

Abstract
Motivation: With the rapid development of high-throughput sequencing technologies, the genome-wide profiling of nucleosome positioning has become increasingly affordable. Many future studies will investigate the dynamic behaviour of nucleosome positioning in cells that have different states or that are exposed to different conditions. However, a robust method to effectively identify the regions of differential nucleosome positioning (RDNPs) has not been previously available.
Results: We describe a novel computational approach, DiNuP, that compares nucleosome profiles generated by high-throughput sequencing under various conditions. DiNuP provides a statistical P-value for each identified RDNP based on the difference of read distributions. DiNuP also empirically estimates the false discovery rate as a cutoff when two samples have different sequencing depths and differentiate reliable RDNPs from the background noise. Evaluation of DiNuP showed it to be both sensitive and specific for the detection of changes in nucleosome location, occupancy and fuzziness. RDNPs that were identified using publicly available datasets revealed that nucleosome positioning dynamics are closely related to the epigenetic regulation of transcription.
Availability and implementation: DiNuP is implemented in Python and is freely available at http://www.tongji.edu.cn/~zhanglab/DiNuP.
Contact: yzhang@tongji.edu.cn
Supplementary Information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
As a basic unit of the eukaryotic genome, the nucleosome is formed by an octamer of histones and the surrounding 147 bp of DNA (Kornberg and Lorch, 1999; Luger et al., 1997). Nucleosomes play an important role in the epigenetic regulation of diverse cellular processes through covalent modifications of histone tails (Heintzman et al., 2007; Liu et al., 2005) and positioning of nucleosomes (Jiang and Pugh, 2009; Li et al., 2007). Although previous studies have focused primarily on the former mechanism, the relative location of the DNA and the histone octamer, or the nucleosome positioning, is also a determining mechanism for epigenetic regulation through controlling the accessibility of transcription factor binding sites (Mellor, 2006; Workman and Kingston, 1998). In addition, in the process of gene transcription, the frequency of nucleosome unwrapping and formation can reflect the rate of Pol II elongation (Luger, 2006; Schwabish and Struhl, 2004). As a result, if genome-wide nucleosome profiles for cells exposed to different conditions are known, then researchers can better understand the dynamic behaviours of the transcriptional machinery.
With the rapid development of high-throughput sequencing technologies, genome-wide nucleosome profiles have been generated for several organisms at a single-nucleotide resolution (Kaplan et al., 2009; Mavrich et al., 2008; Schones et al., 2008; Shivaswamy et al., 2008; Valouev et al., 2008, 2011), which provides an opportunity to study nucleosome positioning dynamics in cells that are in different states or that are exposed to different conditions. To identify regions of differential nucleosome positioning (RDNPs), Shivaswamy et al. (2008) introduced the concept of a nucleosome score to indicate the stability of the nucleosome position and then compared scores for yeast samples before and after heat shock. In addition, a fold change calculation can be an intuitive method for identifying regions with sequencing read number changes. However, those approaches have several limitations. First, to the best of our knowledge, none of the previous studies provided a statistical measurement, e.g. a P-value, to evaluate the significance of the difference in the nucleosome positioning changes. Second, both the fold change calculation and the nucleosome score comparison are sequencing-depth independent, which would affect the robustness of the results, especially when the sequencing depth is low.
To address these limitations, we present a novel method called differential nucleosome positioning (DiNuP) in this article. DiNuP takes advantage of the single-nucleotide resolution of nucleosome profiles, and it directly compares the distributions of sequenced nucleosome-DNA centres along the genome between different samples to detect genomic regions with differential nucleosome positioning without introducing any intermediate concepts (such as the nucleosome score or the positioned nucleosome). DiNuP also calculates P-values and empirically estimates the false discovery rate (FDR), to evaluate the statistical significance of the identified difference. Moreover, DiNuP provides various parameters with which to characterize the physical properties of those identified regions. When applied to publicly available nucleosome profiles for yeast (Kaplan et al., 2009), DiNuP reliably detected differences in nucleosome positioning in a sequencing-depth-dependent manner, and the detection of nucleosome position differences in the functionally important regions implies a close relationship between nucleosome positioning and eukaryotic transcriptional regulation.
2 METHODS
2.1 Estimation of the FDR
We first randomly choose 1% of all of the sliding windows to be the estimating region. Next, the reads of each paired window were combined and then re-sampled based on the initial ratio of that window's number of reads. Most of the re-sampled windows are expected to have no differences except for the differences in the sequencing depths. The Kolmogorov–Smirnov (K–S) test is then used to calculate the difference between these re-sampled windows, and the P-values are ranked from small to large. Finally, a specific percentile of the ranked P-value is used as an FDR estimation. The FDR that is obtained by this method accounts for both the sequencing depth and the background noise, giving a robust cutoff when identifying RDNPs.
2.2 Evaluation in simulated datasets
To evaluate the reliability and accuracy of DiNuP, we simulated different types of differential nucleosome positioning and used DiNuP to detect the simulated regions. First, to facilitate the computation, we chose a 20 kb region as the control and simulated the nucleosome profile region. Data were also obtained from (Kaplan et al., 2009). Second, regions with a length of 200 bp were randomly selected to simulate repositioned variation from 0 to 100 bp, an occupancy change percentage from 0 to 100% and a positioning degree change from 0 to 0.5. The background noise was then added by applying a coordinate disturbance of 3 bp to the residual part of the 20 kb region. Third, DiNuP was used to identify the region of differential nucleosome positioning between the simulated region and the original region, repeating every degree of the simulation 1000 times. If the detected region was in accordance with the simulated region, then it was a true-positive hit; otherwise, it was a false-positive hit. The FDR was calculated as the ratio of the number of true-positive hits to the number of all positive hits.
2.3 Evaluation in real datasets
Three physical properties that we defined, the repositioned variation, the occupancy change and the positioning degree change, were used to characterize three major types of differential nucleosome positioning: changes in nucleosome location, changes in occupancy and changes in fuzziness. We calculated those three properties in each 200 bp sliding window (10 bp as a step) separately. For each property, windows with values that were larger than a specific cutoff were regarded as the positive condition. Positive windows identified by DiNuP as differential windows were regarded as true positives; otherwise, they were regarded as false negatives. Windows with no obvious change (i.e. 5 bp for repositioned variation, 20% for occupancy change and 0.05 for positioning degree change) were then regarded as the negative condition. The negative windows identified by DiNuP as differential windows were then regarded as false positives; otherwise, they were regarded as true negatives. Since the number of true negatives and false positives are constant values in our definition, we used the ratio between false positives and outcome positives as the FDR.
2.4 Physical properties
Zhang et al. (2009) defined the nucleosome positioning degree of a certain genomic site as the ratio of the number of reads for a 20 bp window to that of a 160 bp window centred around that location. A positioning degree of 1.0 indicates that this site is a nucleosome that is perfectly positioned, whereas a positioning degree of 0.05 indicates that this site is a nucleosome that is poorly positioned. The change in the positioning degree can be obtained by calculating the average difference in the positioning degree between samples. First, each identified RDNP was divided into 160 bp regions with a step of 10 bp. Second, the largest positioning degree of each short region was used to represent the degree of this region. Third, the average degree of each 160 bp region was used to represent the positioning degree of the whole region. Finally, the difference between the samples was defined as the change in the positioning degree.
2.5 Analysis of identified RDNPs
Because the identified regions can overlap more than one gene, we assigned a summit (the candidate driver location) of each RDNP to its corresponding genomic feature. Yeast promoters were defined as the region from -350 bp upstream from the transcription start site (TSS) to +50 bp downstream from the TSS. Using this definition, 22% of the RDNPs should occur randomly within promoters. The ratio of real hits to random hits was used to represent the enrichment. The P-value significance was calculated using the binomial test. Moreover, if one identified RDNP overlapped with one gene, we then put this gene into a gene list and used DAVID to perform GO analysis. In addition, we used Transcription factor (TF) data with intermediate-confidence conservation and a binding criterion of 0.005. The significance of TF enrichment was calculated using the binomial test. Gene expression data for yeast grown in YPD medium and YPGal medium were obtained from (Komili et al., 2007; Verstrepen et al., 2008). The significance of the overlapping between genes proximal to RDNPs and genes that were differentially expressed was calculated by a hypergeometric test.
2.6 Software implementation
DiNuP is implemented in Python and is freely available. It runs from a command line and inputs the following parameters: -t for the first file of nucleosome profiles; -c for the second file of nucleosome profiles; --name for the name of the run; -f for enabling DiNuP to calculate physical properties; --windowsize for the size of the sliding window (default 200 bp); --fdr for the FDR cutoff to detect RDNPs; --pvalue for setting a P-value cutoff; --region for the minimum length of the identified RDNPs (default 70 bp); --format for the format of the input file; --bias for the simulation of the experimental bias (default 3 bp); --times for the number of times to calculate the K–S test and take the average P-value as the significance of the difference (Default 3); --wig for whether to save significant P-values into the wiggle file; -a for setting the average nucleosomal DNA length for Sample A and -b for setting the average nucleosomal DNA length for Sample B; --fold for additionally applying fold change method; --fcutoff for the cutoff of fold change method (Default 2).
3 RESULTS
3.1 Strategy to detect RDNP
There are three major types of differential nucleosome positioning: changes in nucleosome location, occupancy and fuzziness (Fig. 1A). Our strategy was designed to capture all three types based solely on the changes in the distribution of nucleosome sequencing reads. To date, all publicly available nucleosome profiles that were generated using high-throughput sequencing technology include sequences for only one end of nucleosome-DNA fragments. To represent the whole nucleosome, each read was extended toward its 3′ end by 147 bp, as described previously (Zhang et al., 2008, 2009). We took the centre of the extended read (dyad) to represent the precise nucleosome location in the following steps (Fig. 1A).
Fig. 1.
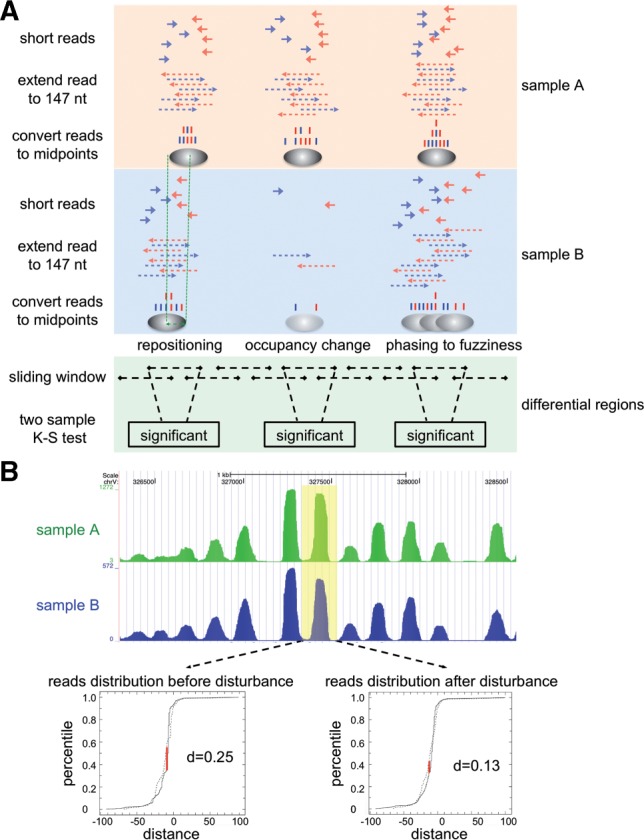
Approach for identifying RDNPs. (A) Schematic of DiNuP. (B) An example of reducing experimental bias by giving coordinate disturbances. The read distribution is obtained from reads within the sliding window (yellow). D is the largest distance between the cumulative distributions
For two samples, we scanned the whole genome with a sliding window (200 bp as the default window size and 10 bp as the default step), and for each sample, the location coordinates (relative to the window's midpoint) of the derived dyads in each window were treated as a numeric list. Since the two-sample K–S test is a non-parametric approach for determining whether two numeric lists are derived from the same distribution, we applied this approach to test whether the nucleosome positions in each window were different between the two samples. In this way, the centre of each window is assigned a P-value to indicate the significance of the difference. To evaluate and eliminate the effect of sequencing depth and background noise, we used a sampling method to empirically estimate the FDR, which provided a robust cutoff for comparing the different pairs (see Section 2). Consecutive regions with P-values smaller than the cutoff and having a certain length (5% as the default FDR and 70 bp as the default minimum length) are identified as candidate RDNPs. The final length of each identified region will, thus, be the default minimum length plus the size of the sliding window.
To analyse the performance of this strategy, we checked the consistency between the regions of change in the profile and the identified candidate regions. As expected, most of the candidate regions have obvious differences in the nucleosome profiles (Supplementary Fig. S1). However, we also observed some candidate regions with similar profiles between samples. An example region is shown in Figure 1B. Nucleosome profiles for Sample A and Sample B are generated in the same way as described in a previous study (Zhang et al., 2008). In the window chr5: 327, 340–327, 540 (yellow), Sample A has 248 dyads at location 327, 443 (24.3% of the dyads) in the window, while Sample B has only 3 dyads at location 327, with 443 (0.7% of the dyads) in the window. Although our strategy provides high sensitivity to detect even single-nucleotide changes between samples, such differences may not be biologically meaningful. Potential artifacts that arise from MNase treatment, PCR amplification or sequencing biases (Zhang and Pugh, 2011) can cause such small changes. To eliminate these artifacts, we introduced coordinate disturbances (3 bp as the default) by adding a random number to the location of the dyads for each sliding window within the candidate region, and then we re-calculated the differences. This step filters out the majority of artifacts from the predicted RDNPs because the read distribution of the two samples has been transformed to be more similar (Fig. 1B). Finally, our strategy identifies a list of genomic regions with significantly different nucleosome profiles.
3.2 Method evaluation
To systematically inspect the performance of DiNuP when identifying different types of nucleosome positioning dynamics, including changes in location, occupancy and fuzziness, we used computational simulations to evaluate the performance of our method by comparing simulated datasets with a real dataset that has an equivalent genomic coverage of 200× (see Section 2). Since our approach has a very low false-positive rate, we use a true-positive rate and a FDR as measurements of performance. In the simulation of the repositioning, DiNuP has a sensitivity of 96.6% and an FDR of 1.2% when the simulated variation is only 20 bp (Fig. 2A and B). This result indicates that our approach can identify even mild nucleosome repositioning. In addition, DiNuP has a sensitivity of 83.4% and an FDR of 3.9% with a 2-fold occupancy change (Fig. 2C and D), demonstrating that our approach can also detect this type of change. We use the positioning degree, which has been described previously (Zhang et al., 2009), to represent the nucleosome fuzziness. The results of the positioning degree simulation show that the DiNuP has a high sensitivity and a low FDR when detecting changes in the positioning degree. For example, when the positioning degree change is 0.2, the sensitivity of DiNuP is 98.7% and the FDR is 1.9% (Fig. 2E and F). In general, the simulation results demonstrate that DiNuP performs well in identifying different types of differential nucleosome positioning.
Fig. 2.
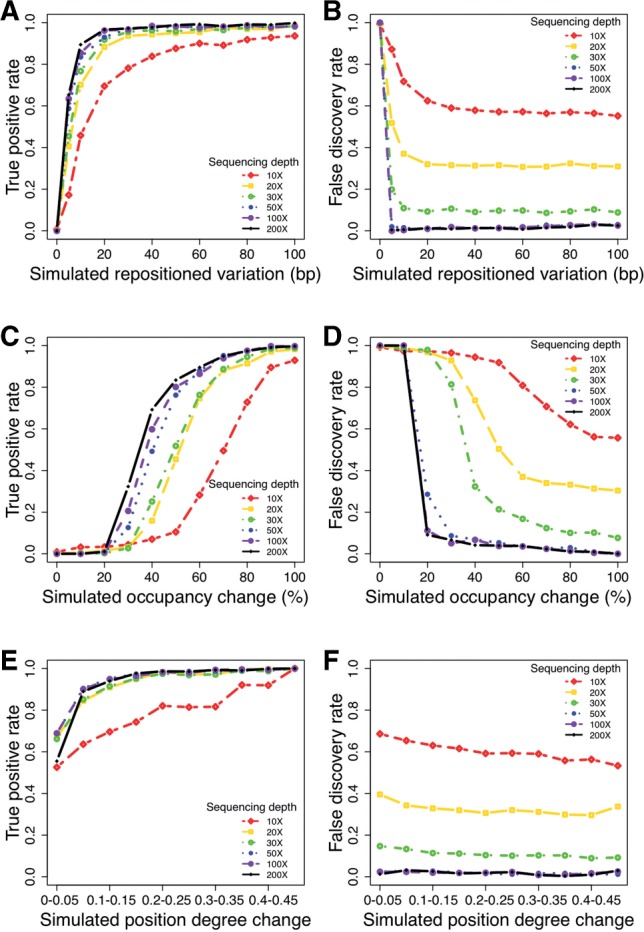
Sensitivity and FDR of DiNuP evaluated by the simulation method. (A) Sensitivity for the detection of the repositioned variation. (B) FDR for the detection of the repositioned variation. (C) Sensitivity for the detection of the occupancy change. (D) FDR for the detection of the occupancy change. (E) Sensitivity for the detection of the positioning degree change. (F) FDR for the detection of the positioning degree change
In addition to characterizing the reliability of DiNuP, we also assessed the effect of the sequencing depth on the ability to identify RDNPs by read sampling (Fig. 2A–F). The results indicate that the sequencing depth affects the performance of DiNuP to a large extent; for example, when the sequencing depth decreases from 200 to 10×, the sensitivity for identifying repositioning by 30 bp decreases from 97.2 to 78.1%, the sensitivity for identifying a 3-fold occupancy change drops from 94.9 to 49.5%, and the sensitivity for identifying a positioning degree change of 0.1 decreases from 94.0 to 58.6%. In other words, the ability of DiNuP to detect RDNPs improves with increasing sequencing depth.
We also evaluated the performance of DiNuP under different cutoffs in real datasets (see Section 2). The evaluation results show that DiNuP has a high sensitivity for the identification of different types of differential nucleosome positioning (Fig. 3A, C and E; Supplementary Fig. S3A, C and E). In addition, the evaluated specificity of DiNuP is also very high (more than 98%). We then compared DiNuP with the fold change method based on both the simulation and the real biological datasets. Except for the occupancy change (where the standard was defined by the fold change), the fold change method performs much worse than DiNuP for both changes in nucleosome location and fuzziness (Fig. 3B, D and Figs; Supplementary Figs. S2 and S3B, D and F). Furthermore, the FDR of DiNuP is also lower than the fold change method under different cutoffs (Supplementary Table S1). This superiority of DiNuP arises from its natural characteristics, and an example region is shown in Supplementary Figure S4.
Fig. 3.

Sensitivity of DiNuP and the fold change method evaluated in Kaplan's YPD-YPEtOH paired samples under different cutoffs. (A) Sensitivity of DiNuP for the detection of repositioned variation. (B) Sensitivity of the fold change method for the detection of repositioned variation. (C) Sensitivity of DiNuP for the detection of occupancy change. (D) Sensitivity of the fold change method for the detection of occupancy change. (E) Sensitivity of DiNuP for the detection of the positioning degree change. (F) Sensitivity of the fold change method for the detection of the positioning degree change
3.3 Physical properties of RDNPs
A previous study has defined some physical properties of single nucleosomes, such as the positioning location, the occupancy and the fuzziness (Mavrich et al., 2008), all of which may change under different conditions. To obtain a better understanding of RDNPs, we defined three parameters, the repositioned variation, the occupancy change and the change in the positioning degree, to characterize the identified regions (Table 1). The mean location difference of nucleosomal dyads between samples within a certain RDNP was defined as the repositioned variation. In addition, nucleosome occupancy changes or the change in the number of bound nucleosomes were measured by calculating the fold change in the number of reads between samples. Finally, to represent the change from fuzzy nucleosomes to phased nucleosomes, a parameter for the change in the nucleosome positioning degree was introduced (see Section 2).
Table 1.
Physical properties of the RDNPs
| Property | General description | Technical description |
|---|---|---|
| Repositioned variation | Change in the nucleosome location | Mean location change for the nucleosomal dyads within an RDNP |
| Occupancy change | Change in the number of bound nucleosomes | Fold change in the number of reads |
| Positioning degree change | A measure of the change in the delocalization of nucleosomes | Difference in the positioning degree between samples |
| Effective width | Effectiveness of the differential nucleosome positioning | Width of the RDNP |
| Candidate driver location | Locations that may drive differential nucleosome positioning | Location with the most significant P-value within an RDNP |
Assuming that there is a mechanistic driving force (e.g. TF binding or Pol II elongation) behind each RDNP, we defined two additional parameters to characterize potential mechanisms. To identify the location that might be related to the cause of the differential nucleosome positioning, the genomic site with the most significant P-value calculated by DiNuP was defined as the candidate driver location. Based on the perspective that different drivers could cause changes with different ranges, we merged adjacent RDNPs and used the length of the merged region to represent the effective width (Table 1). In addition to analysing the RDNPs in a quantitative way using the parameters provided above, we were also able to classify the regions into different groups based on one or several physical properties. For example, the parameter for occupancy change can classify regions into three groups, i.e. groups with increased, equal or decreased occupancy. This classification is useful when researchers are interested only in specific types of RDNPs.
3.4 Use of DiNuP to analyse public datasets
To our knowledge, Kaplan et al. (2009) generated nucleosome profiles with the greatest sequencing depth among the publicly available datasets for yeast grown in three culture media. These media were YPD, YPGal and YPEtOH, and the sequencing had genomic coverage of 294×, 152× and 187×, respectively. We applied DiNuP to compare the nucleosome profiles of pairs of these three datasets. When comparing the YPD medium samples and the YPGal medium samples, 698 RDNPs were identified using an FDR cutoff of 5%, which is equivalent to ~ 2.2% of the yeast genome. After assigning each region to its relevant genomic feature, 228 regions are found to be within promoters (Fig. 4A), with a fold enrichment of 1.54 and a P-value significance of 8.9 × 10−11 relative to the background (see Section 2). Based on the assumption that dynamic nucleosome positioning is related to genes that are responsive to different environmental signals, we also assigned each identified region to its neighbouring genes and performed gene ontology (GO) analysis using DAVID (Huang da et al., 2009). As expected, the identified RDNPs are proximal to genes that are significantly enriched in GO terms, including oxidative phosphorylation, the galactose metabolic process and the generation of precursor metabolites and energy (Fig. 4B). Moreover, 9 out of 11 genes with the GO annotation of the galactose metabolic process were identified as having differential nucleosome positioning.
Fig. 4.

Identified RDNPs are related to transcriptional regulation. (A) Genomic distribution of the identified RDNPs. (B) Enriched GO terms (biological process) among the genes that are proximal to the RDNPs. The Benjamini adjusted P-value is listed above each bar. (C) Percentage of genes surrounding the RDNPs with differential gene expression. The group ‘all’ includes the genes that are proximal to all of the identified RDNPs. Group 1 includes the genes that are proximal to RDNPs that have an effective width shorter than 400 bp, Group 2 includes the genes proximal to RDNPs that have an effective width longer than 400 bp but shorter than 700 bp and Group 3 includes genes proximal to RDNPs that have an effective width longer than 700 bp. (D) Venn diagram of the RDNPs obtained for the YPD_YPGal pair and RDNPs obtained for the YPD_YPEtOH pair
We next examined whether differential nucleosome positioning is closely related to the binding of transcription factors. An enrichment score was calculated by comparing the number of functional cis-elements measured by ChIP-chip (MacIsaac et al., 2006) within RDNPs and the number that would be within the elements by chance. We observed that several transcription factors with important roles in regulating the glucose and galactose metabolic processes were significantly enriched in the identified RDNPs (see Section 2) (Supplementary Table S2). For example, among the 20 conserved binding sites of GAL4, 7 of them are within RDNPs, with a fold enrichment of 16.1. Moreover, when the relationship between RDNPs and differential gene expression was examined, 43% of the genes that were proximal to the RDNPs were differentially expressed (Fig. 4C), with a significant P-value of 4.8 × 10−5 (see Section 2). However, for more than half of the genes that were proximal to RDNPs, their expression levels in YPD medium and YPGal medium were almost the same. This result suggests that nucleosome positioning dynamics has a close relationship with gene transcription but cannot determine the absolute expression level.
To assess the properties of the identified RDNPs, the parameters that are defined in Table 1 were used to classify these regions into groups. We found that most of the regions were in the unrepositioned group (reposition variation smaller than 20 bp) and that six out of nine identified galactose response genes were accompanied by severe positioning degree changes. In accordance with a previous report that nucleosome remodelling is always restricted to one or two individual nucleosomes (Shivaswamy et al., 2008), we observed that more than 80% of the identified RDNPs had effective widths that were shorter than 400 bp. Interestingly, there was an increase in the percentage of differentially expressed genes among genes that were proximal to RDNPs when the effective width of the RDNPs was increased (Fig. 4C). For example, 25 out of 37 of the genes that were proximal to RDNPs with an effective width that was longer than 700 bp were differentially expressed, whereas only 264 out of the 610 genes that were proximal to RDNPs with an effective width shorter than 400 bp were differentially expressed. This result suggests that extensive changes in nucleosome positioning might be more directly and closely associated with gene expression than slight nucleosome positioning changes.
We further compared the nucleosome profiles of yeast that was grown in YPD medium and YPEtOH medium. The results show that the identified RDNPs are also slightly enriched in promoters (Supplementary Fig. S5A) and that the genes surrounding RDNPs are significantly enriched in GO terms, including the vacuolar protein catabolic process, oxidation reduction and the monosaccharide metabolic process (Supplementary Fig. S5B). After obtaining two lists of RDNPs for different pairs, we then asked whether differential nucleosome positioning is cell-type specific. By comparing the RDNPs of the YPD-YPGal pair and the YPD-YPEtOH pair, unique differential regions in each pair were obtained (Fig. 4D). Seven out of nine galactose response genes were found to be specifically proximal to RDNPs for the YPD-YPGal pair. Genes that were proximal to unique RDNP in the YPD-YPGal pair also included a larger percentage of differentially expressed genes than the genes that were not proximal to these regions. In summary, our analysis shows that the identified RDNPs are within functionally important regions and that differential nucleosome positioning is closely related to the epigenetic regulation of transcription. The identified RDNPs for this dataset can be obtained in Supplementary Table S3.
It is reported that nucleosome profiles may vary between biological replicates in some genomic regions (Zhang and Pugh, 2011). To systematically inspect the role of the variation between replicates, we applied DiNuP to all possible 15 pair-wise comparisons of the six biological replicates of YPD medium. Even with a stringent cutoff (FDR 0.01), dozens of or even hundreds of RDNPs can still be identified. After carefully checking the identified regions, we found that nucleosome profiles in these regions are largely different (Supplementary Fig. S6). To check whether the variability between replicates shows any biological meaning, we picked genes that were proximal to the identified RDNPs and analysed the functional enrichment of those genes. As a result, none of the gene lists obtained from the 15 pairs is enriched in any of the GO terms. We further checked whether those identified RDNPs are randomly distributed by summarizing the number of shared RDNPs among 15 pairs (Supplementary Fig. S7). Then, 75% of the identified RDNPs were in only one or two pairs, indicating that the variability between biological replicates is largely random. This variation could be mainly caused by a bias in the MNase treatment, PCR amplification or sequencing, which cannot be corrected solely based on a computational method. Considering the experimental bias, when comparing nucleosome profiles between samples, some nucleosome positioning changes might not be biologically meaningful. However, if the compared samples were treated with the same experimental strategy of nucleosome profiling, then this variability would not largely affect the identification of RDNPs with real biological meaning.
4 DISCUSSION
When comparing the nucleosome profiles of different samples, it is important to first make the samples comparable to each other. DiNuP uses the K–S test to calculate statistical P-values between sliding windows and estimates the FDR from the background P-value for use as the cutoff to achieve this goal. After reducing the number of potential artifacts, genomic regions with P-values consecutively lower than the cutoff were then identified as RDNPs. It is worth noting that, when using different FDRs or minimum length cutoffs, the number of identified RDNPs can differ greatly. Therefore, the cutoff chosen depends on the features of the identified regions that are preferred by the user.
Because DiNuP is designed to capture the most significant read distribution changes, it should strike a balance between the detection of different types of nucleosome positioning changes at a certain FDR cutoff. In the identification of short-range nucleosome occupancy change, although regions with different occupancy levels may have similar read distributions, the K–S test can identify the differences in both boundaries around the occupancy change region. After combining the differential signals on the boundary of sharp nucleosome occupancy changes, regions with differential occupancy levels can be identified. However, in terms of long-range nucleosome occupancy change, as the differential signals on the boundaries can be too far away from each other to be combined as an RDNP, our method will have limitations in the accurate identification of regions with long-range occupancy changes. As a result, we implemented the fold change method into the software package of DiNuP and provided an option to users who are especially interested in detecting occupancy changes. Nevertheless, DiNuP is, in general, an effective and robust method for identifying RDNPs.
Another important factor that influences the detection of differential nucleosome positioning is sequencing depth. To evaluate this effect, we sampled datasets with different levels of genomic coverage and used DiNuP to detect RDNPs. Assuming that the results obtained from the datasets with the deepest sequencing depth were the most reliable results, we used these RDNPs as a standard to determine the level of consistency between the regions identified using datasets with lower sequencing depths and those identified using the standard dataset (Supplementary Fig. S8). With an FDR cutoff of 0.01, the consistency percentage dropped to ~ 60% with a genomic coverage of 50, indicating that the sequencing depth indeed affects the detection of RDNP to a large extent. From this perspective, we argue that a minimum sequencing depth, i.e. a genomic coverage of 20 or 30, is required for the accurate identification of differential nucleosome positioning; otherwise, genomic regions that are identified as different may not be truly different but instead may appear different as a result of random discrepancies or background noise.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Lin Liu, Yiqian Zhang, Chenfei Wang, Hanfei Sun, Meng Zhou, Qian Zhao, and Hongtao Sun for their kind help and insightful discussions. We also thank the anonymous reviewers for their constructive suggestions.
Funding: The National Basic Research Program of China (973 Program; No. 2010CB944904 and 2011CB965104), the National Natural Science Foundation of China (31071114), the Shanghai Rising-Star Program (10QA1407300), the New Century Excellent Talents in the University of China (NCET-11-0389) and the Innovative Research Team Program Ministry of Education of China (IRT1168).
Conflict of Interest: none declared.
REFERENCES
- Heintzman N.D., et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- Huang da,W., et al. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Jiang C., Pugh B.F. Nucleosome positioning and gene regulation: advances through genomics. Nat. Rev. Genet. 2009;10:161–172. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N., et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009;458:362–366. doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komili S., et al. Functional specificity among ribosomal proteins regulates gene expression. Cell. 2007;131:557–571. doi: 10.1016/j.cell.2007.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg R.D., Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–294. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- Li B., et al. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Liu C.L., et al. Single-nucleosome mapping of histone modifications in S. cerevisiae. PLoS Biol. 2005;3:1753–1769. doi: 10.1371/journal.pbio.0030328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K. Dynamic nucleosomes. Chromosome Res. 2006;14:5–16. doi: 10.1007/s10577-005-1026-1. [DOI] [PubMed] [Google Scholar]
- Luger K., et al. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- MacIsaac K.D., et al. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinformatics. 2006;7:113. doi: 10.1186/1471-2105-7-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrich T.N., et al. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res. 2008;18:1073–1083. doi: 10.1101/gr.078261.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor J. Dynamic nucleosomes and gene transcription. Trends Genet. 2006;22:320–329. doi: 10.1016/j.tig.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Schones D.E., et al. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132:887–898. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabish M.A., Struhl K. Evidence for eviction and rapid deposition of histones upon transcriptional elongation by RNA polymerase II. Mol. Cell Biol. 2004;24:10111–10117. doi: 10.1128/MCB.24.23.10111-10117.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivaswamy S., et al. Dynamic remodeling of individual nucleosomes across a eukaryotic genome in response to transcriptional perturbation. PLoS Biol. 2008;6:e65. doi: 10.1371/journal.pbio.0060065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev A., et al. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res. 2008;18:1051–1063. doi: 10.1101/gr.076463.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev A., et al. Determinants of nucleosome organization in primary human cells. Nature. 2011;474:516–520. doi: 10.1038/nature10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstrepen K.J., et al. FLO1 is a variable green beard gene that drives biofilm-like cooperation in budding yeast. Cell. 2008;135:726–737. doi: 10.1016/j.cell.2008.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman J.L., Kingston R.E. Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annu. Rev. Biochem. 1998;67:545–579. doi: 10.1146/annurev.biochem.67.1.545. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Pugh B.F. High-resolution genome-wide mapping of the primary structure of chromatin. Cell. 2011;144:175–186. doi: 10.1016/j.cell.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., et al. Identifying positioned nucleosomes with epigenetic marks in human from ChIP-Seq. BMC Genomics. 2008;9:537. doi: 10.1186/1471-2164-9-537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., et al. Intrinsic histone-DNA interactions are not the major determinant of nucleosome positions in vivo. Nat. Struct. Mol. Biol. 2009;16:847–852. doi: 10.1038/nsmb.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.