

Abstract
Cajal bands are cytoplasmic channels flanked by appositions where the abaxonal surface of Schwann cell myelin apposes and adheres to the overlying plasma membrane. These appositions contain a dystroglycan complex that includes periaxin and dystrophin-related protein 2 (Drp2). Loss of periaxin disrupts appositions and Cajal bands in Schwann cells and causes a severe demyelinating neuropathy in mouse and human. Here, we investigated the role of mouse Drp2 in apposition assembly and Cajal band function and compared it with periaxin. We show that periaxin and Drp2 are not only both required to form appositions, but they must also interact. Periaxin–Drp2 interaction is also required for Drp2 phosphorylation, but phosphorylation is not required for the assembly of appositions. Drp2 loss causes corresponding increases in Dystrophin family members, utrophin and dystrophin Dp116, although dystroglycan remains unchanged. We also show that all dystroglycan complexes in Schwann cells use the uncleaved form of β-dystroglycan. Drp2-null Schwann cells have disrupted appositions and Cajal bands, and they undergo focal hypermyelination and concomitant demyelination. Nevertheless, they do not have the short internodal lengths and associated reduced nerve conduction velocity seen in the absence of periaxin, showing that periaxin regulates Schwann cell elongation independent of its role in the dystroglycan complex. We conclude that the primary role of the dystroglycan complex in appositions is to stabilize and limit the radial growth of myelin.
Introduction
Dystroglycan complexes link extracellular matrix proteins such as laminin with the actin cytoskeleton by means of the extracellular α-subunit and the transmembrane β-subunit of dystroglycan (Ervasti and Campbell, 1993). In skeletal muscle, dystroglycan complexes involving dystrophin family members dystrophin and utrophin (dystrophin-related protein 1) regulate the segregation of the plasma membrane into distinct domains (Ibraghimov-Beskrovnaya et al., 1992; Tinsley et al., 1993; Colognato et al., 1999; Cohn and Campbell, 2000). Dystroglycan complexes are also critical for the establishment of basal membrane polarity in epithelial cells (Durbeej et al., 1995; Li et al., 2003).
Dystroglycan complexes in myelinating Schwann cells of the vertebrate peripheral nervous system (PNS) use dystrophin (Dp116), utrophin, or dystrophin-related protein 2 (Drp2) (Matsumura et al., 1993; Yamada et al., 1994; Saito et al., 1999; Imamura et al., 2000; Sherman et al., 2001; Albrecht et al., 2008; Masaki and Matsumura, 2010). Drp2 interacts with periaxin to form the periaxin–Drp2–dystroglycan (PDG) complex, which is concentrated at zones of adhesion between the abaxonal surface of compact peripheral myelin and the Schwann cell plasma membrane (Gillespie et al., 1994, 2000; Sherman et al., 2001; Court et al., 2004) (see Fig. 1A). These appositions define the boundaries of cytoplasm-filled channels that were first described histologically at the beginning of the 20th century (Nageotte, 1910; Nemiloff, 1910; Ramón y Cajal, 1912), and more recently by scanning EM (Ushiki and Ide, 1987). These channels have been named Cajal bands and may have a biosynthetic role in myelin assembly (Ramón y Cajal, 1933; Ushiki and Ide, 1987; Court et al., 2004). Correlation between their integrity and the longitudinal growth of Schwann cells has implicated Cajal bands in defining internodal length (Court et al., 2004, 2009). Recently, a model has been proposed whereby regulated cleavage of β-dystroglycan to a 31 kDa form might control the relative sizes of appositions and Cajal bands thereby influencing internodal length (Court et al., 2011).
Figure 1.
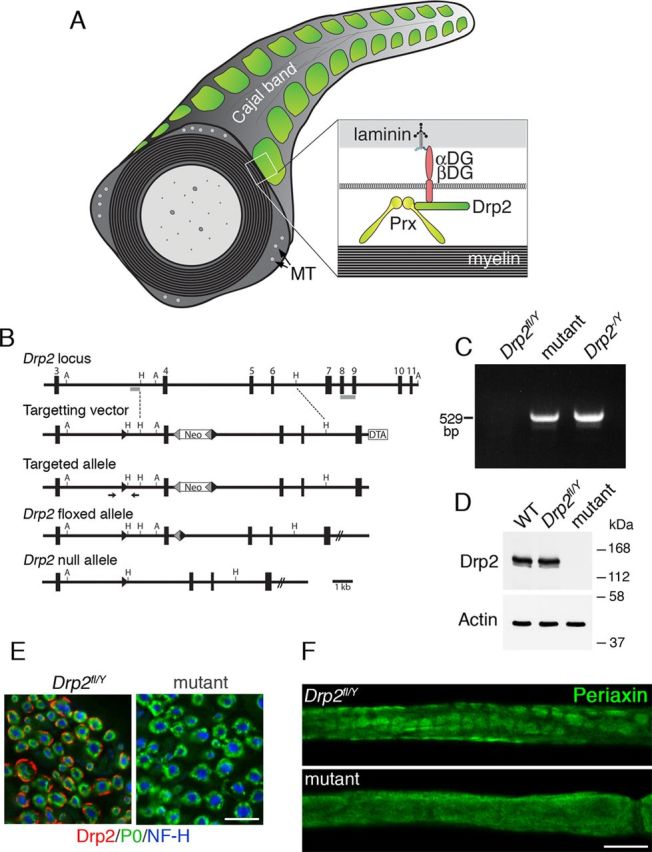
Disruption of the Drp2 gene causes loss of Drp2 and mislocalization of Periaxin. A, Diagram showing the relationship between the periaxin (Prx)–Drp2–dystroglycan (αDG and βDG) complex, appositions and Cajal bands in a myelinated peripheral nerve. MT, Microtubules. B, Gene targeting strategy to generate a floxed Drp2 allele. Exons are numbered and the position of probes for Southern blotting are shown in gray. DTA, Diphtheria toxin A chain; Neo, neomycin selection cassette; forward arrowheads are LoxP sites, and reverse arrowheads are Frt sites. A, AvrII; H, HindIII. The arrows show the position of primers used for routine genotyping. C, Genomic PCR used to detect recombination in sciatic nerves of Drp2fl/Y/Cnp1-Cre+/− (mutant) and Drp2−/y mice. D, Western blot analysis of nerve lysates from WT, Drp2fl/Y, and Drp2fl/Y/Cnp1-Cre+/− (mutant) mice, respectively. Actin was the loading control. E, Immunofluorescence analysis of transverse sections of nerve showing Drp2 (red) is not detectable in the mutant. Myelin is labeled with P0 antibody (green) and axons with Neurofilament protein, NF-H antibody (blue). Scale bar, 10 μm. F, Immunofluorescence staining shows that loss of Drp2 causes mislocalization of periaxin. Scale bar, 10 μm.
Loss of periaxin causes demyelination in mice and a recessive form of a severe demyelinating neuropathy in humans, CMT4F (Charcot–Marie–Tooth 4F) (Gillespie et al., 2000; Boerkoel et al., 2001; Guilbot et al., 2001). However, it is not clear whether this disease is primarily the result of the disruption of appositions, and therefore Cajal bands; it is also not certain whether the reduced internodal lengths observed in periaxin-null Schwann cells are an additional consequence of the disruption of Cajal bands. To shed further light on the mechanisms of assembly and the function of Cajal bands, we sought to disrupt the PDG complex by an alternative approach, namely by eliminating Drp2. Here, we show that, although periaxin and Drp2 cooperate to form appositions and Cajal bands, they have distinct functions in myelinating Schwann cells. Importantly, we show that the primary role of appositions and Cajal bands is to limit radial myelin growth rather than regulate Schwann cell elongation.
Materials and Methods
Mice.
All animal work conformed to United Kingdom legislation (Scientific Procedures) Act 1986, and to the University of Edinburgh Ethical Review Committee policy. Cnp1-Cre mice have been described and shown to be effective in promoting Cre-mediated recombination in mouse embryonic peripheral nerves (Lappe-Siefke et al., 2003; Grove et al., 2007; Sherman et al., 2012). Hprt-Cre mice (Su et al., 2002) (from John Mason, University of Edinburgh, Edinburgh, UK) were used to drive germ line recombination and inactivation of the Drp2 gene and FLPe mice (Wallace et al., 2007) (from Andrew Smith, University of Edinburgh, Edinburgh, UK) were used to drive recombination and excision between Frt sites. Mice carrying a Drp2 floxed allele were generated by homologous recombination in E14-TG2a.IV (129/Ola strain) ES cells by methods described previously (Sherman et al., 2005). The strategy used for gene targeting is summarized in Figure 1B. The replacement targeting vector contained two loxP sequences in the same orientation. The first loxP sequence was inserted at a SmaI site in intron 3, 1990 bp upstream of exon 4, and the other loxP sequence was at the 3′ of a frt/PGKpA/neo/EM7/PGK/frt/loxP cassette (gift from Dr. Andrew Smith, University of Edinburgh, Edinburgh, UK), which was introduced at an NsiI site in intron 4 located 520 bp downstream of the end of exon 4. A HindIII–XhoI fragment containing MC1-DTA-PGKpA from pMC1DTApA (a gift from Dr. Takeshi Yagi, Osaka University, Osaka, Japan) was ligated to the 3′ end of the targeting clone. For the initial targeting, ES cells were selected in G418 only and resistant clones screened by correct targeting was confirmed by Southern blot analysis of AvrII-digested genomic DNA by hybridization with a 3′ probe external to the homology region and using a probe 5′ to exon 4 to show the presence of the LoxP site with HindIII-digested DNA. The 5′ probe detected a 7.8 kb fragment corresponding to the wild-type (WT) allele and a 1.3 kb fragment corresponding to the targeted allele and the 3′ probe detected a 9.6 kb fragment corresponding to the wild-type allele, and an 11.4 kb fragment corresponding to the targeted allele. Two correctly targeted clones were injected into blastocysts. Both clones gave good germ line transmitting chimeras in test crosses with the C57BL/6 strain, and offspring heterozygous for the correctly targeted allele were subsequently identified by ear biopsy and PCR analysis. One of these lines was crossed with the FLPeR deleter mouse to remove the neo cassette. Heterozygotes were then backcrossed onto the C57BL/6 background for at least 10 generations before experimental analysis. Genotyping of Drp2fl/Y/Cnp1-Cre+/− mice by successive PCRs of genomic DNA extracted from ear biopsies was using the following primers, first 5′-GCTATTCTTGTAGGCGGACCT-3′ and 5′-GGGTCACTGGAGGTCACTGT-3′, yielding a 471 bp product, which after HindIII digestion yielded a 309 and a 162 bp fragment for the floxed allele but remained undigested for the wild-type allele. Second, the presence of Cre was detected as described previously (Lappe-Siefke et al., 2003). Detection of the recombined allele in Drp2−/y or Drp2−/− mice was by PCR using 5′-TCCTAACACAAGTGATGCCTGTAGC-3′ and 5′-TTCAGCAAGGGGATGAGAGTCC-3′ and yielded a 529 bp product. Mice lacking Drp2 in Schwann cells were generated by crossing male mice carrying a floxed allele of Drp2 (Drp2 is on the X chromosome) with Cnp1-Cre mice. We have shown previously that the Cnp1-Cre driver line is effective in Schwann cells during embryonic development before the radial sorting of axons (Grove et al., 2007; Sherman et al., 2012).
Transgenic mice were generated by pronuclear injection using methods previously described (Sherman and Brophy, 2000). We used a well characterized promoter generously provided by John Bermingham (McLaughlin Research Institute, Great Falls, MT) and Steven Scherer (University of Pennsylvania School of Medicine, Philadelphia, PA) comprising a 1.1 kb fragment containing the rat Mpz promoter fused to the human GJB1 gene, which is known to drive expression specifically in Schwann cells (Abel et al., 1999; Scherer et al., 2005; Jeng et al., 2006). Transgenic mice expressed a cDNA encoding mouse Drp2 obtained from the IMAGE consortium with a FLAG tag sequence fused at its 3′ end (Drp2Tg). The codons encoding S748 and T910 were mutated to alanine codons in the Drp2 cDNA by site-directed mutagenesis using QuikChange (Stratagene) to generate mice expressing nonphosphorylated Drp2 (Drp2ΔTg). This transgenic protein also had a C-terminal FLAG tag. A transgenic mouse expressing the cDNA encoding mouse periaxin (PrxTg) was generated by RT-PCR and a FLAG tag fused at its 5′ end. A transgenic that expressed periaxin lacking the Drp2 binding site (PrxΔTg) was generated after deleting the sequence encoding amino acids 130–199 by a PCR-ligation technique as described previously (Sherman and Brophy, 2000). This also had an N-terminal FLAG tag. After backcrossing to C57BL/6 for at least six generations, these mice were interbred with either Drp2−/y or Prx−/− mice so that the transgenes were expressed on either Drp2- or periaxin-null backgrounds, respectively. To facilitate breeding for the transgenic studies, instead of using mice in which Drp2 was inactivated conditionally in Schwann cells, Drp2−/y mice were derived using the Hprt-Cre deleter mouse, since we had previously shown that Drp2 is predominantly if not exclusively expressed in myelinating Schwann cells in the murine nervous system (Sherman et al., 2001). Utr−/− mice were on a DBA/C57BL/6 background and were a generous gift from Kay Davies (Oxford University, Oxford, UK) (Deconinck et al., 1997).
Immunostaining, Western blotting, histology, and in situ hybridization.
All histology and immunofluorescence analyses were performed on quadriceps nerves unless specified otherwise. The perineurium was removed before immunostaining of teased fibers. Further preparation and the method for immunostaining of cryosections or teased fiber preparations were as described previously (Sherman et al., 2005, 2012; Grove et al., 2007). Primary antibodies were used at the following dilutions for immunostaining or Western blotting as indicated. Chicken anti-Po (Aves Labs; 1:200 for immunostaining), mouse anti-β-dystroglycan (Novocastra; 43DAG1/8D5; 1:50 for Western blotting), rabbit anti-Drp2.2222 (anti-CWNEIKKKSHNLRARLEA in exon 4; 1:200 for immunostaining), rat anti-E-cadherin (Zymed; for immunostaining; 1:1000). All other primary and secondary antibodies and nuclear stains have been described previously (Tait et al., 2000; Sherman et al., 2001, 2012). Conventional fluorescence microscopy was performed using an Olympus BX60 microscope, and images were captured using a Hamamatsu Orca-ER camera and Improvision Openlab software. For confocal microscopy, we used a Leica TCL-SL confocal microscope with either a 40× or a 63× objective, 1.4 NA, and Leica proprietary software. The acquired stacks were assembled using the maximum projection tool, and all figures were prepared using Adobe Photoshop CS4 extended, version 11, and were not subjected to any subsequent image processing. Western blotting was performed as described previously (Sherman et al., 2005) on sciatic nerve lysates prepared in 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm sodium orthovanadate, 2 mm sodium pyrophosphate, 10 mm NaF with proteinase inhibitors, 1 mm benzamidine, 0.5 mm 7-amino-1-chloro-3-tosylamido-2-heptanone (TLCK), 1 mm PMSF, and leupeptin and antipain, both 10 mg/ml. For electron microscopy, mice were perfused intravascularly with 2.5% glutaraldehyde and 4% paraformaldehyde in 0.1 m sodium cacodylate buffer, pH 7.4. Nerves were removed, fixed for 2 h at room temperature, then 18 h at 4°C in the same fixative, postfixed in OsO4, and embedded in Araldite. Ultrathin sections were stained with uranyl acetate and lead citrate and examined on a Phillips BioTwin CM120 electron microscope equipped with a Gatan Orius camera. Light micrographs were of toluidine blue-stained transverse sections of quadriceps nerve. In situ hybridization for myelin basic protein (MBP) mRNA was performed as described previously (Court et al., 2004), and surface intensities were plotted using NIH ImageJ.
Phosphopeptide mapping, GST pull downs, kinase and phosphatase assays.
The rabbit anti-Drp2.2222 antibody was used to immunoprecipitate Drp2 from mouse peripheral nerve lysates. The immunoprecipitate was resolved by SDS-PAGE and the Drp2 band was identified using SimplyBlue (Invitrogen). The band was cut out, washed with acetonitrile, and frozen. Following in-gel digestion by trypsin, chymotrypsin, and endoproteinase AspN, sequence analysis of proteolytic fragments was performed by microcapillary reverse-phase HPLC nano-electrospray tandem mass spectrometry (ALC/MS/MS) on a Finnigan LCQ DECA XP Plus quadrupole ion trap mass spectrometer at the Microchemistry and Proteomics Analysis Facility (Harvard University, Cambridge, MA).
GST-fusion proteins were expressed and purified as described previously (Sherman et al., 2001). The GST sequence was ligated with a cDNA encoding amino acids 723–957 in the C terminus of Drp2 and the codons encoding S748 and T910 were mutated to alanine codons in the Drp2 cDNA by site-directed mutagenesis using QuikChange (Stratagene). The purified proteins (30 μg) were incubated while attached to glutathione-Sepharose beads (GE Healthcare) with 250 μl of peripheral nerve lysate and incubated for 3 h at 4°C. The beads were then washed three times with lysis buffer (1% Triton X-100, 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, phosphatase inhibitors, 1 mm sodium orthovanadate, 2 mm sodium pyrophosphate, 10 mm NaF) with proteinase inhibitors (1 mm benzamidine, 0.5 mm TLCK, 1 mm PMSF, and leupeptin and antipain, both 10 μg/ml). The beads were washed twice with kinase buffer, 20 mm HEPES, pH 7.5, 20 mm MgCl2, phosphatase inhibitors, 2 mm DTT, and then resuspended in 30 μl of the same buffer with 33 μm ATP and 5 μCi of [γ-32P]ATP at 30°C for 20 min. Reactions were stopped by SDS-PAGE sample buffer, and proteins were resolved by SDS-PAGE. Phosphorylation was visualized by autoradiography.
For dephosphorylation of Drp2, sciatic nerve extracts were extracted in 1% Triton X-100, 100 mm NaCl, 10 mm MgCl2, 50 mm Tris-HCl, pH 8.0, with proteinase inhibitors and incubated with 20 U of calf intestinal phosphatase (New England Biolabs) in the presence or absence of phosphatase inhibitors at 37°C in a total volume of 25 μl.
Crush injury, electrophysiology, and Rotarod.
Crush injury of sciatic nerves was performed similar to as described previously (Williams and Brophy, 2002). Two-month-old mice were anesthetized, and the right sciatic nerve was exposed under anesthesia and crushed distal to the sciatic notch. Crush was performed for 3 s three times with a number 5 fine forceps. Acutely prepared quadriceps nerves, taken from 3-month- and 6-month-old control and mutant mice, were placed in oxygenated mammalian HEPES physiological solution (137 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 5.5 mm d-glucose, and 5 mm HEPES, pH 7.2–7.4) and nerve conduction velocities measured as described previously (Court et al., 2004). Accelerating Rotarod analysis of mice at 3 and 6 months (minimum of 13 per group) was performed as described previously (Zonta et al., 2011).
Morphometry.
The diameters of axons and axons plus myelin were measured in electron micrographs by first measuring the circumference of each by hand-tracing using ImageJ (NIH), and then by calculating areas from which diameters were derived. Their g ratios were calculated from these diameters. A minimum of 100 axons was counted per animal and a minimum of three animals was used per condition. Internodal lengths were measured as described using Openlab (PerkinElmer) (Court et al., 2004; Sherman et al., 2012). A minimum of 100 fibers was counted per mouse for each condition to quantitate appositions from electron micrographs. Numbers of Schmidt-Lanterman incisures were similarly measured after staining teased fibers with antibodies against Neurofascin (green) and E-cadherin (red). A minimum of 50 measurements was made per animal.
Statistical analysis.
Statistical analysis was by unpaired Student's t tests with GraphPad Prism 5.0c software.
Results
Loss of Drp2 disrupts appositions and Cajal bands
We generated mice without Drp2 in Schwann cells as described in Materials and Methods, and we refer to mice with the genotype Drp2fl/Y/Cnp1-Cre+/− as mutants. The DNA fragment generated by PCR of peripheral nerve genomic DNA after Cre-mediated recombination showed effective inactivation of the Drp2 gene (Fig. 1C). Loss of Drp2 expression in myelinating Schwann cells was highly efficient, as shown by Western blot and immunofluorescence microscopy (Fig. 1D,E). Levels of Drp2 in WT and Drp2fl/Y mice were identical, but Drp2fl/Y mice were considered to be the more appropriate control not only genotypically but also because they were littermates of mutants (Fig. 1D). Drp2 is mislocalized in the absence of periaxin (Sherman et al., 2001), and similarly, periaxin in Drp2-null Schwann cells was no longer concentrated in patches known to colocalize with appositions (Fig. 1F). Hence, loss of Drp2 disrupted both the PDG complex and appositions (Fig. 2A–C). Since it was no longer compartmentalized, cytoplasm now formed an annulus under the plasma membrane of the Schwann cell (Fig. 2B). Derangement of Cajal bands affected the accumulation of MBP mRNA at distal paranodal extremities (Fig. 2D), suggesting that microtubule-based transport was affected, as has been proposed for periaxin-null Schwann cells (Court et al., 2004).
Figure 2.

Loss of Drp2 causes disruption of appositions. A, Immunofluorescence for S100 protein (green) and staining with Alexa Fluor 568-phalloidin for microfilaments (red) shows that loss of Drp2 disrupts Cajal bands. Scale bar, 10 μm. B, Electron microscopy of nerve transverse sections shows loss of appositions in mutants. Scale bar, 2 μm. C, Quantitation of appositions from Drp2fl/Y and mutant mice at P21 and P42 expressed as a frequency distribution. Values for the frequency of each number of appositions are means ± SEM (n = 3 mice). D, Pseudocolor surface plot of mean internodal signal intensity shows that MBP mRNA in Drp2fl/Y Schwann cells (n = 15) accumulates in the perinuclear and paranodal domains, whereas in DRP2 mutants lacking Cajal bands the signal declines from the perinuclear region toward the paranodes (n = 17) as observed in periaxin-null (Prx−/−) Schwann cells (n = 17). Nuclei and nodes of Ranvier are indicated by asterisks and arrowheads, respectively.
In contrast to periaxin-null Schwann cells, and despite their disrupted Cajal bands, Drp2 mutants had normal internodal lengths, conduction velocities, and motor behavior (Fig. 3A–C). Nevertheless, abnormal profiles in the mutant increased with age, and at 6 months they comprised 6.3 ± 0.6% (mean value ± SEM) of mutant nerve fibers (Fig. 4D). These profiles comprised myelin outfoldings, focal hypermyelination, and onion bulbs with thin myelin and supernumerary Schwann cells indicative of remyelination (Fig. 4A–C). In contrast, periaxin-null peripheral nerves are more severely affected, and normal myelin profiles at 6 months are very rare (Gillespie et al., 2000). Although g ratios in the mutant were not significantly different at P21 (0.63 ± 0.01 and 0.65 ± 0.02; mean values ± SEM; n = 3 for control and mutant, respectively) as we have found previously for periaxin-null quadriceps nerves at the same age (Court et al., 2004), by 3.5 months overproduction of myelin was reflected in a decrease in g ratio, which was recapitulated after crush injury. (Fig. 4E). Similar hypermyelination has been observed in periaxin-null Schwann cells and seems to be a prelude to demyelination (Gillespie et al., 2000). These differences in g ratio were not accompanied by any significant difference in axon diameter between control and mutant nerves at 3 weeks (2.47 ± 0.10 and 2.76 ± 0.07 μm; mean values ± SEM; n = 3, respectively) or at 3.5 months (3.75 ± 0.32 and 3.50 ± 0.19 μm; mean values ± SEM; n = 3, respectively) in sciatic nerves.
Figure 3.

Loss of Cajal bands does not cause functional deficits. A, Internodal lengths of control (Drp2fl/Y) quadriceps nerves compared with mutants at P21 (n = 3) and P90 (n = 4). Values are means ± SEM. B, Nerve conduction velocities were measured in control and mutant mice at P90 (n = 4) and at P180 (n = 8). Values are means ± SEM. C, Latency to fall on the accelerating Rotarod was measured during successive trials for control and mutant mice at P90 and P180. Values are means ± SEM (n = 13 mice for each condition). For A–C, control and mutant values for each condition were not significantly different.
Figure 4.

Disrupted myelination in Drp2 mutant mice. A, Transverse sections showed increasing numbers of aberrant myelin profiles in the mutant with age. Scale bar, 10 μm. B, C, Electron micrographs of mutant nerves showing hypermyelination and onion bulbs. Scale bars: B, 1 μm; C, 2 μm. D, Percentage of abnormal myelinated axon profiles with age in control and mutant quadriceps nerves. Values are means ± SEM, where n = 3 mice for each condition; **p < 0.005; ***p < 0.0005. E, Mean values of g ratios in uncrushed (contralateral) and crushed (distal) sciatic nerves from 3.5-month-old mice 6 weeks after crush. Mutant nerves have lower g ratios. Values are means ± SEM, where n = 3 mice for each condition; *p < 0.05; **p < 0.005.
Unlike periaxin, Drp2 is not a component of Schmidt-Lanterman incisures, the cytoplasm-filled channels in PNS myelin. Nevertheless, since loss of Drp2 caused changes in the organization of the Schwann cell cytoplasm in the Cajal bands, we wondered whether Schmidt-Lanterman incisures were also affected. Surprisingly, at 6 weeks of age, we found that these incisures are more numerous in Drp2 mutant Schwann cells (2.24 ± 0.07 100 μm−1 and 3.67 ± 0.02 100 μm−1; mean values ± SEM; n ≧ 2 for control and Drp2 mutant, respectively; p < 0.0001). The number of incisures did not change with further development when measured at 9 weeks (2.25 ± 0.01 100 μm−1 and 3.69 ± 0.02 100 μm−1; mean values ± SEM; n = 3 for control and Drp2 mutant, respectively; p < 0.0001). This is in contrast to periaxin-null Schwann cells in which Schmidt-Lanterman incisures are lost (Gillespie et al., 1994; Scherer et al., 1995) (Fig. 5). This underlines the fact that, although both are components of the dystroglycan complex, loss of Drp2 and periaxin can have differential effects on peripheral nerve structure and function. Insofar as Schmidt-Lanterman incisures may serve as conduits for communication between the Schwann cell body and the inner myelin tongue, the fact that they are not reduced in number suggests that they are not malfunctioning and thus contributing to the pathology of the Drp2-null mouse.
Figure 5.

Immunofluorescence staining of Schmidt-Lanterman incisures in teased fibers from Drp2fl/Y, Drp2 mutant, and periaxin-null mice. Incisures are stained with antibodies against Neurofascin (green) and E-cadherin (red). Scale bar, 25 μm.
Loss of Drp2 causes upregulation of other Dystrophins but not enhanced cleavage of β-dystroglycan
Loss of dystrophin in skeletal muscle leads to a compensatory twofold increase in utrophin, and transgenically expressed utrophin can significantly reduce dystrophic pathology in the skeletal muscles of mice lacking dystrophin (Tinsley et al., 1996; Moghadaszadeh et al., 2003). Hence, we asked whether there was increased expression of the dystrophin family members Dp116 and utrophin in Drp2-null Schwann cells. We did observe increased levels of Dp116 and utrophin, but not α- or β-dystroglycan, in Drp2 mutant nerves (Fig. 6A) (Albrecht et al., 2008); similar increases were not observed in periaxin-null mice, possibly because they still express Drp2 (Fig. 6A).
Figure 6.

Composition of Schwann cell dystroglycan complexes in the absence of either Drp2 or periaxin. A, Western blot of sciatic nerve lysates shows that loss of Drp2 causes complementary increases in utrophin and the dystrophin isoform, Dp116. Only traces of the cleaved form of β-dystroglycan (β-DG 31) were detected in the presence of the MMP inhibitor 1,10-phenanthroline (5 mm). B, Preparation of sciatic nerve lysates with or without 1,10-phenanthroline (PHEN) showed that β-dystroglycan is susceptible to proteolytic breakdown during sample preparation. Actin was the loading control.
A model has been proposed in which Dp116 and utrophin in the plasma membrane domains associated with Cajal bands preferentially associate with the cleaved form of β-dystroglycan, whereas the PDG complex in Schwann cell appositions contains intact β-dystroglycan (Court et al., 2011). Certainly, β-dystroglycan is very susceptible to cleavage to a 31 kDa form by two matrix metalloproteinases, MMP2 and MMP9 (Yamada et al., 2001; Matsumura et al., 2005; Hnia et al., 2006; Zhong et al., 2006). However, we did not detect significant amounts of this cleaved form in control, mutant or periaxin-null nerves when nerves were lysed in the presence of 1,10-phenanthroline, a highly effective inhibitor of MMPs (Fig. 6A). Neither was this cleaved species present in the pellet after vigorous centrifugation of the lysate. A faint but variable signal of the breakdown product could be detected (Fig. 6A), but in five different experiments there was no correlation between the genotype of the mouse and the amount of 31 kDa β-dystroglycan, and it never represented >1–2% of total β-dystroglycan. Hence, disruption of the PDG complex does not appear to activate the MMPs. We compared the amounts of 31 kDa β-dystroglycan in peripheral nerve lysates with and without 1,10-phenanthroline. This confirmed that the intact protein that migrates at 43 kDa is the normal species in peripheral nerves but that it is very susceptible to proteolysis in the absence of the MMP inhibitor (Fig. 6A,B).
Drp2 phosphorylation and recruitment to appositions requires interaction with periaxin
Loss of periaxin in Schwann cells results in increased Drp2 mobility in SDS-PAGE (Fig. 6A). We asked whether this shift reflected a change in phosphorylation status and, if so, phosphorylation might play a role in apposition formation. Treatment of peripheral nerve extracts from wild-type mice with calf intestinal phosphatase in the presence or absence of phosphatase inhibitors showed that Drp2 is phosphorylated (Fig. 7A). The two major sites of phosphorylation were shown by mass spectrometry to be S748 and T910 (Fig. 7B), and these were confirmed by in vitro kinase assays (Fig. 7C).
Figure 7.

Drp2 is phosphorylated. A, Western blots of nerve lysates treated with calf intestinal phosphatase (CIP) with or without phosphatase inhibitors (PI) shows that Drp2 is phosphorylated. B, Phosphopeptide mapping of Drp2 by mass spectrometry identified S748 and T910 as the primary sites of phosphorylation. C, In vitro phosphorylation of GST fusion proteins containing the C-terminal region of Drp2 (amino acids 723–957) with peripheral nerve lysates in the presence of [γ-32P]ATP. Mutation of either S748 or T910 to alanine reduced phosphorylation. Coomassie Blue staining shows the amounts of the GST proteins were comparable.
Phosphorylation at these sites survived lysis in the absence of phosphatase inhibitors suggesting that they are protected, possibly through interaction with periaxin in vivo. This was supported by the fact that Drp2 from periaxin-null nerves was dephosphorylated even when lysates were prepared with phosphatase inhibitors (Fig. 8A). The role of periaxin in promoting Drp2 phosphorylation was also demonstrated in periaxin heterozygous-null nerves, which have reduced levels of periaxin (Gillespie et al., 2000). As the animals get older, periaxin levels increase with a concomitant shift toward more phosphorylated Drp2 (Fig. 8B).
Figure 8.
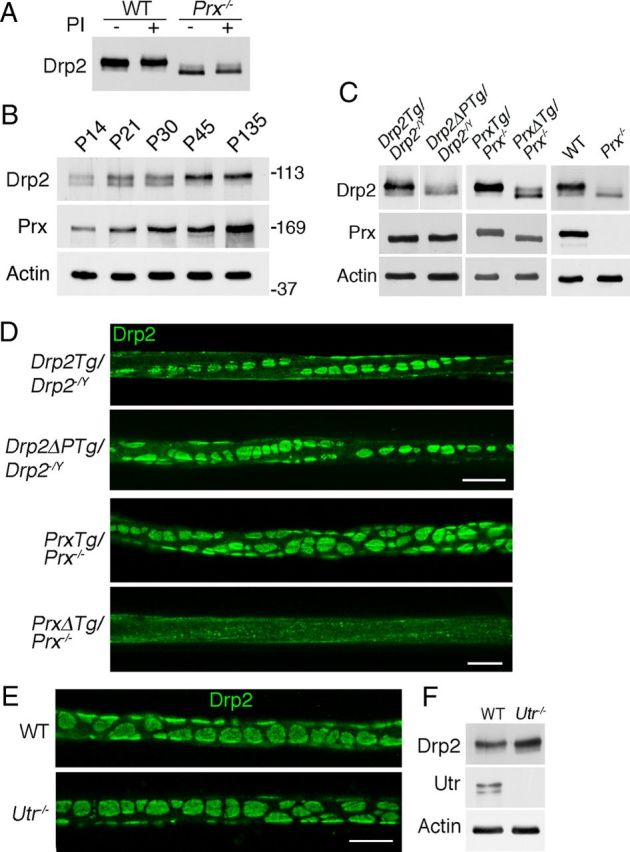
Drp2 is phosphorylated when complexed with Periaxin in appositions. A, Preparation of nerve lysates from either WT or periaxin-null nerves (Prx−/−) in the presence of phosphatase inhibitors showed that Drp2 is not dephosphorylated during extraction. B, Mice heterozygous null for Prx accumulate periaxin with age with a concomitant shift of Drp2 to a more phosphorylated state. Actin was used as a loading control. C, Western blots showing the expression and phosphorylation state of Drp2 in nerves from transgenic mice expressing wild-type Drp2 (Drp2Tg/Drp2−/−) or the phosphorylation site mutant (Drp2ΔPTg/Drp2−/−) on a Drp2-null background, and from transgenic wild-type periaxin (PrxTg/Prx−/−) or a mutant lacking the Drp2 binding domain (PrxΔTg/Prx−/−) on a periaxin-null background. Control lanes show periaxin and Drp2 from WT and periaxin-null (Prx−/−) nerves, respectively. Actin was used as a loading control. D, Immunofluorescence of Drp2 localization in teased quadriceps fibers from Drp2Tg/Drp2−/− and Drp2ΔPTg/Drp2−/− mice and from PrxTg/Prx−/− and PrxΔTg/Prx−/− mice. Scale bar, 10 μm. E, F, Immunofluorescence localization of Drp2 in quadriceps nerves from 6-week-old utrophin-null mice (Utr−/−) was indistinguishable from controls. Western blot analysis showed that Drp2 is also phosphorylated. Scale bar, 10 μm.
Since loss of appositions is associated with a shift to the dephosphorylated form of Drp2, we asked whether nonphosphorylated Drp2 could still assemble into appositions. We generated transgenic mice that expressed wild-type and mutant Drp2 in which the two major phosphorylation sites were mutated on a Drp2-null background. We used mice expressing wild-type Drp2 on a Drp2-null background rather than wild-type mice to control for the presence of the FLAG tag and the timing of expression using a heterologous promoter. Western blots showed that the levels of wild-type and mutant Drp2 and the amounts of periaxin in the respective transgenics were similar (Fig. 8C). The blots also showed that the mobility of the mutant nonphosphorylated Drp2 was increased as predicted (Fig. 8C). Immunofluorescence analysis of Drp2 showed that recruitment to appositions did not require phosphorylation at these sites (Fig. 8D).
We next wanted to determine whether direct interaction between periaxin and Drp2 was necessary for apposition formation and Drp2 phosphorylation. Transgenic mice were generated on a Prx−/− background expressing either wild-type periaxin or a periaxin mutant lacking the basic domain (amino acids 130–199), which is known to be essential for interaction with Drp2 (Sherman et al., 2001). Levels of transgenically encoded wild-type and mutant periaxin were similar (Fig. 8C). Loss of periaxin interaction with Drp2 reduced the phosphorylation of Drp2, and Drp2-positive appositions were disrupted (Fig. 8C,D). The effect of complete loss of periaxin in Prx−/− mutant sciatic nerves compared with WT mice is shown for comparison (Fig. 8C). Despite the loss of Cajal bands, internodal lengths in the periaxin mutant lacking the Drp2 binding domain were normal (PrxTG/Prx−/−, 498.4 ± 10.0 μm; PrxΔTG/Prx−/−, 528.0 ± 6.8 μm; mean values ± SEM; n ≥ 3). Hence, from these data we concluded that, although phosphorylation of Drp2 was not essential for the assembly of appositions, and therefore Cajal bands, it was an excellent index of periaxin–Drp2 interaction in appositions.
To further explore the relationship between the formation of appositions and Drp2 phosphorylation, we wanted to analyze peripheral nerves from another mutant where appositions might be either abnormal or absent. Mice lacking utrophin were of interest since we had observed upregulation of utrophin in the absence of Drp2 (Fig. 6A) and these mice have Schwann cells that have been reported to have disrupted appositions (Court et al., 2009). However, we found their Drp2-stained appositions to be normal (Fig. 8E). Furthermore, and in accord with predictions from the data above, Drp2 was phosphorylated in utrophin-null nerves, which provided an independent measure of the intactness of appositions in addition to immunofluorescence (Fig. 8F).
Discussion
We have shown by disrupting appositions in myelinating Schwann cells through the loss of Drp2 that these structures and their associated Cajal bands are required to regulate the radial growth of myelin and prevent focal hypermyelination. This work has also demonstrated that Cajal bands do not have an essential role in the regulation of Schwann cell elongation and thus in determining internodal length. We have also been able to evaluate a recent model proposed for the regulation of Cajal band size (Court et al., 2011).
Recent quantitative studies have shown that periaxin comprises 16% by weight of PNS myelin protein, whereas Drp2 represents ∼0.2% (Patzig et al., 2011). These values are not consistent with an exclusive stoichiometric relationship between periaxin and Drp2 in a dystroglycan complex and suggest that periaxin has roles beyond its participation in forming the PDG complex and appositions. Loss of periaxin disrupts appositions, impairs Schwann cell elongation, reduces nerve conduction velocity, and causes focal hypermyelination and demyelination (Gillespie et al., 2000; Sherman et al., 2001; Court et al., 2004). Furthermore, mutations in the Periaxin gene in humans cause Charcot–Marie–Tooth disease 4F (Boerkoel et al., 2001; Guilbot et al., 2001; Takashima et al., 2002). In contrast, the loss of Drp2 or the disruption of dystroglycan complexes by Schwann cell-specific loss of dystroglycan have much less severe phenotypes (Saito et al., 2003).
A very clear difference between peripheral nerves lacking Drp2 or periaxin is the effect on Schmidt-Lanterman incisures. Although their function is still not completely understood, it is assumed that they act as conduits for the delivery of nutrients and/or myelin components to the inner layers of the myelin sheath. Periaxin is a constituent of the incisures but Drp2 is not (Gillespie et al., 2000; Sherman et al., 2001). This may explain why the incisures are deranged in periaxin-null Schwann cells, whereas they are actually increased in number in the absence of Drp2. In Drp2 mutants, excess periaxin no longer interacting with Drp2 might be available for participating in incisure formation, so generating an increased number of incisures.
In similar vein, in Drp2 mutant nerves, we observed increased levels of Dp116 and utrophin, but not α- or β-dystroglycan. This suggests that, in the absence of Drp2, other dystrophin family members supplant Drp2 in dystroglycan complexes, while the number of these complexes remains relatively constant. However, since the Dp116 and utrophin complexes are concentrated in the plasma membrane contiguous with Cajal bands, they would not be able to functionally replace Drp2 in forming appositions (Albrecht et al., 2008). Periaxin-null Schwann cells do not display the same increased levels of of Dp116 and utrophin, probably because they still express Drp2.
The cleaved 31 kDa form of β-dystroglycan has been reported to represent a substantial fraction of total β-dystroglycan in peripheral nerves (Yamada et al., 2001; Zaccaria et al., 2001; Court et al., 2011), although it is only significant in skeletal muscle or brain under pathological conditions (Yamada et al., 2001; Zaccaria et al., 2001; Matsumura et al., 2005; Shang et al., 2008). A model has been proposed recently suggesting that dystroglycan complexes containing Dp116 and utrophin in the plasma membrane domains associated with Cajal bands preferentially associate with the cleaved 31 kDa form of β-dystroglycan, whereas the PDG complex in Schwann cell appositions contains intact β-dystroglycan (Court et al., 2011). Regulated conversion of β-dystroglycan to its cleaved form would then control the relative sizes of appositions and therefore Cajal bands.
The proteinases responsible for cleavage of dystroglycan, MMP2 and MMP9, are highly susceptible to activation by detergents (Birkedal-Hansen and Taylor, 1982; Birkedal-Hansen et al., 1993), and SDS-mediated activation of proteolysis as a result of substrate unfolding is also a potential complication (Linderstrom-Lang and Moller, 1953; Reynolds and Tanford, 1970; Beynon, 1988). Interestingly, when we used 2% SDS instead of our normal lysis buffer, degradation of β-dystroglycan to the cleaved form was considerably accentuated in the absence of the MMP inhibitor 1,10-phenanthroline (data not shown), which could also have been caused by more effective activation of the MMPs by SDS. Importantly, we found that inclusion of 1,10-phenanthroline effectively prevented generation of the cleaved β-dystroglycan during nerve lysis. This suggests that high activities of MMP2 and MMP9 are intrinsic to the nerve and do not result from the invasion of macrophages that normally accompanies pathology (Li et al., 2009).
From our work and that of others, we conclude that the model by which Cajal band segregation is regulated by means of MMPs (Court et al., 2011) is unlikely for the following reasons. In this model, appositions are proposed to be enriched in intact β-dystroglycan, whereas the β-dystroglycan found outside appositions in Cajal bands is proposed to be the proteolysed, cleaved form. Hence, a prediction from the model is that loss of appositions in Drp2- and periaxin-null nerves would lead to a reduction in intact β-dystroglycan and a substantial increase in cleaved β-dystroglycan. However, this is not the case (Fig. 6A). Second, the model predicts that Dp116 in Cajal bands would preferentially interact with the cleaved form of β-dystroglycan. We have indeed previously shown that Dp116 is segregated to Cajal bands (Sherman et al., 2001; Albrecht et al., 2008), and this was confirmed by Court et al. (2011). However, it has been previously demonstrated experimentally by Mornet's group (Hnia et al., 2006) that Dp116 in Schwann cells preferentially interacts with uncleaved, intact β-dystroglycan and not the cleaved form. Finally, and we believe most conclusively, we have shown that the generation of cleaved β-dystroglycan in peripheral nerve extracts is an adventitious consequence of proteolysis during nerve lysis (Fig. 6B).
Although we have shown that β-dystroglycan is uncleaved in myelinating Schwann cells, it is still unclear how the PDG complex segregates from the two other dystroglycan complexes that use utrophin and the dystrophin isoform Dp116. In contrast to Court et al. (2009), we find that Schwann cells lacking utrophin have normal appositions, Cajal bands, and normally phosphorylated Drp2. Hence, it seems unlikely that the utrophin–dystroglycan complex has a role in cytoplasmic compartmentalization or that the upregulation of utrophin seen in Drp2-null Schwann cells compensates functionally for loss of Drp2. Although Drp2 is normally phosphorylated in the PDG complex when Drp2 and periaxin interact at appositions, recruitment of Drp2 to appositions did not require phosphorylation at the major sites that we identified. However, we cannot exclude possible compensatory phosphorylation at new sites. The role of phosphorylation will be the subject of future work.
Periaxin and Drp2 are not only essential to form the PDG complex but they must also interact. Nevertheless, the presence of periaxin, even without the Drp2 binding domain, ensures that Schwann cell elongation is normal and further underlines the importance of this protein in the regulation of Schwann cell growth independent of its role in forming Cajal bands. This experiment provides further support for our conclusion that internodal lengths are not regulated by appositions and Cajal bands. Crucially, our ability to uncouple Cajal band disruption from the regulation of Schwann cell elongation has revealed a distinct and vital role for periaxin in peripheral nerve development.
How might disruption of Cajal bands cause focal hypermyelination? The close association of the PDG complex with the myelin sheath, the Schwann cell plasma membrane, and the interaction with basement membrane is likely to promote the mechanical stability of peripheral nerves to prevent their damage as has been found for the dystroglycan complexes of skeletal muscle (Han et al., 2009). This is likely to be important in the PNS because it is vulnerable to quotidian insult. Although we have shown that loss of Cajal bands in Drp2-null nerves does not prevent remyelination after nerve crush, we propose that disruption of this complex that mechanically links the extracellular milieu with the myelin sheath could lead to cumulative long-term instability that is reflected in the age-related accumulation of abnormal myelin profiles and focal hypermyelination.
Footnotes
This work was supported by the Wellcome Trust. We thank Heather Anderson, Stuart Fleming, and Linda Ferguson for excellent technical support. Austin Smith (Cambridge University, Cambridge, UK), Andrew Smith (Edinburgh University, Edinburgh, UK), and Vladimir Buchman (Cardiff University, Cardiff, UK) are also thanked for valuable advice on gene targeting. We also thank Kay Davies (Oxford University, Oxford, UK) for providing utrophin-null mice and Klaus-Armin Nave (Max Planck Institute for Experimental Medicine, Göttingen, Germany) for the Cnp1-Cre mice.
References
- Abel A, Bone LJ, Messing A, Scherer SS, Fischbeck KH. Studies in transgenic mice indicate a loss of connexin32 function in X-linked Charcot–Marie–Tooth disease. J Neuropathol Exp Neurol. 1999;58:702–710. doi: 10.1097/00005072-199907000-00004. [DOI] [PubMed] [Google Scholar]
- Albrecht DE, Sherman DL, Brophy PJ, Froehner SC. The ABCA1 cholesterol transporter associates with one of two distinct dystrophin-based scaffolds in Schwann cells. Glia. 2008;56:611–618. doi: 10.1002/glia.20636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beynon RJ. Prevention of unwanted proteolysis. Methods Mol Biol. 1988;3:1–23. doi: 10.1385/0-89603-126-8:1. [DOI] [PubMed] [Google Scholar]
- Birkedal-Hansen H, Taylor RE. Detergent-activation of latent collagenase and resolution of its component molecules. Biochem Biophys Res Commun. 1982;107:1173–1178. doi: 10.1016/s0006-291x(82)80120-4. [DOI] [PubMed] [Google Scholar]
- Birkedal-Hansen H, Moore WG, Bodden MK, Windsor LJ, Birkedal-Hansen B, DeCarlo A, Engler JA. Matrix metalloproteinases: a review. Crit Rev Oral Biol Med. 1993;4:197–250. doi: 10.1177/10454411930040020401. [DOI] [PubMed] [Google Scholar]
- Boerkoel CF, Takashima H, Stankiewicz P, Garcia CA, Leber SM, Rhee-Morris L, Lupski JR. Periaxin mutations cause recessive Dejerine-Sottas neuropathy. Am J Hum Genet. 2001;68:325–333. doi: 10.1086/318208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn RD, Campbell KP. Molecular basis of muscular dystrophies. Muscle Nerve. 2000;23:1456–1471. doi: 10.1002/1097-4598(200010)23:10<1456::aid-mus2>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Colognato H, Winkelmann DA, Yurchenco PD. Laminin polymerization induces a receptor-cytoskeleton network. J Cell Biol. 1999;145:619–631. doi: 10.1083/jcb.145.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court FA, Sherman DL, Pratt T, Garry EM, Ribchester RR, Cottrell DF, Fleetwood-Walker SM, Brophy PJ. Restricted growth of Schwann cells lacking Cajal bands slows conduction in myelinated nerves. Nature. 2004;431:191–195. doi: 10.1038/nature02841. [DOI] [PubMed] [Google Scholar]
- Court FA, Hewitt JE, Davies K, Patton BL, Uncini A, Wrabetz L, Feltri ML. A laminin-2, dystroglycan, utrophin axis is required for compartmentalization and elongation of myelin segments. J Neurosci. 2009;29:3908–3919. doi: 10.1523/JNEUROSCI.5672-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court FA, Zambroni D, Pavoni E, Colombelli C, Baragli C, Figlia G, Sorokin L, Ching W, Salzer JL, Wrabetz L, Feltri ML. MMP2-9 cleavage of dystroglycan alters the size and molecular composition of Schwann cell domains. J Neurosci. 2011;31:12208–12217. doi: 10.1523/JNEUROSCI.0141-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deconinck AE, Potter AC, Tinsley JM, Wood SJ, Vater R, Young C, Metzinger L, Vincent A, Slater CR, Davies KE. Postsynaptic abnormalities at the neuromuscular junctions of utrophin-deficient mice. J Cell Biol. 1997;136:883–894. doi: 10.1083/jcb.136.4.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbeej M, Larsson E, Ibraghimov-Beskrovnaya O, Roberds SL, Campbell KP, Ekblom P. Non-muscle alpha-dystroglycan is involved in epithelial development. J Cell Biol. 1995;130:79–91. doi: 10.1083/jcb.130.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie CS, Sherman DL, Blair GE, Brophy PJ. Periaxin, a novel protein of myelinating Schwann cells with a possible role in axonal ensheathment. Neuron. 1994;12:497–508. doi: 10.1016/0896-6273(94)90208-9. [DOI] [PubMed] [Google Scholar]
- Gillespie CS, Sherman DL, Fleetwood-Walker SM, Cottrell DF, Tait S, Garry EM, Wallace VC, Ure J, Griffiths IR, Smith A, Brophy PJ. Peripheral demyelination and neuropathic pain behavior in periaxin-deficient mice. Neuron. 2000;26:523–531. doi: 10.1016/s0896-6273(00)81184-8. [DOI] [PubMed] [Google Scholar]
- Grove M, Komiyama NH, Nave KA, Grant SG, Sherman DL, Brophy PJ. FAK is required for axonal sorting by Schwann cells. J Cell Biol. 2007;176:277–282. doi: 10.1083/jcb.200609021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilbot A, Williams A, Ravisé N, Verny C, Brice A, Sherman DL, Brophy PJ, LeGuern E, Delague V, Bareil C, Mégarbané A, Claustres M. A mutation in periaxin is responsible for CMT4F, an autosomal recessive form of Charcot–Marie–Tooth disease. Hum Mol Genet. 2001;10:415–421. doi: 10.1093/hmg/10.4.415. [DOI] [PubMed] [Google Scholar]
- Han R, Kanagawa M, Yoshida-Moriguchi T, Rader EP, Ng RA, Michele DE, Muirhead DE, Kunz S, Moore SA, Iannaccone ST, Miyake K, McNeil PL, Mayer U, Oldstone MB, Faulkner JA, Campbell KP. Basal lamina strengthens cell membrane integrity via the laminin G domain-binding motif of alpha-dystroglycan. Proc Natl Acad Sci U S A. 2009;106:12573–12579. doi: 10.1073/pnas.0906545106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnia K, Hugon G, Masmoudi A, Mercier J, Rivier F, Mornet D. Effect of beta-dystroglycan processing on utrophin/Dp116 anchorage in normal and mdx mouse Schwann cell membrane. Neuroscience. 2006;141:607–620. doi: 10.1016/j.neuroscience.2006.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- Imamura M, Araishi K, Noguchi S, Ozawa E. A sarcoglycan-dystroglycan complex anchors Dp116 and utrophin in the peripheral nervous system. Hum Mol Genet. 2000;9:3091–3100. doi: 10.1093/hmg/9.20.3091. [DOI] [PubMed] [Google Scholar]
- Jeng LJ, Balice-Gordon RJ, Messing A, Fischbeck KH, Scherer SS. The effects of a dominant connexin32 mutant in myelinating Schwann cells. Mol Cell Neurosci. 2006;32:283–298. doi: 10.1016/j.mcn.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Li H, Mittal A, Makonchuk DY, Bhatnagar S, Kumar A. Matrix metalloproteinase-9 inhibition ameliorates pathogenesis and improves skeletal muscle regeneration in muscular dystrophy. Hum Mol Genet. 2009;18:2584–2598. doi: 10.1093/hmg/ddp191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Edgar D, Fässler R, Wadsworth W, Yurchenco PD. The role of laminin in embryonic cell polarization and tissue organization. Dev Cell. 2003;4:613–624. doi: 10.1016/s1534-5807(03)00128-x. [DOI] [PubMed] [Google Scholar]
- Linderstrøm-Lang K, Møller KM. Proteolytic enzymes. Annu Rev Biochem. 1953;22:57–84. doi: 10.1146/annurev.bi.22.070153.000421. [DOI] [PubMed] [Google Scholar]
- Masaki T, Matsumura K. Biological role of dystroglycan in Schwann cell function and its implications in peripheral nervous system diseases. J Biomed Biotechnol. 2010;2010:740403. doi: 10.1155/2010/740403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura K, Yamada H, Shimizu T, Campbell KP. Differential expression of dystrophin, utrophin and dystrophin-associated proteins in peripheral nerve. FEBS Lett. 1993;334:281–285. doi: 10.1016/0014-5793(93)80695-q. [DOI] [PubMed] [Google Scholar]
- Matsumura K, Zhong D, Saito F, Arai K, Adachi K, Kawai H, Higuchi I, Nishino I, Shimizu T. Proteolysis of beta-dystroglycan in muscular diseases. Neuromuscul Disord. 2005;15:336–341. doi: 10.1016/j.nmd.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Moghadaszadeh B, Albrechtsen R, Guo LT, Zaik M, Kawaguchi N, Borup RH, Kronqvist P, Schroder HD, Davies KE, Voit T, Nielsen FC, Engvall E, Wewer UM. Compensation for dystrophin-deficiency: ADAM12 overexpression in skeletal muscle results in increased alpha 7 integrin, utrophin and associated glycoproteins. Hum Mol Genet. 2003;12:2467–2479. doi: 10.1093/hmg/ddg264. [DOI] [PubMed] [Google Scholar]
- Nageotte J. Les étranglements de Ranvier et les espaces interannulaires des fibres nerveuses a myéline. Comptes Rendus de l'Association des Anatomistes. 1910;12:30–45. [Google Scholar]
- Nemiloff A. Über die Beziehung der sog. “Zellen der Schwannschen Scheide” zum Myelin in den Nervenfasen von Säugetieren. Archiv für Mikroskopische Anatomie und Entwicklungsgeschichte. 1910;76:329–348. [Google Scholar]
- Patzig J, Jahn O, Tenzer S, Wichert SP, de Monasterio-Schrader P, Rosfa S, Kuharev J, Yan K, Bormuth I, Bremer J, Aguzzi A, Orfaniotou F, Hesse D, Schwab MH, Möbius W, Nave KA, Werner HB. Quantitative and integrative proteome analysis of peripheral nerve myelin identifies novel myelin proteins and candidate neuropathy loci. J Neurosci. 2011;31:16369–16386. doi: 10.1523/JNEUROSCI.4016-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramón y Cajal S. El aparato endocelular de Golgi de la célula de Schwann y algunas observaciones sobre la estructura de los tubos nerviosos. Trabajos del Laboratorio de Investigaciones Biológicas de la Universidad de Madrid. 1912;10:221–246. [Google Scholar]
- Ramón y Cajal S. Histology. London: Bailliere, Tindall and Cox; 1933. [Google Scholar]
- Reynolds JA, Tanford C. Binding of dodecyl sulfate to proteins at high binding ratios. Possible implications for the state of proteins in biological membranes. Proc Natl Acad Sci U S A. 1970;66:1002–1007. doi: 10.1073/pnas.66.3.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito F, Masaki T, Kamakura K, Anderson LV, Fujita S, Fukuta-Ohi H, Sunada Y, Shimizu T, Matsumura K. Characterization of the transmembrane molecular architecture of the dystroglycan complex in schwann cells. J Biol Chem. 1999;274:8240–8246. doi: 10.1074/jbc.274.12.8240. [DOI] [PubMed] [Google Scholar]
- Saito F, Moore SA, Barresi R, Henry MD, Messing A, Ross-Barta SE, Cohn RD, Williamson RA, Sluka KA, Sherman DL, Brophy PJ, Schmelzer JD, Low PA, Wrabetz L, Feltri ML, Campbell KP. Unique role of dystroglycan in peripheral nerve myelination, nodal structure, and sodium channel stabilization. Neuron. 2003;38:747–758. doi: 10.1016/s0896-6273(03)00301-5. [DOI] [PubMed] [Google Scholar]
- Scherer SS, Xu YT, Bannerman PG, Sherman DL, Brophy PJ. Periaxin expression in myelinating Schwann cells: modulation by axon-glial interactions and polarized localization during development. Development. 1995;121:4265–4273. doi: 10.1242/dev.121.12.4265. [DOI] [PubMed] [Google Scholar]
- Scherer SS, Xu YT, Messing A, Willecke K, Fischbeck KH, Jeng LJ. Transgenic expression of human connexin32 in myelinating Schwann cells prevents demyelination in connexin32-null mice. J Neurosci. 2005;25:1550–1559. doi: 10.1523/JNEUROSCI.3082-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang ZJ, Ethunandan M, Górecki DC, Brennan PA. Aberrant expression of beta-dystroglycan may be due to processing by matrix metalloproteinases-2 and -9 in oral squamous cell carcinoma. Oral Oncol. 2008;44:1139–1146. doi: 10.1016/j.oraloncology.2008.02.016. [DOI] [PubMed] [Google Scholar]
- Sherman DL, Brophy PJ. A tripartite nuclear localization signal in the PDZ-domain protein L-periaxin. J Biol Chem. 2000;275:4537–4540. doi: 10.1074/jbc.275.7.4537. [DOI] [PubMed] [Google Scholar]
- Sherman DL, Fabrizi C, Gillespie CS, Brophy PJ. Specific disruption of a Schwann cell dystrophin-related protein complex in a demyelinating neuropathy. Neuron. 2001;30:677–687. doi: 10.1016/s0896-6273(01)00327-0. [DOI] [PubMed] [Google Scholar]
- Sherman DL, Tait S, Melrose S, Johnson R, Zonta B, Court FA, Macklin WB, Meek S, Smith AJ, Cottrell DF, Brophy PJ. Neurofascins are required to establish axonal domains for saltatory conduction. Neuron. 2005;48:737–742. doi: 10.1016/j.neuron.2005.10.019. [DOI] [PubMed] [Google Scholar]
- Sherman DL, Krols M, Wu LM, Grove M, Nave KA, Gangloff YG, Brophy PJ. Arrest of myelination and reduced axon growth when Schwann cells lack mTOR. J Neurosci. 2012;32:1817–1825. doi: 10.1523/JNEUROSCI.4814-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, Mills AA, Wang X, Bradley A. A targeted X-linked CMV-Cre line. Genesis. 2002;32:187–188. doi: 10.1002/gene.10043. [DOI] [PubMed] [Google Scholar]
- Tait S, Gunn-Moore F, Collinson JM, Huang J, Lubetzki C, Pedraza L, Sherman DL, Colman DR, Brophy PJ. An oligodendrocyte cell adhesion molecule at the site of assembly of the paranodal axo-glial junction. J Cell Biol. 2000;150:657–666. doi: 10.1083/jcb.150.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima H, Boerkoel CF, De Jonghe P, Ceuterick C, Martin JJ, Voit T, Schröder JM, Williams A, Brophy PJ, Timmerman V, Lupski JR. Periaxin mutations cause a broad spectrum of demyelinating neuropathies. Ann Neurol. 2002;51:709–715. doi: 10.1002/ana.10213. [DOI] [PubMed] [Google Scholar]
- Tinsley JM, Blake DJ, Pearce M, Knight AE, Kendrick-Jones J, Davies KE. Dystrophin and related proteins. Curr Opin Genet Dev. 1993;3:484–490. doi: 10.1016/0959-437x(93)90124-8. [DOI] [PubMed] [Google Scholar]
- Tinsley JM, Potter AC, Phelps SR, Fisher R, Trickett JI, Davies KE. Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature. 1996;384:349–353. doi: 10.1038/384349a0. [DOI] [PubMed] [Google Scholar]
- Ushiki T, Ide C. Scanning electron microscopic studies of the myelinated nerve fibres of the mouse sciatic nerve with special reference to the Schwann cell cytoplasmic network external to the myelin sheath. J Neurocytol. 1987;16:737–747. doi: 10.1007/BF01611982. [DOI] [PubMed] [Google Scholar]
- Wallace HA, Marques-Kranc F, Richardson M, Luna-Crespo F, Sharpe JA, Hughes J, Wood WG, Higgs DR, Smith AJ. Manipulating the mouse genome to engineer precise functional syntenic replacements with human sequence. Cell. 2007;128:197–209. doi: 10.1016/j.cell.2006.11.044. [DOI] [PubMed] [Google Scholar]
- Williams AC, Brophy PJ. The function of the Periaxin gene during nerve repair in a model of CMT4F. J Anat. 2002;200:323–330. doi: 10.1046/j.1469-7580.2002.00038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada H, Shimizu T, Tanaka T, Campbell KP, Matsumura K. Dystroglycan is a binding protein of laminin and merosin in peripheral nerve. FEBS Lett. 1994;352:49–53. doi: 10.1016/0014-5793(94)00917-1. [DOI] [PubMed] [Google Scholar]
- Yamada H, Saito F, Fukuta-Ohi H, Zhong D, Hase A, Arai K, Okuyama A, Maekawa R, Shimizu T, Matsumura K. Processing of beta-dystroglycan by matrix metalloproteinase disrupts the link between the extracellular matrix and cell membrane via the dystroglycan complex. Hum Mol Genet. 2001;10:1563–1569. doi: 10.1093/hmg/10.15.1563. [DOI] [PubMed] [Google Scholar]
- Zaccaria ML, Di Tommaso F, Brancaccio A, Paggi P, Petrucci TC. Dystroglycan distribution in adult mouse brain: a light and electron microscopy study. Neuroscience. 2001;104:311–324. doi: 10.1016/s0306-4522(01)00092-6. [DOI] [PubMed] [Google Scholar]
- Zhong D, Saito F, Saito Y, Nakamura A, Shimizu T, Matsumura K. Characterization of the protease activity that cleaves the extracellular domain of beta-dystroglycan. Biochem Biophys Res Commun. 2006;345:867–871. doi: 10.1016/j.bbrc.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Zonta B, Desmazieres A, Rinaldi A, Tait S, Sherman DL, Nolan MF, Brophy PJ. A critical role for Neurofascin in regulating action potential initiation through maintenance of the axon initial segment. Neuron. 2011;69:945–956. doi: 10.1016/j.neuron.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]