

Abstract
Summary: Alternative Splicing Effects ASsessment Tools (AS-EAST) is an online tool for the functional annotation of putative proteins encoded by transcripts generated by alternative splicing (AS). When provided with a transcript sequence, AS-EAST identifies regions altered by AS events in the putative protein sequence encoded by the transcript. Users can evaluate the predicted function of the putative protein by inspecting whether functional domains are included in the altered regions. Moreover, users can infer the loss of inter-molecular interactions in the protein network according to whether the AS events affect interaction residues observed in the 3D structure of the reference isoform. The information obtained from AS-EAST will help to design experimental analyses for the functional significance of novel splice isoforms.
Availability: The online tool is freely available at http://as-alps.nagahama-i-bio.ac.jp/ASEAST/.
Contact: m_shionyu@nagahama-i-bio.ac.jp
1 INTRODUCTION
In higher eukaryotes, genes often produce alternatively spliced transcripts (AS transcripts). Many AS events have been actively detected with high-throughput experimental methods, such as RNA-Seq and microarrays (Hallegger et al., 2010). However, the functions of putative proteins encoded by AS transcripts (termed ‘AS isoforms’) have not been experimentally analyzed in many cases. Functional annotation tools for transcripts with novel patterns of splicing are desirable to infer the functional significance of AS isoforms. There are few tools for analyzing AS isoforms translated from novel transcripts queried by users. AltAnalyze (http://www.altanalyze.org) identifies AS events using RNA-Seq or microarray data and shows how these events may affect domain composition. However, it does not provide information on the effects of AS on the 3D structures of AS isoforms. MAISTAS (Floris et al., 2011) assesses whether user-queried AS isoforms are structurally plausible proteins, but explicit functional annotations are not provided.
Previously, we developed a pipeline that detects regions altered by AS events (termed ‘AS regions’) in AS isoforms using genome sequences and full-length transcript data (Yura et al., 2006). The pipeline then evaluates the impact of AS events on the interactions between the AS isoforms and other molecules by identifying interaction residues from 3D structure data of relevant molecular complexes. All of the data derived from the pipeline are provided in the AS-ALPS database (Shionyu et al., 2009). In this article, we describe AS-EAST that annotates and analyzes user-uploaded transcript sequences using AS-ALPS. AS-EAST determines whether the transcript encodes a novel AS isoform and annotates such functional sites in the AS isoform as residues interacting with other molecules. We provide an example: AS-EAST predicts that a novel AS isoform of mitogen-activated protein kinase 1 (MAPK1) in human skeletal muscle inhibits the signaling pathway by removing residues that interact with ATP and substrate proteins.
2 OVERVIEW OF AS-EAST
2.1 Input data
To detect and annotate AS events in a user-submitted transcript (termed ‘query transcript’), AS-EAST accepts a FASTA-formatted transcript sequence. A novel pattern of splicing detected with RNA-Seq or exon junction microarray often determines whether a certain exon in a known transcript model is skipped. AS-EAST has a user interface for generating an exon-skipped transcript sequence from the known transcript sequences stored in AS-ALPS. For example, RNA-Seq data (Wang et al., 2008) shows that the fourth exon of the MAPK1 transcript tends to be skipped in human skeletal muscle (Fig. 1a). This splicing pattern is novel because no fourth exon-skipped transcript is found in RefSeq or Ensembl transcript datasets stored in AS-ALPS. Users can build a fourth exon-skipped transcript sequence by checking the checkbox of ‘exon 4’ and selecting the ‘Generate’ button. Then, the transcript sequence excluding the fourth exon is shown in FASTA format.
Fig. 1.
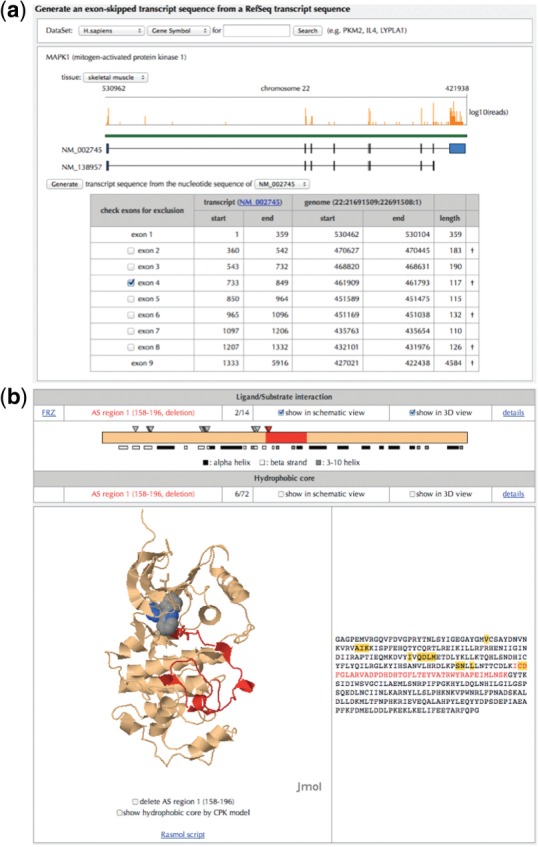
(a) User interface for generating query transcript data. As a reference of exon-skipped transcript sequence generation, the mapping results of RNA-Seq data from Wang et al. (2008) are shown at the top of the schematic figure in the human dataset. Mapping of the RNA-Seq data was performed using the TopHat program (Trapnell et al., 2009) with default parameters. (b) Interaction residues obtained from 3D structure information assigned to a reference isoform. The deletion-type AS region is shown in red. Interaction residues are denoted with arrowheads in the schematic view and stick models in the 3D structure view. The ligand is shown in a space-filling model. The 3D structure is shown with Jmol (http://www.jmol.org/)
2.2 AS region detection
First, a genome contig sequence aligned to the query transcript sequence with the largest value of both length coverage and sequence identity is selected using MEGABLAST (Altschul et al., 1997). An alignment of the query transcript and the contig sequence is performed using SPLIGN (Kapustin et al., 2008). AS-EAST searches the AS-ALPS database for transcripts mapped on the same region of the contig as the query transcript. The user can select one of the transcripts as a reference transcript. Second, the protein-coding sequence (CDS) of the query transcript is predicted by identifying the longest open reading frame (ORF) or FrameDP program (Gouzy et al., 2009). Users can also use the ORF starting with the users' chosen first codon as the CDS. By comparing the genomic regions corresponding to the CDSs of a reference transcript and the query transcript, AS-EAST identifies CDSs changed through AS (termed ‘AS CDSs’). The AS CDSs of a reference transcript that have no corresponding region in the query transcript are classified as deletions, and the AS CDSs of the query transcript that have no corresponding region in a reference transcript are classified as insertions.
From the AS CDSs, AS-EAST identifies amino acid sequence regions changed through AS, termed ‘AS regions’. An amino acid sequence encoded by a deletion AS CDS whose length is a multiple of 3 is identified as a deletion-type AS region. In addition, an amino acid sequence encoded by an insertion AS CDS whose length is a multiple of 3 is identified as an insertion-type AS region. When the length of an AS CDS is not a multiple of 3 and the reading frame of the 3′-flanking CDS is shifted, the amino acid sequence encoded by the AS CDS and the 3′-flanking CDS is identified as a substitution-type AS region.
2.3 Functional annotation
To analyze the functional effect of AS on a putative protein encoded by the query transcript (query isoform), annotations of the query isoform with functional regions are performed using InterProScan (Zdobnov and Apweiler, 2001). From the results of InterProScan, functional domains from the Pfam, Gene3D and SUPERFAMILY databases and transmembrane regions predicted with TMHMM are shown in AS-EAST. Moreover, AS-EAST annotates the query isoform using functional amino acid residue information (Fig. 1b). Amino acid residues interacting with other molecules (interaction residues) are identified from the 3D structure data of protein complexes (Yura et al., 2006). AS-EAST assigns 3D structures to the query isoform with BLASTP (Altschul et al., 1997). AS-EAST determines whether 3D structures are assigned to AS regions on the basis of the criteria by Yura et al. (2006). According to the alignment of the sequences of the reference isoform and the assigned 3D structures, interaction residues are projected to the corresponding residues in the AS regions. In the MAPK1 example, the query isoform has a protein kinase domain, according to the results of InterProScan. However, the query isoform lacks a region that has some residues constituting the ATP-binding pocket and interaction residues and is predicted to lose protein kinase activity. Therefore, the expression of the AS isoform encoded by the fourth exon-skipped transcript in human skeletal muscle might inhibit the MAP kinase signaling pathway and regulate cell proliferation/differentiation as a result.
ACKNOWLEDGEMENTS
The authors thank Akihiro Yamaguchi, Kei Iida, Atsushi Hijikata, Kazuki Shinoda and Kei Yura for helpful comments and discussions.
Funding: Grant-in-Aid for Scientific Research (B) #18370061 (to M.G.) and the Targeted Proteins Research Program (TPRP) and a Grant-in-Aid for Young Scientists (B) #23770187 (to M.S.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Conflict of Interest: none declared.
REFERENCES
- Altschul S., et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floris M., et al. MAISTAS: a tool for automatic structural evaluation of alternative splicing products. Bioinformatics. 2011;27:1625–1629. doi: 10.1093/bioinformatics/btr198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouzy J., et al. FrameDP: sensitive peptide detection on noisy matured sequences. Bioinformatics. 2009;25:670–671. doi: 10.1093/bioinformatics/btp024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallegger M., et al. Alternative splicing: global insights. FEBS J. 2010;277:856–866. doi: 10.1111/j.1742-4658.2009.07521.x. [DOI] [PubMed] [Google Scholar]
- Kapustin Y., et al. Splign: algorithms for computing spliced alignments with identification of paralogs. Biol. Direct. 2008;3:20. doi: 10.1186/1745-6150-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shionyu M., et al. AS-ALPS: a database for analyzing the effects of alternative splicing on protein structure, interaction and network in human and mouse. Nucleic Acids Res. 2009;37:D305–D309. doi: 10.1093/nar/gkn869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., et al. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E.T., et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yura K., et al. Alternative splicing in human transcriptome: functional and structural influence on proteins. Gene. 2006;380:63–71. doi: 10.1016/j.gene.2006.05.015. [DOI] [PubMed] [Google Scholar]
- Zdobnov E.M., Apweiler R. InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]