

Abstract
Motivation: LibSBGN is a software library for reading, writing and manipulating Systems Biology Graphical Notation (SBGN) maps stored using the recently developed SBGN-ML file format. The library (available in C++ and Java) makes it easy for developers to add SBGN support to their tools, whereas the file format facilitates the exchange of maps between compatible software applications. The library also supports validation of maps, which simplifies the task of ensuring compliance with the detailed SBGN specifications. With this effort we hope to increase the adoption of SBGN in bioinformatics tools, ultimately enabling more researchers to visualize biological knowledge in a precise and unambiguous manner.
Availability and implementation: Milestone 2 was released in December 2011. Source code, example files and binaries are freely available under the terms of either the LGPL v2.1+ or Apache v2.0 open source licenses from http://libsbgn.sourceforge.net.
Contact: sbgn-libsbgn@lists.sourceforge.net
1 INTRODUCTION
The Systems Biology Graphical Notation (SBGN, Le Novère et al., 2009) facilitates the representation and exchange of complex biological knowledge in a concise and unambiguous manner: as standardized pathway maps. It has been developed and supported by a vibrant community of biologists, biochemists, software developers, bioinformaticians and pathway databases experts.
SBGN is described in detail in the online specifications (see http://sbgn.org/Documents/Specifications). Here we summarize its concepts only briefly. SBGN defines three orthogonal visual languages: Process Description (PD), Entity Relationship (ER) and Activity Flow (AF). SBGN maps must follow the visual vocabulary, syntax and layout rules of one of these languages. The choice of language depends on the type of pathway or process being depicted and the amount of available information. The PD language, which originates from Kitano's Process Diagrams (Kitano et al., 2005) and the related CellDesigner tool (Funahashi et al., 2008), is equivalent to a bipartite graph (with a few exceptions) with one type of nodes representing pools of biological entities, and a second type of nodes representing biological processes such as biochemical reactions, transport, binding and degradation. Arcs represent consumption, production or control, and can only connect nodes of differing types. The PD language is very suitable for metabolic pathways, but struggles to concisely depict the combinatorial complexity of certain proteins with many phosphorylation states. The ER language, on the other hand, is inspired by Kohn's Molecular Interaction Maps (Kohn et al., 2006), and describes relations between biomolecules. In ER, two entities can be linked with an interaction arc. The outcome of an interaction (for example, a protein complex), is considered an entity in itself, represented by a black dot, which can engage in further interactions. Thus ER represents dependencies between interactions, or putting it differently, it can represent which interaction is necessary for another one to take place. Interactions are possible between two or more entities, which make ER maps roughly equivalent to a hypergraph in which an arc can connect more than two nodes. ER is more concise than PD when it comes to representing protein modifications and protein interactions, although it is less capable when it comes to presenting biochemical reactions. Finally, the third language in the SBGN family is AF, which represents the activities of biomolecules at a higher conceptual level. AF is suitable to represent the flow of causality between biomolecules even when detailed knowledge on biological processes is missing.
Efficient integration of the SBGN standard into the research cycle requires adoption by visualization and modeling software. Encouragingly, a growing number of pathway tools (see http://sbgn.org/SBGN_Software) offer some form of SBGN compatibility. However, current software implementations of SBGN are often incomplete and sometimes incorrect. This is not surprising: as SBGN covers a broad spectrum of biological phenomena, complete and accurate implementation of the full SBGN specifications represents a complex, error-prone and time-consuming task for individual tool developers. This development step could be simplified, and redundant implementation efforts avoided, by accurately translating the full SBGN specifications into a single software library, available freely for any tool developer to reuse in their own project. Moreover, the maps produced by any given tool usually cannot be reused in another tool, because SBGN only defines how biological information should be visualized, but not how the maps should be stored electronically. Related community standards for exchanging pathway knowledge, namely BioPAX (Demir et al., 2010) and SBML (Hucka et al., 2003), have proved insufficient for this role (more on this topic in Section 4). Therefore, we observed a second need, for a dedicated, standardized SBGN file format.
Following these observations, we started a community effort with two goals: to encourage the adoption of SBGN by facilitating its implementation in pathway tools, and to increase interoperability between SBGN-compatible software. This has resulted in a file format called SBGN-ML and a software library called LibSBGN. Each of these two components will be explained separately in the next sections.
2 THE SBGN-ML FILE FORMAT
SBGN-ML is a dedicated lightweight XML-based file format describing the overall geometry of SBGN maps, while also preserving their underlying biological meaning. SBGN-ML is designed to fulfill two basic requirements:
easy to draw (as a machine) and read (as a human) and
easy to interpret (as a machine).
The first set of requirement deals with the graphical aspect of SBGN. It means it should be easy to render a SBGN-ML file to the screen. Therefore, the format stores all necessary information, such as coordinates, to draw the map faithfully, so that rendering tools do not have to perform any complex calculations. Incidentally, this implies the layout of the whole SBGN map has to be expressed explicitly: the size and position of each graphical object and the path of each arc. Various efforts have shown that generating a layout for heterogeneous biological pathways is a computationally hard problem, so a good layout is always worth preserving, if only from a computational perspective. Besides, the choice of a specific layout by the author of a map is often driven by concerns related to aesthetics, readability or to reinforce ideas of chronology or proximity. This information might be lost with automated layouts. Layout conventions predate SBGN, and are not part of any standard, but they nonetheless play a large role in making it easier for other human beings to understand the biological system being described.
The second requirement encompasses two perpendicular characteristics of SBGN as a language: semantics and syntax. Beyond the picture itself, the format should capture the biological meaning of an SBGN map. Therefore, SBGN-ML specifies the nature of graphical elements (glyphs), following the SBGN terminology (e.g., macromolecule, process, etc.). For example, we can distinguish between a ‘logic arc’ and a ‘consumption arc’ even though they have the same visual appearance. Supporting tools refer to this terminology and draw the glyph according to the SBGN specifications. In terms of syntax, SBGN-ML encodes information on relationships between the various SBGN objects: the glyphs at both ends of an arc, the components of a complex, the members of a compartment and the ‘decorations’ (such as unit of information and state variable) belonging to specific glyphs and arcs. This semantic and syntactic information is essential to a number of automated tasks, such as map validation, or network analysis (as the topology of the underlying biological network can be inferred from the various relationships encoded by the format).
To explain the syntax of SBGN-ML in more detail, consider the example in Figure 1. This figure shows a PD map describing the import of glucose by GLUT4, followed by the first step of the glycolysis. The root element is named ‘sbgn’ (line 1). Below that, there is a ‘map’ element with an attribute indicating that the PD language is used. Below the map element, one finds a series of glyph and arc elements. Each glyph carries a ‘class’ attribute to denote the meaning in SBGN terms. In this example, there is a glyph with class ‘process’ (lines 14–18, in orange). Each glyph also carries an ‘id’ attribute that can be referred from elsewhere in the document, thus storing the network topology (in this case merely the letter ‘f’ for the sake of brevity). Each glyph must define a ‘bbox’ or bounding box, which allows the glyph to be placed at the correct position. Its coordinates denote the smallest rectangle that completely encompasses the glyph. Consumption and production arcs connect to process nodes at a so-called ‘port’ just outside the glyph. ‘Port’ elements are part of the network topology, so they carry identifiers as well (lines 16 and 17). Another glyph in this example represents the active form of hexokinase (lines 24–31). It carries a label element, which should be positioned in the center of the parent glyph, unless otherwise defined. Hexokinase also contains a sub-glyph for a state variable (lines 27–30, in blue) to indicate that it is the allosterically active form of the enzyme. ATP (lines 19–23, in green) is a simple chemical, and uses a circle as its shape, as opposed to macromolecules that use a rounded rectangle shape. Small molecules often occur multiple times in a map, in which case they must carry a clone marker, a black bottom half. In SBGN-ML this is represented by the ‘clone’ element (line 21). Cellular compartments are represented by glyphs as well (lines 32–35, in yellow). Entities refer to their surrounding compartment using a ‘compartmentRef’ attribute.
Fig. 1.
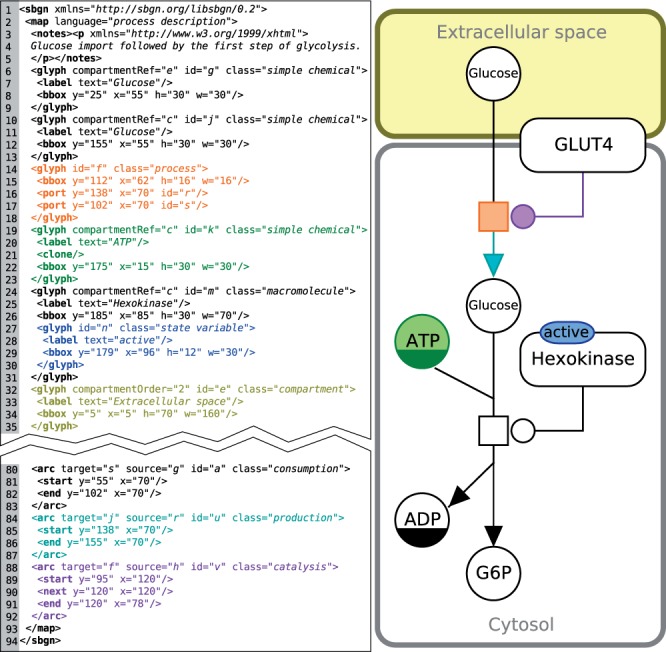
An example PD map (right) with the corresponding SBGN-ML code (left). This example shows the import of glucose followed by the first step of glycolysis. The colors used have no special meaning in SBGN, here they merely indicate the relation between each SBGN glyph and its SBGN-ML representation; a process node in orange, a simple chemical (ATP) in green, a production arc in cyan, a catalysis arc in purple, a compartment in yellow and a state variable in blue
Just like glyphs, arcs must define a ‘class’ attribute and an ‘id’ attribute. See for example the production arc (lines 84–87, in cyan). Each arc must have a source attribute, referring to the identifier of a glyph that the arc points from, as well as a target attribute, referring to the identifier of the glyph that the arc points to. Source and target may refer to identifiers of either glyphs or ports. Arcs must also define start and end coordinates. Arcs can optionally include waypoints for path routing as with the ‘catalysis’ arc (lines 88–92, in purple). It is not possible to deduce the start and end coordinates from the source and target glyphs, as there may be some white space between the end of the arc and the border of the glyph.
Each element can be freely annotated with notes encoded with valid XHTML elements (lines 3–5). Each SBGN-ML can also be extended with elements in proprietary namespaces to add additional features (not shown in this example).
3 THE LIBSBGN LIBRARY
A software library called LibSBGN complements the file format. It consists of two parallel implementations in Java and C++. The libraries share the same object model, so that algorithms operating on it can be easily translated to different programming languages.
The primary goal of LibSBGN is to simplify the work for developers of existing pathway tools. To reach this goal we followed three design principles. First, we avoided tool-specific implementation details. Implementation artifacts that are specific for one bioinformatics tool would impose difficulties for adoption by others. We sought input from several tool developers into the LibSBGN effort early on.
Second, we do not want to force the use of a single rendering implementation (meaning the software routine that translates from memory objects to screen or graphic format). Early in the development of LibSBGN, it became clear that for most pathway drawing tools, the rendering engine is an integral part that is not easily replaced by a common library. The typical usage scenario is therefore to let LibSBGN handle input and output, but to translate to the application's own object model, and display using the application's own rendering engine. Enforcing a common rendering library would hamper adoption of LibSBGN. We instead opted to build a render comparison pipeline to ensure consistency between various renderers (this pipeline is described in more detail in Section 3.2).
Third, we wish to provide optimal libraries for each development environment. For both the C++ and Java versions, code is automatically generated based on the XML Schema definition (XSD). The method of generating code from XSD has reduced the effort needed to keep the Java and C++ versions synchronized during development. The generated Java code plus helper classes form a pure Java library. The alternative possibility, to create a single C++ library and a Java wrapper around that, is not preferable because it complicates multi-platform installation and testing. Our experience with a related project, LibSBML (Bornstein et al., 2008), is that the community has a need for a pure Java library in spite of existing Java bindings for C++, which has led to the development of the pure Java JSBML (Dräger et al., 2011) as an alternative. Although both LibSBML and JSBML are successful projects, the maintenance of two similar projects in different languages is costly in terms of developer time. By generating native libraries for both environments automatically, we hope to avoid that extra cost.
3.1 Code sample
See Figure 2 for an example of usage of LibSBGN in practice. The Java library contains convenient helper functions for reading, writing and validation. In the case of this example the function readFromFile from the SbgnUtil class is used. The source package contains example programs for common operations, and the LibSBGN wiki includes a developer tutorial (see http://sourceforge.net/apps/mediawiki/libsbgn/index.php?title=Developer_tutorial) aimed at developers who want to include LibSBGN into an existing bioinformatics application.
Fig. 2.

Example of reading a file using the Java version of LibSBGN. Here an SBGN-ML file named ‘adh.sbgn’ (included in the LibSBGN source distribution) is read, and some basic information about each glyph in that file is printed to standard output. The complete program can be found as ReadExample.java in the LibSBGN source distribution
3.2 Rendering comparison
We created dozens of test-cases for each of the three languages of SBGN, covering all aspects of the syntax. Each test-case consists of a reference diagram in PNG format and a corresponding SBGN-ML file. To test our software, all SBGN-ML files are automatically rendered by the participating programs, currently SBGN-ED (Czauderna et al., 2010), PathVisio (van Iersel et al., 2008) and SBML Layout (Deckard et al., 2006). The resulting images are viewable side-by-side with the reference map. An example of this can be found in Figure 3.
Fig. 3.
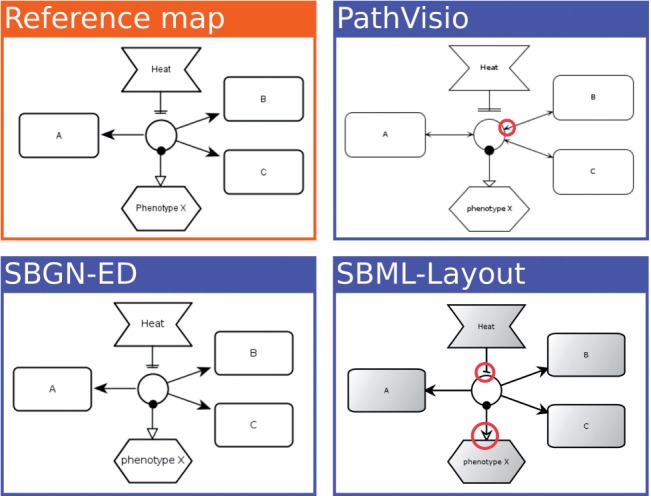
Rendering comparison. A series of test-cases is rendered by all supported tools in an automated render comparison pipeline. The rendering results are compared with the reference map (top-left), in this case an ER map. A couple of significant differences have been highlighted with red circles. In the PathVisio case (top-right), arrowheads are drawn where none is expected. In the SBML Layout example (bottom-right), the wrong arrowheads are drawn for absolute inhibition and stimulation arcs. Note that these are historical images for illustration purposes, and the highlighted issues have already been fixed
This pipeline was of tremendous value during development. Typically, an observed difference between a given rendering and the reference diagram could lead to several possible outcomes. Most commonly, the difference indicated a mistake in the participating renderer, which had to be fixed by the author of that software. A second possibility is that the mistake is due to an ambiguity in the interpretation of SBGN-ML. This could lead to a correction in the specification or a clarification in the documentation, so that all involved are in agreement. In several instances, the source of ambiguity was derived not from SBGN-ML but from the SBGN specification. This way, LibSBGN has led to feedback on SBGN itself. A final possibility is that the difference was deemed insignificant. Certain differences in use of color, background shading and line thickness are not meaningful in terms of biological interpretation of the SBGN map. An exception here is differences in layout. As mentioned before, we consider layout valuable to preserve even though it is not semantically significant. This pipeline is now fully automated, and runs automatically, whenever new test-cases are added to the source repository. It can be viewed online at http://libsbgn.sourceforge.net/render_comparison/. We encourage developers of software to contact us to add their tool to the gallery.
3.3 Validation
For syntactic validation of SBGN-ML documents, we created an XML Schema definition (XSD). Unfortunately, XSD is not sufficient to validate the many semantic rules defined in the SBGN specification. To solve this we also developed higher level, semantic validation using the Schematron (http://www.schematron.com) language.
To give a few examples: in PD, a production arc should point from a process towards an entity pool node. It is not allowed to draw the arc in the other direction, or to connect two entity pools directly without an intermediate process (see Figure 4). In ER, outcome glyphs may be drawn on interaction arcs but not on influence arcs. If such a rule were violated, the meaning of the map would be ambiguous or contradictory.
Fig. 4.
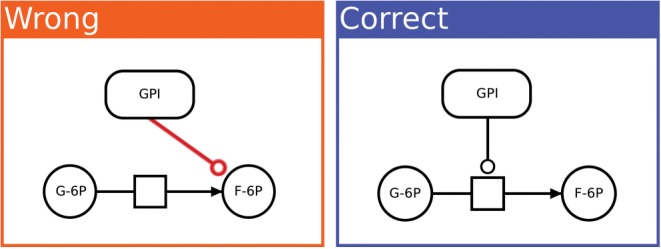
Typical validator benchmark. This particular example tests the software for rule pd10110: in PD maps, catalysis arcs must point to a process node (not to an entity pool node). In the negative test-case on the left, the enzyme GPI appears to ‘catalyze’ a molecule rather than a reaction. This is a logical impossibility. The positive test-case on the right shows correctly how the enzyme GPI catalyzes the reaction from glucose-6P to fructose-6P. Taken together, these test-cases help to prevent bugs in the validation software
LibSBGN provides functionality for users and developers to validate diagrams against these rules. This validation capability is built using Schematron language which has been previously used for Molecular Interaction Map diagram validation (Luna et al., 2011). Schematron rules are assertion tests written using XPath syntax. Each rule possesses a role to denote the severity of failure, a human-readable message and diagnostic elements to identify the source of the error or warning. Rules in Schematron can be grouped in phases; this feature can be used to denote subsets of rules to be activated during validation. Schematron makes use of XML stylesheet transformations (XSLT) and the validation process occurs in two steps. The first step is the transformation of the rule sets written in the Schematron language to an XSLT stylesheet, and the second step is the transformation of an SBGN-ML file using the XSLT stylesheet from the first step. The product of this second transformation is a validation report that uses the Schematron Validation Report Language (SVRL). The usage of Schematron rule sets allows for validation to be flexibly incorporated into various environments and using any programming language with an XSLT processor. Command-line validation can be done using XSLT processors such as Saxon (http://saxon.sourceforge.net/) by performing the two transformation steps mentioned above. Alternatively, validation can also be incorporated into automated pipelines using the Ant task for Schematron (http://code.google.com/p/schematron/); an example of this is provided in the distributed files. Lastly, validation can be incorporated into projects by using provided utility Java classes found in the LibSBGN API. The PathVisio-Validator plugin (Chandan et al., 2011) is an example of diagram validation using LibSBGN and Schematron.
There are three rule sets for SBGN-ML, one for each of the SBGN languages. These rule sets validate syntactic correctness of SBGN maps. An example validation is shown in Figure 4, where a stimulation arc is incorrectly drawn by pointing to an entity pool node, rather than a process node.
Unfortunately software can have bugs, and if the validation routine does not report any validity errors, this could indicate that either the diagram is indeed correct (true negative), or that there is a bug in the software encoding the rules (false negative). To ensure correctness of the validation rules themselves, we have created benchmarks for each of them. For each rule there is a positive test-case, for which the rule should pass, and a negative one, for which the rule should fail, similar to the example given in Figure 4.
3.4 Supporting tools
As mentioned earlier, we seek support from a wide community of tool developers. The following tools are already using LibSBGN: PathVisio (van Iersel et al., 2008), SBGN-ED (Czauderna et al., 2010), SBML Layout (Deckard et al., 2006) and VISIBIOweb (Dilek et al., 2010). We are aware of two other tools with LibSBGN support in development: Arcadia (Villéger et al., 2010) and CellDesigner (Funahashi et al., 2008). Desktop applications using LibSBGN are shown in Figure 5.
Fig. 5.
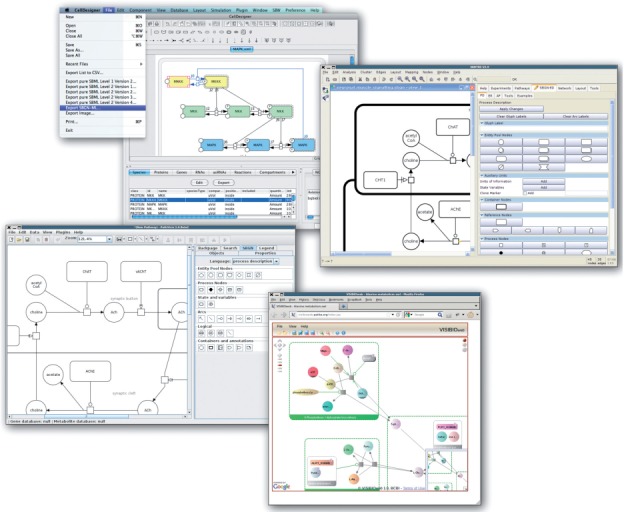
Screenshots of a number of tools that use LibSBGN. Clockwise, from the top: CellDesigner, SBGN-ED, VISIBIOweb and PathVisio. These tools are able to use SBGN-ML for import, export or both. At the time of writing, for some of these tools a version with SBGN support has not been officially released, but is expected soon
4 DISCUSSION
We have set out to fulfill the dual goals of simplifying SBGN support as well as standardizing electronic exchange of SBGN. The first goal has been addressed with an open-source software library, which can be used to read, write, manipulate and validate SBGN. The second goal has been addressed with a file format named SBGN-ML.
SBGN-ML fills a pragmatic need for a format that can be mapped directly to concepts from the SBGN specification. We see the rapid adoption of SBGN-ML by a number of tools as proof of the pragmatic need for it.
A potential criticism of SBGN-ML is the addition of yet another file format to the repertoire of file formats in systems biology. Different approaches have been explored for electronically representing SBGN: from graphical file formats such as SVG, or graph representation stored as GraphML files, to additional information on top of an existing model, such as the Systems Biology Markup Language (SBML) layout extension (Gauges et al., 2006). All these approaches have limitations, as they have been developed independently of SBGN. A new format was needed to support all characteristics of SBGN maps (graphics, relationships and semantics). The other formats could be extended to cover these concepts, but at the expense of brevity and clarity.
So we created a new format for the following reasons. First, SBGN-ML focuses on the domain of visualization of SBGN concepts. This sets it apart from existing exchange formats for pathways. BioPAX is a pathway exchange format that occupies the domain of knowledge management, and has close relations to the semantic web. SBML occupies the domain of computational modeling of systems biology. The latter two could be extended to accommodate SBGN concepts, but there is not a straight one-to-one mapping. For example, there is no good equivalent for the AND/OR gates which can be drawn in SBGN. Furthermore, omitted/uncertain processes can be drawn in SBGN but have no direct equivalent in BioPAX.
Second, SBGN-ML is easier to validate against the SBGN specification. As mentioned before, the complexity of SBGN makes software support for validation a must. Rules describing validation of SBGN-ML are simpler and more concise than they would be if they were encoded on top of an existing format.
Third, the rendering comparison pipeline has ensured that conversion of SBGN-ML to graphical formats is straightforward. On the other hand, conversion from a graphical format such as SVG to SBGN-ML requires inferring the meaning of lines, glyphs and symbols, which is bound to lead to loss of information.
Fourth, by making SBGN independent, it is not tied to either the SBML, BioPAX or any other research community. We observe that currently LibSBGN is being used by both BioPAX-oriented tools such as ChIBE and PaxTools as well as SBML-oriented tools such as CellDesigner or GraphML-oriented tools such as SBGN-ED.
SBGN-ML is officially endorsed by the SBGN scientific committee as a reference implementation and the best way to exchange diagrams between applications. It is orthogonal to specific formats used to represent pathways and models such as BioPAX (Demir et al., 2010) and SBML (Hucka et al., 2003), and thus follows the vision of the COMBINE initiative (http://co.mbine.org/about).
In the field of bioinformatics, it occurs all too often that the lack of a feature in an existing piece of software is used to justify the development of a complete new bioinformatics tool, which will in its turn lack features in another area. The end result is the current state of affairs: a balkanization of bioinformatics tools, or in other words, many fragmented tools that integrate poorly. One of the goals of LibSBGN is to improve existing software. LibSBGN could serve as a model to counter the balkanization trend. We prefer to see the development of software libraries instead of incomplete tools. Libraries, especially if they are open source, can be shared, re-used and adopted by developers.
5 CONCLUSION
The SBGN-ML file format and LibSBGN library provide open-source software support for SBGN maps. They have been adopted by several tools already, and development is ongoing. It is expected that the availability of a community-supported API will significantly expedite SBGN's adoption. We use the word ‘Milestone’ for versioning purposes—the latest release is Milestone 2, which was released in December 2011.
LibSBGN is primarily focused on exchanging between SBGN software. Other functionalities, such as conversion to other formats, or generating suitable layout, are not currently supported. It is certainly likely that some or all of these functionalities will be added in the future as optional modules. SBGN-ML will likely see the addition of fine-grained graphics specification, support for linking between files, and improved usage of ontologies. Additionally, LibSBGN will see expansion to other programming languages beyond Java and C++, such as for example Javascript.
The SBGN-ML file format is represented as an XML schema (SBGN.XSD). Examples are available as test files (XML, PNG). The accompanying documentation reflects the content of the schema, and clarifies a number of additional rules and conventions (e.g., coordinate system). This set of resources constitutes the SBGN-ML specifications. The LibSBGN library (in C++ and Java) and the file format have been released on Sourceforge, under a dual license: the Lesser General Public Licence (LGPL) version 2.1 or later, and Apache version 2.0.
The development process is an active community effort, organized around: regular online meetings, discussions on the mailing list, and development tools on Sourceforge (bug tracker, SVN repository and documentation wiki). New developers are very welcome.
ACKNOWLEDGEMENTS
The authors thank their individual sources of funding. Authors are grateful for useful feedback from the Path2Models project.
Funding: This work was in part supported by the Biotechnology and Biological Sciences Research Council (BBSRC); the Netherlands Consortium for Systems Biology (NCSB), which is part of the Netherlands Genomics Initiative/Netherlands Organisation for Scientific Research; BioPreDyn which is a grant within the Seventh Framework Programme of the EU, the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research; and the German Ministry of Education and Research (BMBF).
Conflict of Interest: none declared.
REFERENCES
- Bornstein B.J., et al. LibSBML: an API library for SBML. Bioinformatics. 2008;24:880–881. doi: 10.1093/bioinformatics/btn051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandan K., et al. PathVisio-Validator: A rule-based validation plugin for graphical pathway notations. Bioinformatics. 2011;28:889–890. doi: 10.1093/bioinformatics/btr694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czauderna T., et al. Editing, validating, and translating of SBGN maps. Bioinformatics. 2010;26:2340–2341. doi: 10.1093/bioinformatics/btq407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckard A., et al. Supporting the SBML layout extension. Bioinformatics. 2006;22:2966–2967. doi: 10.1093/bioinformatics/btl520. [DOI] [PubMed] [Google Scholar]
- Demir E., et al. The BioPAX community standard for pathway data sharing. Nat. Biotechnol. 2010;28:935–942. doi: 10.1038/nbt.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilek A., et al. VISIBIOweb: visualization and layout services for BioPAX pathway models. Nucleic Acids Res. 2010;38:W150–W154. doi: 10.1093/nar/gkq352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dräger A., et al. JSBML: a flexible Java library for working with SBML. Bioinformatics. 2011;27:2167–2168. doi: 10.1093/bioinformatics/btr361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funahashi A., et al. CellDesigner 3.5: a versatile modeling tool for biochemical networks. Proc. IEEE. 2008;96:1254–1265. [Google Scholar]
- Gauges R., et al. A model diagram layout extension for SBML. Bioinformatics. 2006;22:1879–1885. doi: 10.1093/bioinformatics/btl195. [DOI] [PubMed] [Google Scholar]
- Hucka M., et al. The Systems Biology Markup Language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics. 2003;9:524–531. doi: 10.1093/bioinformatics/btg015. [DOI] [PubMed] [Google Scholar]
- Kitano H., et al. Using process diagrams for the graphical representation of biological networks. Nat. Biotechnol. 2005;23:961–966. doi: 10.1038/nbt1111. [DOI] [PubMed] [Google Scholar]
- Kohn K.W., et al. Molecular interaction maps of bioregulatory networks: a general rubric for systems biology. Mol. Biol. Cell. 2006;17:1–13. doi: 10.1091/mbc.E05-09-0824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Novère N., et al. The systems biology graphical notation. Nat. Biotechnol. 2009;27:753–741. doi: 10.1038/nbt.1558. [DOI] [PubMed] [Google Scholar]
- Luna A., et al. A formal MIM specification and tools for the common exchange of MIM diagrams: an XML-Based format, an API, and a validation method. BMC Bioinformatics. 2011;12:167. doi: 10.1186/1471-2105-12-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Iersel M.P., et al. Presenting and exploring biological pathways with PathVisio. BMC Bioinformatics. 2008;9:399. doi: 10.1186/1471-2105-9-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villéger A.C., et al. Arcadia: a visualization tool for metabolic pathways. Bioinformatics. 2010;26:1470–1471. doi: 10.1093/bioinformatics/btq154. [DOI] [PMC free article] [PubMed] [Google Scholar]