonso A, Ragsdale DS. Increased persistent sodium currents in rat entorhinal cortex layer V neurons in a post-status epilepticus model of temporal lobe epilepsy. Epilepsia. 2003;44:1601–1604. doi: 10.1111/j.0013-9580.2003.23103.x. [DOI] [PubMed] [Google Scholar]
- Baines RA. Postsynaptic protein kinase A reduces neuronal excitability in response to increased synaptic excitation in the Drosophila CNS. J Neurosci. 2003;23:8664–8672. doi: 10.1523/JNEUROSCI.23-25-08664.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines RA, Bate M. Electrophysiological development of central neurons in the Drosophila embryo. J Neurosci. 1998;18:4673–4683. doi: 10.1523/JNEUROSCI.18-12-04673.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines RA, Uhler JP, Thompson A, Sweeney ST, Bate M. Altered electrical properties in Drosophila neurons developing without synaptic transmission. J Neurosci. 2001;21:1523–1531. doi: 10.1523/JNEUROSCI.21-05-01523.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenfeld H, Klein JP, Schridde U, Vestal M, Rice T, Khera DS, Bashyal C, Giblin K, Paul-Laughinghouse C, Wang F, Phadke A, Mission J, Agarwal RK, Englot DJ, Motelow J, Nersesyan H, Waxman SG, Levin AR. Early treatment suppresses the development of spike-wave epilepsy in a rat model. Epilepsia. 2008;49:400–409. doi: 10.1111/j.1528-1167.2007.01458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borodinsky LN, Root CM, Cronin JA, Sann SB, Gu X, Spitzer NC. Activity-dependent homeostatic specification of transmitter expression in embryonic neurons. Nature. 2004;429:523–530. doi: 10.1038/nature02518. [DOI] [PubMed] [Google Scholar]
- Brooks AN, Yang L, Duff MO, Hansen KD, Park JW, Dudoit S, Brenner SE, Graveley BR. Conservation of an RNA regulatory map between Drosophila and mammals. Genome Res. 2011;21:193–202. doi: 10.1101/gr.108662.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaghan DA, Schwark WS. Pharmacological modification of amygdaloid-kindled seizures. Neuropharmacology. 1980;19:1131–1136. doi: 10.1016/0028-3908(80)90113-6. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Ion channel voltage sensors: structure, function, and pathophysiology. Neuron. 2010;67:915–928. doi: 10.1016/j.neuron.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Su H, Yue C, Remy S, Royeck M, Sochivko D, Opitz T, Beck H, Yaari Y. An increase in persistent sodium current contributes to intrinsic neuronal bursting after status epilepticus. J Neurophysiol. 2011;105:117–129. doi: 10.1152/jn.00184.2010. [DOI] [PubMed] [Google Scholar]
- Darbon P, Yvon C, Legrand JC, Streit J. INaP underlies intrinsic spiking and rhythm generation in networks of cultured rat spinal cord neurons. Eur J Neurosci. 2004;20:976–988. doi: 10.1111/j.1460-9568.2004.03565.x. [DOI] [PubMed] [Google Scholar]
- Demarque M, Spitzer NC. Neurotransmitter phenotype plasticity: an unexpected mechanism in the toolbox of network activity homeostasis. Dev Neurobiol. 2012;72:22–32. doi: 10.1002/dneu.20909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diss JK, Archer SN, Hirano J, Fraser SP, Djamgoz MB. Expression profiles of voltage-gated Na+ channel alpha-subunit genes in rat and human prostate cancer cell lines. Prostate. 2001;48:165–178. doi: 10.1002/pros.1095. [DOI] [PubMed] [Google Scholar]
- Ermentrout B. Simulating, analyzing, and animating dynamical systems. Philadelphia: Society for Industrial and Applied Mathematics; 2002. [Google Scholar]
- Estacion M, Gasser A, Dib-Hajj SD, Waxman SG. A sodium channel mutation linked to epilepsy increases ramp and persistent current of Nav1.3 and induces hyperexcitability in hippocampal neurons. Exp Neurol. 2010;224:362–368. doi: 10.1016/j.expneurol.2010.04.012. [DOI] [PubMed] [Google Scholar]
- Feng G, Deák P, Chopra M, Hall LM. Cloning and functional analysis of TipE, a novel membrane protein that enhances Drosophila para sodium channel function. Cell. 1995;82:1001–1011. doi: 10.1016/0092-8674(95)90279-1. [DOI] [PubMed] [Google Scholar]
- Heinzen EL, Yoon W, Weale ME, Sen A, Wood NW, Burke JR, Welsh-Bohmer KA, Hulette CM, Sisodiya SM, Goldstein DB. Alternative ion channel splicing in mesial temporal lobe epilepsy and Alzheimer's disease. Genome Biol. 2007;8:R32. doi: 10.1186/gb-2007-8-3-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki Y, Tang W, Zhang L, Du G, Xu W, Kokudo N. Novel aminopeptidase N (APN/CD13) inhibitor 24F can suppress invasion of hepatocellular carcinoma cells as well as angiogenesis. Biosci Trends. 2010;4:56–60. [PubMed] [Google Scholar]
- Jelen N, Ule J, Zivin M. Cholinergic regulation of striatal Nova mRNAs. Neuroscience. 2010;169:619–627. doi: 10.1016/j.neuroscience.2010.05.013. [DOI] [PubMed] [Google Scholar]
- Kahlig KM, Misra SN, George AL., Jr Impaired inactivation gate stabilization predicts increased persistent current for an epilepsy-associated SCN1A mutation. J Neurosci. 2006;26:10958–10966. doi: 10.1523/JNEUROSCI.3378-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss T. Persistent Na-channels: origin and function. A review. Acta Biol Hung [Suppl] 2008;59:1–12. doi: 10.1556/ABiol.59.2008.Suppl.1. [DOI] [PubMed] [Google Scholar]
- Lampl I, Schwindt P, Crill W. Reduction of cortical pyramidal neuron excitability by the action of phenytoin on persistent Na+ current. J Pharmacol Exp Ther. 1998;284:228–237. [PubMed] [Google Scholar]
- Landgraf M, Thor S. Development of Drosophila motoneurons: specification and morphology. Semin Cell Dev Biol. 2006;17:3–11. doi: 10.1016/j.semcdb.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Li Q, Lee JA, Black DL. Neuronal regulation of alternative pre-mRNA splicing. Nat Rev Neurosci. 2007;8:819–831. doi: 10.1038/nrn2237. [DOI] [PubMed] [Google Scholar]
- Li Y, Gorassini MA, Bennett DJ. Role of persistent sodium and calcium currents in motoneuron firing and spasticity in chronic spinal rats. J Neurophysiol. 2004;91:767–783. doi: 10.1152/jn.00788.2003. [DOI] [PubMed] [Google Scholar]
- Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, Clark TA, Schweitzer AC, Blume JE, Wang X, Darnell JC, Darnell RB. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456:464–469. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin WH, Wright DE, Muraro NI, Baines RA. Alternative splicing in the voltage-gated sodium channel DmNav regulates activation, inactivation, and persistent current. J Neurophysiol. 2009;102:1994–2006. doi: 10.1152/jn.00613.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marley R, Baines RA. Increased persistent Na+ current contributes to seizure in the slamdance bang-sensitive Drosophila mutant. J Neurophysiol. 2011;106:18–29. doi: 10.1152/jn.00808.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mee CJ, Pym EC, Moffat KG, Baines RA. Regulation of neuronal excitability through pumilio-dependent control of a sodium channel gene. J Neurosci. 2004;24:8695–8703. doi: 10.1523/JNEUROSCI.2282-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu Y, Otsuka T, Horton AC, Scott DB, Ehlers MD. Activity-dependent mRNA splicing controls ER export and synaptic delivery of NMDA receptors. Neuron. 2003;40:581–594. doi: 10.1016/s0896-6273(03)00676-7. [DOI] [PubMed] [Google Scholar]
- Muraro NI, Weston AJ, Gerber AP, Luschnig S, Moffat KG, Baines RA. Pumilio binds para mRNA and requires Nanos and Brat to regulate sodium current in Drosophila motoneurons. J Neurosci. 2008;28:2099–2109. doi: 10.1523/JNEUROSCI.5092-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Dowd DK, Aldrich RW. Voltage-clamp analysis of sodium channels in wild-type and mutant Drosophila neurons. J Neurosci. 1998;8:3633–3643. doi: 10.1523/JNEUROSCI.08-10-03633.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh Y, Waxman SG. Novel splice variants of the voltage-sensitive sodium channel alpha subunit. Neuroreport. 1998;9:1267–1272. doi: 10.1097/00001756-199805110-00002. [DOI] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- Park JW, Parisky K, Celotto AM, Reenan RA, Graveley BR. Identification of alternative splicing regulators by RNA interference in Drosophila. Proc Natl Acad Sci U S A. 2004;101:15974–15979. doi: 10.1073/pnas.0407004101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plummer NW, Galt J, Jones JM, Burgess DL, Sprunger LK, Kohrman DC, Meisler MH. Exon organization, coding sequence, physical mapping, and polymorphic intragenic markers for the human neuronal sodium channel gene SCN8A. Genomics. 1998;54:287–296. doi: 10.1006/geno.1998.5550. [DOI] [PubMed] [Google Scholar]
- Ragsdale DS. How do mutant Nav1.1 sodium channels cause epilepsy? Brain Res Rev. 2008;58:149–159. doi: 10.1016/j.brainresrev.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Rhodes TH, Lossin C, Vanoye CG, Wang DW, George AL., Jr Noninactivating voltage-gated sodium channels in severe myoclonic epilepsy of infancy. Proc Natl Acad Sci U S A. 2004;101:11147–11152. doi: 10.1073/pnas.0402482101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rundfeldt C, Hönack D, Löscher W. Phenytoin potently increases the threshold for focal seizures in amygdala-kindled rats. Neuropharmacology. 1990;29:845–851. doi: 10.1016/0028-3908(90)90159-o. [DOI] [PubMed] [Google Scholar]
- Schlachter K, Gruber-Sedlmayr U, Stogmann E, Lausecker M, Hotzy C, Balzar J, Schuh E, Baumgartner C, Mueller JC, Illig T, Wichmann HE, Lichtner P, Meitinger T, Strom TM, Zimprich A, Zimprich F. A splice site variant in the sodium channel gene SCN1A confers risk of febrile seizures. Neurology. 2009;72:974–978. doi: 10.1212/01.wnl.0000344401.02915.00. [DOI] [PubMed] [Google Scholar]
- Segal MM, Douglas AF. Late sodium channel openings underlying epileptiform activity are preferentially diminished by the anticonvulsant phenytoin. J Neurophysiol. 1997;77:3021–3034. doi: 10.1152/jn.1997.77.6.3021. [DOI] [PubMed] [Google Scholar]
- Spitzer NC, Borodinsky LN. Implications of activity-dependent neurotransmitter-receptor matching. Philos Trans R Soc Lond B Biol Sci. 2008;363:1393–1399. doi: 10.1098/rstb.2007.2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer NC, Vincent A, Lautermilch NJ. Differentiation of electrical excitability in motoneurons. Brain Res Bull. 2000;53:547–552. doi: 10.1016/s0361-9230(00)00388-9. [DOI] [PubMed] [Google Scholar]
- Spitzer NC, Kingston PA, Manning TJ, Conklin MW. Outside and in: development of neuronal excitability. Curr Opin Neurobiol. 2002;12:315–323. doi: 10.1016/s0959-4388(02)00330-6. [DOI] [PubMed] [Google Scholar]
- Stafstrom CE. Persistent sodium current and its role in epilepsy. Epilepsy Curr. 2007;7:15–22. doi: 10.1111/j.1535-7511.2007.00156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stragier B, De Bundel D, Sarre S, Smolders I, Vauquelin G, Dupont A, Michotte Y, Vanderheyden P. Involvement of insulin-regulated aminopeptidase in the effects of the renin-angiotensin fragment angiotensin IV: a review. Heart Fail Rev. 2008;13:321–337. doi: 10.1007/s10741-007-9062-x. [DOI] [PubMed] [Google Scholar]
- Thompson CH, Kahlig KM, George AL., Jr SCN1A splice variants exhibit divergent sensitivity to commonly used antiepileptic drugs. Epilepsia. 2011;52:1000–1009. doi: 10.1111/j.1528-1167.2011.03040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Ule J, Stefani G, Mele A, Ruggiu M, Wang X, Taneri B, Gaasterland T, Blencowe BJ, Darnell RB. An RNA map predicting Nova-dependent splicing regulation. Nature. 2006;444:580–586. doi: 10.1038/nature05304. [DOI] [PubMed] [Google Scholar]
- Vervaeke K, Hu H, Graham LJ, Storm JF. Contrasting effects of the persistent Na+ current on neuronal excitability and spike timing. Neuron. 2006;49:257–270. doi: 10.1016/j.neuron.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, McCobb DP. Control of alternative splicing of potassium channels by stress hormones. Science. 1998;280:443–446. doi: 10.1126/science.280.5362.443. [DOI] [PubMed] [Google Scholar]
- Zhang H, Tan J, Reynolds E, Kuebler D, Faulhaber S, Tanouye M. The Drosophila slamdance gene: a mutation in an aminopeptidase can cause seizure, paralysis and neuronal failure. Genetics. 2002;162:1283–1299. doi: 10.1093/genetics/162.3.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Jiao YY, Sun QQ. Developmental maturation of excitation and inhibition balance in principal neurons across four layers of somatosensory cortex. Neuroscience. 2011;174:10–25. doi: 10.1016/j.neuroscience.2010.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]