

Summary
Background
The conversion of fibrinogen to fibrin and its cross-linking to form a stable clot are key events in providing effective hemostasis.
Objectives
To evaluate the relationship of fibrinopeptide release and factor XIII activation in hemophilia whole blood.
Patients/Methods
We investigated fibrinopeptide (FP)A and FPB release, factor(F) XIII activation and fibrin mass in tissue factor-initiated coagulation in whole blood from individuals with hemophilia and healthy subjects.
Results
In hemophilia, the rates of fibrin formation are delayed compared to healthy individuals. FPA/FPB release and FXIII activation are decreased in hemophilia versus healthy, 5.4±0.7μM/min to 1.7±0.4μM/min (p=0.003), 2.3±0.6μM/min to 0.5±0.1μM/min (p=0.025) and 12.1±0.7nM/min to 3.1±0.7nM/min (p<0.0005), respectively. More FPA is released in hemophilia (6.6±1.2μM) prior to clot time (CT) than in healthy individuals (2.6±0.4μM,p=0.013); whereas FPB and activated FXIII levels remain comparable.FXIII activation, which normally coincides with FPA release, is delayed in hemophilia. At CT in normal blood, the FPA concentration is 2.6-fold higher than FPB release (p=0.003), while in hemophiliacs this ratio is increased to 6.6-fold (p=0.001).
Conclusions
These data suggest essential dynamic correlations exist between the presentations of fibrin I, fibrin II and FXIIIa. The “discordance” of fibrin formation, in hemophilia, results in clots that are more soluble than normal (43% lower mass, p=0.02). The resulting poor physical clot strength probably plays a crucial role in the pathology of hemophilia.
Keywords: fibrin, factor XIII, fibrinopeptides, hemophilia
Introduction
Hemophilia A and B are the most common congenital bleeding disorders. They are characterized by the decrease or absence of functional factor (F) VIII (hemophilia A) and FIX (hemophilia B)[1–3]. In recent years, the prognosis for individuals with hemophilia has improved greatly, and nearly normal lifespans can be expected. However, significant pathologies still attend replacement therapy ranging from hemearthrosis to inhibitor development to infection.
In the absence of FVIII or FIX, the blood coagulation cascade is defective at the level of FXa generation which is critical to produce the amplification and propagation of thrombin generation through the prothrombinase complex[4–7]. Low levels of thrombin are generated during the initiation phase of tissue factor- activated blood coagulation, in which the cleavage of fibrinogen to fibrin and the activation of platelets, FV, and FXIII all occur[8–12]. The interval prior to clot time, also referred to as the lag or initiation phase of tissue factor-activated thrombin generation is only moderately affected in hemophilia, since <2nM thrombin is required for clotting to occur[12]. The major defect in this bleeding disorder appears to be the ability to generate the major burst of thrombin (>95%) that is required to sustain the clot formation required for sealing the vascular lesion[13]. The intrinsic FXase (FVIIIa-FIXa-Ca++-membrane) and prothrombinase (FVa-FXa-Ca++-membrane) complexes expressed and preserved within the fibrin-platelet clot provide continued activity as additional blood is resupplied to the lesion by flow[13]. With the diminished FXase complex in hemophilia, wound sealing is compromised.
The decreased thrombin generation in hemophilia A depresses the rates of activation of multiple thrombin substrates including those building the fibrin network needed to stem blood loss. The proteins that require activation by thrombin for the normal expression of this fibrin network include: multiple platelet receptors, fibrinogen, FXIII and thrombin activatable fibrinolysis inhibitor (TAFI). Reduced TAFIa formation has been shown to play a role in the premature lysis of clots formed from hemophilic plasma[14]. Although minimal differences in platelet activation between normal and FVIII deficient blood have been shown[7], the overall number of platelets accumulated appear to play a part in the fibrin gel structure in hemophilia[15]. Another study has shown that low FIX delayed the clot formation onset, reduced the fibrin polymerization rate, and produced fibrin fibers thicker than those found in normal controls[16]. In a study on hemophilia B mice, it has been reported that wound healing is abnormal[17]and treatment of back wounds in these mice, with either systemic FIX or FVIIa at levels to produce hemostatic thrombin generation, restored only some of the parameters of wound healing[18]. Other deficiencies in the coagulation pathway have linked reduced rates of thrombin production with decreased levels of TAFIa and premature clot lysis[14,19,20]. Decreased FXIII availability for thrombin and early loss of clot firmness has been identified in patients with unexplained intraoperative bleeding[21]. In general, impaired rates of thrombin generation can translate into an array of defects in the multiple responses to vascular injury.
The conversion of fibrinogen to fibrin and its cross-linking to form a stable clot are key events in effective hemostasis. Previously, we have shown that in corn trypsin inhibited whole blood, the pattern of fibrin formation is different than that observed in systems using citrated plasmas or purified proteins[22]. In tissue factor-activated blood coagulation, partial cleavage of fibrinopeptide (FP) A and clot formation occur just before the propagation phase of thrombin generation[12]. At the point of visual clot formation, virtually all fibrinogen (and some products already cross-linked) disappear from the fluid phase, while only a fraction of FPA and FPB have been released.[22] Thus, the first clot formed is a mixture of fibrin I, some fibrin II and fibrinogen partially cross-linked by FXIIIa. Well after clot formation, the FPB antigen is still found associated with the Bβ-chain in cross-linked products.[22] In contrast, in purified systems, FPB removal precedes the cross-linking reaction. Studies with tissue factor-activated hemophilia blood have shown that FPA and FPB release and fibrin depletion is delayed versus normal blood.[7,23] The insoluble fibrin weight is also reduced compared with normal blood.[7,23] Factor XIII activation in tissue factor-initiated contact pathway suppressed hemophilia whole blood, has not been reported.
In this study, we evaluated the dynamic relationships between thrombin generation, FXIII activation, fibrin I and fibrin II formation and clot stability in hemophilia following tissue factor-activated coagulation and compared the data with that from normal blood.
Materials and Methods
Materials
HEPES, NaCl, Tris-HCl, EDTA, TFA, 1-palmitoyl-2-oleoyl-phosphatidyl serine (PS), 1-palmitoyl-2-oleoyl-phosphatidyl choline (PC) were purchased from Sigma Chemical Co (St. Louis, MO). HPLC grade H2O and CH3CN, HClO4 and KOH were purchased from Fischer Scientific (Pittsburgh, PA). Benzamidine-HCl was purchased from Aldrich, Inc (Milwaukee, WI). Recombinant tissue factor was a gift from Drs. Lundblad and Liu (Hyland division, Baxter Healthcare Corp, Duarte, CA) and was relipidated in PCPS (75% PC: 25% PS) vesicles by a previously described protocol[24]. Corn trypsin inhibitor (CTI) was prepared as previously described.[7,23] D-phenylalanyl-L-prolyl-L-arginine chloromethyl ketone (FPRck) was a gift from Dr. Jenny (Haematologic Technologies, Essex Junction, VT). Rabbit polyclonal α-FXIII (D4679) was a gift from Dr. Bishop (ZymoGenetics, Seattle, WA). Horseradish peroxidase-labeled goat antirabbit IgG antibodies were purchased from Southern Biotechnology (Birmingham, AL). Enzyme-linked immunosorbant assay (ELISA) kits were used to estimate thrombin-antithrombin (TAT) complex formation (Behring, Westwood, MA).
Subjects
Individuals with hemophilia A and B and normal controls were recruited and advised according to a protocol approved by the Institutional Review Board at the University of Vermont Human Studies Committee. All participants gave informed written consent. Blood was obtained from three subjects with hemophilia A (one individual was studied on 4 different occasions) and two subjects with hemophilia B. All hemophilia subjects had normal coagulation profiles for fibrinogen and platelets. One hemophilia A donor (drawn between the ages of 18–21 years), a young man with functionally severe hemophilia (FVIII:C<1%), tested positive for hepatitis C and had a history of bleeding and joint pain and rountinely self-administered recombinant FVIII products when symptomatic. We studied his blood 4 times over the course of 3 years, and individual draws have been incorporated into other studies[7,23]. Each phlebotomy was performed 1 week after his most recent FVIII injection, and FVIII levels were less than 2%. Another hemophilia donor was a 20 year old male with severe FVIII deficiency and the brother of the first donor. He tested positive for hepatitis C, has had recurrent bleeding episodes in his joints, has had bilateral radiosynovectomy of both ankles, and occasionally uses prophylaxis. His FVIII levels at the time of the blood draw were 2%. The third hemophilia A donor was a 35 year old male with severe FVIII deficiency (FVIII:C <0.5%)[7]. He tested positive for HIV and hepatitis C with cirrhosis, has had a knee replacement and was on FVIII prophylaxis as needed. He had received no replacement therapy for 2.5 weeks prior to the whole blood experiments. At the time of his blood draw, his medications included nelfinavir, 3TC, tenofovir, pegylated interferon, ribavirin and Bactrim.
One hemophilia B individual was a 41 year old male with severe FIX deficiency. He tested positive for HIV and hepatitis C and has had a chronic subdural hematoma, seizure disorder and a right middle cerebral artery stroke. His medications at the time of the blood draw included Bactrim, Kaletra, tenofovir, abacavir, Topamax and loperamide. His FIX coagulant level was 2% at the time of the blood draw. A second hemophilia B individual was a 47 year old male with severe FIX deficiency and is the brother of the first hemophilia B individual. He tested positive for hepatitis C, has had multiple bleeds into his right elbow and right hip, and has had a total hip replacement on his right side. His medications include Hyzaar, famotidine, Celebrex. At the time of the blood draw he had <1% FIX.
Normal controls (11 male/9 female) with a mean age of 34±12 years, were selected from a pool of healthy volunteers at the University of Vermont. No individuals had a personal or familial history of thrombosis/hemorrhage or regular aspirin or drug use. Some individuals were studied multiple times. Whole blood from normal individuals was used as contemporaneous controls as well as for the studies of in vitro “acquired hemophilia B” in 2 experiments as described below. Coagulation profiles were performed by Fletcher Allen Hematology Clinic (Burlington, VT) for each experiment.
Whole blood coagulation
Blood was collected by venipuncture at the General Clinical Research Center at the University of Vermont, with a 19 3/4G butterfly needle into a 50mL syringe. Blood was immediately aliquoted (1mL) into polystyrene culture tubes containing CTI (100μg/mL) and tissue factor (functionally 5pM) relipidated in PCPS (1:2000protein/lipid) as previously described[7,12,22,25]. When used, an α-FIX-91 monoclonal antibody (mAb) was added to the tubes prior to the addition of whole blood to produce an “acquired hemophilia B” blood, as previously described[26]. At this concentration, the α-FIX-91 mAb prolonged the activated partial thromboplastin time (aPTT) of normal plasma from 38 to 115 seconds. The aPTT for commercial FIX-deficient plasma (<1% of FIX; George King Biomedical, Overland Park, KS) was 112 seconds. All samples (control individuals, hemophilia A and B individuals and acquired hemophilia blood), were quenched at intervals over the course of 20 minutes, every minute from 0–10 followed by 12-, 14-, 16- and 20-minutes, with a cocktail of inhibitors to a final concentration of: 25mM EDTA and 10mM benzamidine-HCl in HBS (0.15mol/L NaCl, 0.02mol/L HEPES, pH 7.4) and 50μM FPRck in 10mM HCl. The zero time point contained all of the inhibitors prior to the addition of whole blood. A control tube, containing CTI and no tissue factor was used each time. Clot time was determined visually (two observers). After the coagulation reaction was quenched with inhibitors, samples were centrifuged for 15 minutes at 2000 x g and clot material was separated from the solution phase. Solid and solution phases were stored at -80°C for further analysis.
Thrombin generation
The time course of soluble thrombin generation in whole blood, represented as TAT complex, was evaluated by ELISA (Behring, Westwood, MA) measured in duplicate with 5 standards as previously described[12]. The operational sensitivity of the TAT ELISA is ~40pM. Results were analyzed on a Vmax microtiter plate reader (Molecular Devices, Menlo Park, CA) equipped with Softmax version 2.35.
Fibrinopeptide A and B detection
Fibrinopeptide A analyses of each time point was performed by either a commercial ELISA (Asserachrom FPA; Diagnostica Stago/American Bioproducts Parsippany, NJ) performed in duplicate following manufacturer instructions or analyzed using HPLC methodology on a Waters Model 484 Controller and Model 510 Solvent Pumps monitored using a Model 481 detector at 214 nm as previously described[12]. The time course of FPB release was elucidated using HPLC methodology as previously described[12]. The peptides were identified by matrix-assisted laser desorption ionization time of flight mass spectrometry (linear model, PE Applied Biosystems).
Fibrin mass
The insoluble clotted material contained within the whole blood tubes at each time point was analyzed as previously described[22]. Briefly, the insoluble clotted samples were washed 2–3 times with 1mL of 0.15mol/L NaCl and then allowed to incubate in the salt solution (1mL) for 12–15 hours so that soluble material within the clots could diffuse into solution. The clots were rinsed with H2O to remove salt, lyophilized and weighed.
Factor XIII activation
Factor XIII activation was followed by Western analyses of the whole blood quenched time points. SDS-PAGE (4–12%) was performed according to the modified Laemmli procedure[27]. High molecular weight standard mixtures (14 to 200kD) were loaded along with FXIII (50ng/lane) or activated FXIII (FXIIIa) to allow for comparison to the whole blood samples on the immunoblots. The gels were transferred to nitrocellulose membranes (BioRad, Hercules, CA) and subjected to semi-dry transfer for 3 hours at 250mAmps. The primary antibody was a polyclonal α-FXIII used at 5ug/mL, the secondary antibody was goat α-rabbit HRP at 1:5000 dilutions, and the substrate for emitting light was Luminol (Dupont NEN). Comparison of FXIII conversion to FXIIIa was analyzed on the immunoblots.
Statistics
Between group comparisons were done by the Student’s unpaired t test for normally distributed data. A p-value <0.05 was considered statistically significant.
Results
Thrombin generation in hemophilia A and B
Thrombin generation in hemophilia A and B individuals’ and “acquired” hemophilia B blood is represented as TAT in Figure 1 as the mean and the SEM over the time course of 20 minutes. Hemophilia A (n=6) and hemophilia B (n=4) blood lack the propagation phase seen in our control population (n=35). Hemophilia A individuals have an extended initiation phase by ~ 4 min (8.6±1.8min) and in hemophilia B whole blood by ~ 4.5 min (9.2±1.6min). Using the identical tissue factor preparation, the cumulative historical control clot time is 4.7±0.2min[12]. The propagation phase rate of thrombin generation was decreased in hemophilia A by 9.8 fold (9.4±2.9nM/min) and in hemophilia B by 6.5 fold (14.2±2.8nM/min) versus normal controls (92.3±7.0nM/min). The accumulated levels (20 minutes) of thrombin were 135±44nM for hemophilia A individuals and 143±41nM for hemophilia B individuals. This level of thrombin generation is significantly lower (p<0.0005) than the mean for healthy individuals (851±53nM). Although, these final levels of thrombin generation are decreased in hemophilia, they are still sufficient for the activation of the procoagulants ordinarily anticipated to be required to stem blood loss. Since the thrombin generation curves are comparable between hemophilia A and B, we combined all hemophilia blood studies to evaluate the differences between the hemophilia and the healthy populations.
Figure 1. Thrombin generation in hemophilia A and hemophilia B.

Contact pathway inhibited whole blood coagulation is initiated to clot upon addition of 5 pM relipidated tissue factor in hemophilia A (n=6, ▲) and hemophilia B (n=4, ◇) individuals. Thrombin generation is determined as thrombin-antithrombin complex formation (TAT) and compared to a cumulative control of 35 healthy individuals (■)[12]. All data are shown as the mean±SEM. Clot times (CT) are indicated as an arrow below the x-axis.
Factor XIII activation
Western analyses of FXIII activation in quenched time points from a whole blood series are depicted in Figure 2 for a representative healthy control (panel A) and a hemophilia individual (panel B). The Western blot developed using an A chain specific polyclonal α-FXIII antibody, is illustrated with time points from 0 to 20 minutes labeled above the blot. The top band seen in the immunoblot is the A-chain of FXIII (FXIIIA) and the bottom band is seen as the activated form of FXIII (FXIIIAa). The clot time (CT, arrow) for the control individual was 3.3 minutes and 9.8 minutes for the hemophilia individual. The cumulative curves of FXIII activation (panel C) show that FXIII activation proceeded at approximately one fourth the rate in individuals with hemophilia (n=6) than in healthy individuals (n=9), 3.1±0.7nM/min versus 12.1±0.7nM/min (p<0.0005), respectively. In healthy individuals, 51% of FXIII was activated at clot time, 4.4±0.5min and by 20 minutes, FXIIIa concentration reached a maximum value of 58±5nM. In hemophilia blood, with clot time delayed to 8.8±0.7 min, the FXIIIa concentration at 20 minutes was 39±9nM (~43% activated).
Figure 2. Factor XIII activation.
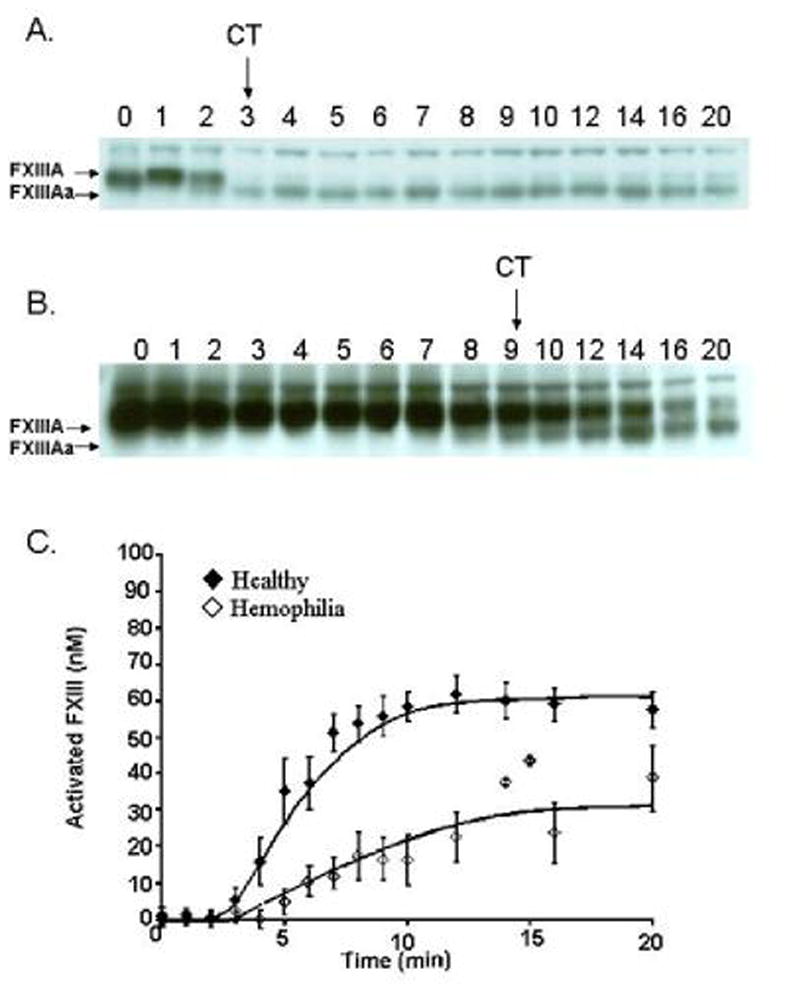
Quenched time points (0–20 minutes) were analyzed for FXIII activation in CTI whole blood from a healthy individual (panel A) and a hemophilia A individual (panel B). The polyclonal antibody used is specific for the unactivated FXIII, Mr=75,000 and activated FXIII species (FXIIIAa, Mr=71,000). Clot time (CT, arrow) in the healthy individual was 3.2 min and 9.8 minutes in the hemophilia individual. C) Quantitation of the appearance of the activated form of FXIII is compared between hemophilia (◇) and healthy (◆) whole blood. The mean clot time (CT) is shown below the graph with an arrow.
Fibrinopeptide release
Fibrinopeptide A release in healthy individuals (n=24) occurred at a rate of 5.4±0.7μM/min with clotting at 4.3±0.2min (Figure 3A). Maximum level of FPA released was 15 ±1.1μM at 20 minutes. With the same tissue factor stimulus, hemophilia blood (hemophilia A and B combined, n=8) clotted at 9.0±0.7min and the rate of FPA release was slower than in healthy (1.7±0.4μM/min; p=0.003). Although the clot time was delayed, the maximum levels of FPA released (13±2μM) in hemophilia approached those seen in healthy individuals. Paradoxically, more FPA was released prior to clot time in the hemophilia blood when compared to healthy individuals, 50% (6.6±1.2μM) versus 17% (2.6±0.4μM; p=0.013), respectively. FPA release in hemophilia blood was also more variable.
Figure 3. Comparison of fibrinopeptide release and fibrin mass in normal and hemophilia whole blood.
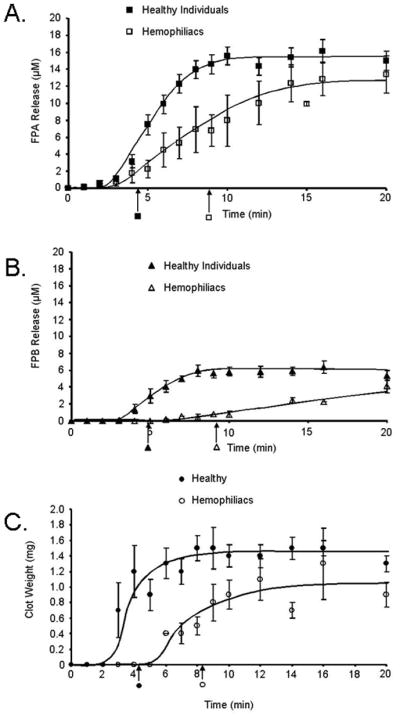
Contact pathway inhibited whole blood was initiated to clot upon addition of 5 pM tissue factor and analyzed for: A) fibrinopeptide A (FPA) release as a function of time in healthy (■) and hemophilia whole blood (□); B) fibrinopeptide B (FPB) release as a function of time in healthy (▲) and hemophilia whole blood (△); C) fibrin mass (clot weights) at each time point in 1 mL of healthy (●) and hemophilia (○) whole blood. All data are shown as the mean±SEM. Clot time is shown below the x-axis as an arrow.
The total FPB released in healthy individuals (n=9) as previously shown[22] represents only 1/3 of the stoichiometric value and reached only 6.4±0.7μM by 20min with a maximum rate of release of 2.3±0.6μM/min (Figure 3B). In hemophilia blood, FPB is released at a maximum rate of 0.5±0.1μM/min (p=0.025) and by 20 minutes obtains a concentration of 4.1±0.8μM. By clot time in healthy individuals (3.8±0.2 min), 0.95±0.2μM FPB was released from fibrinogen and reached maximum levels at 8 minutes. Although, in hemophilia, the clot time was delayed by approximately 5 min (9.2 ±2.1min), the same concentration of FPB (0.9±0.3μM) as in the healthy population was present at clot time.
Fibrin mass
Fibrin clots from healthy individuals (n=15) over the 20 minute time course were consistently heavier than clots from hemophilia individuals (n=6) (Figure 3C). On average, healthy individuals had, at 20 minutes, clot weights of 1.4±0.2mg/mL blood. In hemophilia, the ultimate clot weights were 0.8±0.2mg/mL, significantly less than in healthy (p=0.02). More dense heavier clots represent more extensive fibrin cross-linking and increased strength of the fibrin clot. In hemophilia, the relationship between clot time and the accumulation of fibrin mass was altered. At clot time (4.2±0.2min), clots from healthy individuals were already at 86% (1.2±0.3mg/mL) of their final mass. Whereas in hemophilia clots, only 62% (0.5±0.1mg/mL; p=0.03) of their final mass was present at clot time (8.2±0.4min).
Relationship between fibrinopeptide release and FXIII activation
In healthy individuals FPA release coincides with FXIII activation over the first 6 minutes (panel A, Figure 4). After 6 minutes, FXIII activation slows compared to FPA release, but both reach maximum levels at approximately 10 minutes. When FPA release and FXIII activation are plotted against each other at each time point there is a linear correlation (R2=0.987) (not shown). In individuals with hemophilia, FXIIIa is not generated simultaneously with FPA and FXIII activation is delayed versus FPA release (panel B), achieving a final concentration ~30% lower. The maximum level of FXIII activation is delayed to ~16 minutes. When FPA release and FXIII activation in hemophilia are compared (0–20 minutes), a poorer correlation exists (R2=0.843) (not shown). At early release of FPA, FXIII appears to be activated more slowly, thus is not readily available to the initial fibrin I that is being formed.
Figure 4. Correlative patterns of fibrinopeptide release and FXIII activation.

Fibrinopeptide A (FPA, □) and fibrinopeptide B (FPB, △) release is compared with FXIII activation (◆) for the quenched time points (0–20 minutes) in contact pathway suppressed healthy (panel A) and hemophilia A and B blood (panel B). Data are shown as the mean±SEM with the clot time shown as an arrow below the x-axis. Panel C. Maximum values for the control data for FPA, FPB and FXIII was determined and each time point for controls (closed symbols) and hemophiliacs (open symbols) and was replotted as a percentage of the maximum values.
FPB release trails FPA and FXIII activation during the process of blood coagulation from healthy individuals[22]. In hemophilia, FPB release is delayed relative to FPA and FXIII activation, and never reaches a point of accelerated release seen in the blood from healthy individuals, which occurs between 4 and 7 minutes. The final concentration of FPB achieved in hemophilia blood is only 4.1±0.8μM by 20 minutes. In healthy individuals, the FPA concentration (2.6±0.4μM) is 2.6 fold higher than the FPB concentration (1.0±0.2μM) at clot time (p=0.003). In hemophilic blood at clot time, the FPA (6.6±1.2μM) and FPB (0.9±0.3μM) concentrations give rise to a ratio of 6.6 FPA/ FPB, significantly higher than in healthy (p=0.001).
The aberrant association between the formation of these three products is more clearly observed by replotting the data for each time point as a percentage of the control maximums for FPA, FPB and FXIII activation for hemophiliacs (open symbols) and controls (closed symbols) as shown in Figure 4C.
Discussion
In tissue factor activated hemophilia blood, the patterns of thrombin clot product formation are altered compared to that observed in blood obtained from healthy individuals. FPA and FPB production, and FXIII activation are all decreased in hemophilia. While the clot time is delayed in hemophilia, more FPA is released prior to clot time while FPB release and FXIII activation remain unchanged at clot time compared to whole blood from healthy individuals. This suggests that the clots from hemophiliacs are less robust due to the changed proportions of increased available fibrin monomer units without the compensational support of lateral association and cross-linking.
Fibrin gel structure has been shown to be dependent upon thrombin concentration as well as the rates of thrombin generation[28–30]. Utilizing electron microscopy, it has been shown that clots produced under high thrombin generation conditions are composed of a tight network of thin fibers; whereas under low thrombin generation conditions clots appear turbid, porous and are composed of thick fibrin fibers [30;31]. Accordingly, FVIII and FIX deficient plasma form loose fibrin clots with higher permeability[15]. In our studies the clots from hemophilia blood are less dense (with a 43% decrease in fibrin mass) compared to clots from healthy individuals, which can explain the porosity and flimsiness seen by electron microscopy studies.
The quality of a fibrin/platelet blockade to stem blood loss is associated with the relationships and timing of events in the formation of a cross-linked stable fibrin network. The release of the fibrinopeptides results in the formation of the fibrin monomer, which is molecularly indistinguishable from fibrin. The fibrin monomer elongates from both ends as a two stranded molecule until it reaches approximately 30 monomers when it is identified as a protofibril[32,33]. The linear fibrin assembly is accomplished by the lateral association of the protofibrils to form thicker fibrin fibers[33,34]. It is believed that the forces involved in lateral association are weak and therefore only become “strong” when in the presence of large numbers. The β-chains are thought to be responsible for this aspect of fiber growth[35]. This occurs by subsequent cleavage of the initial fibrin/fibrinogen by thrombin at Bβ-Arg14 which releases FPB. In our study we determined that the overall rate of FPA release from hemophilia blood is decreased, but more FPA is released, and hence more fibrin I formed, prior to clot time than in healthy individuals. This suggests that more protofibrils are available for lateral association at clot time in hemophilia blood. Although, the levels of FPB at clot time are ~6.6 fold lower than those of FPA. Even by 20 minutes the final level of FPB is less than half (4.1±0.8μM), compared to FPA (13±2μM); while ultimate FPA concentrations at 20 minutes are approximately equivalent to those generated in normal blood. FPA and FPB dynamics are also altered when the composition at clot time is examined; FPA concentrations are increased in hemophilia while in healthy individuals FPB concentrations remain the same. Overall, these observations suggest that: (1) the discordant release of FPA relative to FPB in hemophilia can disorder and potentially mask the FPB sites on fibrinogen, which may impede the ability of thrombin cleavage at Bβ-Arg14-Gly15 to produce the Bβ-peptide and fibrin II; (2) although FPA is being released and fibrin monomers are formed, the insoluble clot doesn’t appear until ~1 μM FPB is present; (3) since the ratio of FPA to FPB is disordered in hemophilia, there is diminished lateral association, which can account for the fragility of the clots in hemophilia.
In blood from healthy individuals, when FPA is released from fibrinogen and fibrin monomer units are accruing, FXIIIa is simultaneously available to cross-link these monomers. In hemophilia blood, more FPA is released prior to clot time producing more protofibrils to cross-link, but less FXIIIa is available at clot time and even by 20 minutes (<40%) when compared to healthy individuals. Thus, the overall fibrin clots that are produced are less stable in hemophilia blood, which can contribute to the fragile clot. The diminished levels of FXIIIa probably also affects other cross-linking processes[36,37] including: fibronectin, α2-plasmin inhibitor, collagen vitronectin, vWF, actin, myosin, FV and thrombospondin[38–42].
Normally, there is a dynamic interdependence between the availability of clotting product proteins required for the fibrin network. The interrelationships between the players result in a clot which provides stabilized hemostasis. In hemophilia, the generation of thrombin at a slower rate results in defective relationships between the formation rates of the fibrin network proteins, altering the stability of the fibrin clot. A recent study by Hvas et al.[43] utilizing thromboelastography showed that simultaneous treatment with tranexamic acid and recombinant FVIII significantly improves the clot stability in patients with hemophilia A. Our results also suggest that the availability of FXIIIa in hemophilia may play a crucial role and might have clinical and therapeutic potential.
Acknowledgments
We would like to thank Richard Pouliot and Matthew Whelihan for their technical support. We would also like to thank Dr. Kevin Cawthern for his work on thrombin generation in the FIX deficient population.
Funding source: Supported by grants from the Program Project Grant No. HL 46703 (Project 5 and 1) from the National Institutes of Health (K.BZ and K.G.M) and a Career Development Award from the National Hemophilia Foundation (KBZ).
Footnotes
Presented in abstract form at the 47th Annual ASH meeting, Blood 106: 97a (abstract #321), 2006.
References
- 1.Bolton-Maggs PH, Pasi KJ. Haemophilias A and B. Lancet. 2003;361:1801–1809. doi: 10.1016/S0140-6736(03)13405-8. [DOI] [PubMed] [Google Scholar]
- 2.Hoyer LW. Hemophilia A. N Engl J Med. 1994;330:38–47. doi: 10.1056/NEJM199401063300108. [DOI] [PubMed] [Google Scholar]
- 3.Castaldo G, Nardiello P, Bellitti F, Santamaria R, Rocino A, Coppola A, Di Minno G, Salvatore F. Haemophilia B: from molecular diagnosis to gene therapy. Clin Chem Lab Med. 2003;41:445–451. doi: 10.1515/CCLM.2003.067. [DOI] [PubMed] [Google Scholar]
- 4.Mann KG, Krishnaswamy S, Lawson JH. Surface-dependent hemostasis. Semin Hematol. 1992;29:213–226. [PubMed] [Google Scholar]
- 5.Lawson JH, Mann KG. Cooperative activation of human factor IX by the human extrinsic pathway of blood coagulation. J Biol Chem. 1991;266:11317–11327. [PubMed] [Google Scholar]
- 6.Ahmad SS, Rawala-Sheikh R, Walsh PN. Components and assembly of the factor X activating complex. Semin Thromb Hemost. 1992;18:311–323. doi: 10.1055/s-2007-1002570. [DOI] [PubMed] [Google Scholar]
- 7.Cawthern KM, van 't Veer C, Lock JB, DiLorenzo ME, Branda RF, Mann KG. Blood coagulation in hemophilia A and hemophilia C. Blood. 1998;91:4581–4592. [PubMed] [Google Scholar]
- 8.Bailey K, Bettelheim FR, Lorand L, Middlebrook WR. Action of thrombin in the clotting of fibrinogen. Nature. 1951;167:233–234. doi: 10.1038/167233a0. [DOI] [PubMed] [Google Scholar]
- 9.Davey MG, Luscher EF. Actions of thrombin and other coagulant and proteolytic enzymes on blood platelets. Nature. 1967;216:857–858. doi: 10.1038/216857a0. [DOI] [PubMed] [Google Scholar]
- 10.Lorand L. Factor XIII: structure, activation, and interactions with fibrinogen and fibrin. Ann N Y Acad Sci. 2001;936:291–311. doi: 10.1111/j.1749-6632.2001.tb03516.x. [DOI] [PubMed] [Google Scholar]
- 11.Butenas S, van 't Veer C, Mann KG. Evaluation of the initiation phase of blood coagulation using ultrasensitive assays for serine proteases. J Biol Chem. 1997;272:21527–21533. doi: 10.1074/jbc.272.34.21527. [DOI] [PubMed] [Google Scholar]
- 12.Brummel KE, Paradis SG, Butenas S, Mann KG. Thrombin functions during tissue factor-induced blood coagulation. Blood. 2002;100:148–152. doi: 10.1182/blood.v100.1.148. [DOI] [PubMed] [Google Scholar]
- 13.Orfeo T, Brummel-Ziedins KE, Gissel M, Butenas S, Mann KG. The nature of the stable blood clot procoagulant activities. J Biol Chem. 2008;283:9776–9786. doi: 10.1074/jbc.M707435200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Broze GJ, Jr, Higuchi DA. Coagulation-dependent inhibition of fibrinolysis: role of carboxypeptidase-U and the premature lysis of clots from hemophilic plasma. Blood. 1996;88:3815–3823. [PubMed] [Google Scholar]
- 15.He S, Ekman GJ, Hedner U. The effect of platelets on fibrin gel structure formed in the presence of recombinant factor VIIa in hemophilia plasma and in plasma from a patient with Glanzmann thrombasthenia. J Thromb Haemost. 2005;3:272–279. doi: 10.1111/j.1538-7836.2005.01127.x. [DOI] [PubMed] [Google Scholar]
- 16.Wolberg AS, Allen GA, Monroe DM, Hedner U, Roberts HR, Hoffman M. High dose factor VIIa improves clot structure and stability in a model of haemophilia B. Br J Haematol. 2005;131:645–655. doi: 10.1111/j.1365-2141.2005.05820.x. [DOI] [PubMed] [Google Scholar]
- 17.Hoffman M, Harger A, Lenkowski A, Hedner U, Roberts HR, Monroe DM. Cutaneous wound healing is impaired in hemophilia B. Blood. 2006;108:3053–3060. doi: 10.1182/blood-2006-05-020495. [DOI] [PubMed] [Google Scholar]
- 18.McDonald A, Hoffman M, Hedner U, Roberts HR, Monroe DM. Restoring hemostatic thrombin generation at the time of cutaneous wounding does not normalize healing in hemophilia B. J Thromb Haemost. 2007;5:1577–1583. doi: 10.1111/j.1538-7836.2007.02647.x. [DOI] [PubMed] [Google Scholar]
- 19.Nagashima M, Werner M, Wang M, Zhao L, Light DR, Pagila R, Morser J, Verhallen P. An inhibitor of activated thrombin-activatable fibrinolysis inhibitor potentiates tissue-type plasminogen activator-induced thrombolysis in a rabbit jugular vein thrombolysis model. Thromb Res. 2000;98:333–342. doi: 10.1016/s0049-3848(00)00184-5. [DOI] [PubMed] [Google Scholar]
- 20.Bouma BN, Mosnier LO, Meijers JC, Griffin JH. Factor XI dependent and independent activation of thrombin activatable fibrinolysis inhibitor (TAFI) in plasma associated with clot formation. Thromb Haemost. 1999;82:1703–1708. [PubMed] [Google Scholar]
- 21.Wettstein P, Haeberli A, Stutz M, Rohner M, Corbetta C, Gabi K, Schnider T, Korte W. Decreased factor XIII availability for thrombin and early loss of clot firmness in patients with unexplained intraoperative bleeding. Anesth Analg. 2004;99:1564–1569. doi: 10.1213/01.ANE.0000134800.46276.21. [DOI] [PubMed] [Google Scholar]
- 22.Brummel KE, Butenas S, Mann KG. An integrated study of fibrinogen during blood coagulation. J Biol Chem. 1999;274:22862–22870. doi: 10.1074/jbc.274.32.22862. [DOI] [PubMed] [Google Scholar]
- 23.Butenas S, Brummel KE, Branda RF, Paradis SG, Mann KG. Mechanism of factor VIIa-dependent coagulation in hemophilia blood. Blood. 2002;99:923–930. doi: 10.1182/blood.v99.3.923. [DOI] [PubMed] [Google Scholar]
- 24.Lawson JH, Krishnaswamy S, Butenas S, Mann KG. Extrinsic pathway proteolytic activity. Methods Enzymol. 1993;222:177–195. doi: 10.1016/0076-6879(93)22013-6. [DOI] [PubMed] [Google Scholar]
- 25.Rand MD, Lock JB, van 't Veer C, Gaffney DP, Mann KG. Blood clotting in minimally altered whole blood. Blood. 1996;88:3432–3445. [PubMed] [Google Scholar]
- 26.Butenas S, Brummel KE, Paradis SG, Mann KG. Influence of factor VIIa and phospholipids on coagulation in "acquired" hemophilia. Arterioscler Thromb Vasc Biol. 2003;23:123–129. doi: 10.1161/01.atv.0000042081.57854.a2. [DOI] [PubMed] [Google Scholar]
- 27.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 28.Carr ME, Jr, Shen LL, Hermans J. Mass-length ratio of fibrin fibers from gel permeation and light scattering. Biopolymers. 1977;16:1–15. doi: 10.1002/bip.1977.360160102. [DOI] [PubMed] [Google Scholar]
- 29.Blomback B, Carlsson K, Hessel B, Liljeborg A, Procyk R, Aslund N. Native fibrin gel networks observed by 3D microscopy, permeation and turbidity. Biochim Biophys Acta. 1989;997:96–110. doi: 10.1016/0167-4838(89)90140-4. [DOI] [PubMed] [Google Scholar]
- 30.Blomback B, Carlsson K, Fatah K, Hessel B, Procyk R. Fibrin in human plasma: gel architectures governed by rate and nature of fibrinogen activation. Thromb Res. 1994;75:521–538. doi: 10.1016/0049-3848(94)90227-5. [DOI] [PubMed] [Google Scholar]
- 31.Wolberg AS. Thrombin generation and fibrin clot structure. Blood Rev. 2007;21:131–142. doi: 10.1016/j.blre.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 32.Blomback B, Hessel B, Hogg D, Therkildsen L. A two-step fibrinogen--fibrin transition in blood coagulation. Nature. 1978;275:501–505. doi: 10.1038/275501a0. [DOI] [PubMed] [Google Scholar]
- 33.Hantgan RR, Hermans J. Assembly of fibrin. A light scattering study. J Biol Chem. 1979;254:11272–11281. [PubMed] [Google Scholar]
- 34.Hantgan R, Fowler W, Erickson H, Hermans J. Fibrin assembly: a comparison of electron microscopic and light scattering results. Thromb Haemost. 1980;44:119–124. [PubMed] [Google Scholar]
- 35.Hantgan R, McDonagh J, Hermans J. Fibrin assembly. Ann N Y Acad Sci. 1983;408:344–366. doi: 10.1111/j.1749-6632.1983.tb23256.x. [DOI] [PubMed] [Google Scholar]
- 36.Muszbek L, Yee VC, Hevessy Z. Blood coagulation factor XIII: structure and function. Thromb Res. 1999;94:271–305. doi: 10.1016/s0049-3848(99)00023-7. [DOI] [PubMed] [Google Scholar]
- 37.lowey ag, McDonagh J, Mikkola H, Teller DC, Yee VC. Structure and function of factor XIII. In: Colman RW, Hirsh J, Marder VJ, Clowes AW, George JN, editors. Hemostasis and Thrombosis. Lippincott Williams & Wilkins; 2001. pp. 233–247. [Google Scholar]
- 38.Procyk R, Blomback B. Factor XIII-induced crosslinking in solutions of fibrinogen and fibronectin. Biochim Biophys Acta. 1988;967:304–313. doi: 10.1016/0304-4165(88)90024-4. [DOI] [PubMed] [Google Scholar]
- 39.Tamaki T, Aoki N. Cross-linking of alpha 2-plasmin inhibitor to fibrin catalyzed by activated fibrin-stabilizing factor. J Biol Chem. 1982;257:14767–14772. [PubMed] [Google Scholar]
- 40.Mosher DF, Schad PE. Cross-linking of fibronectin to collagen by blood coagulation Factor XIIIa. J Clin Invest. 1979;64:781–787. doi: 10.1172/JCI109524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mui PT, Ganguly P. Cross-linking of actin and fibrin by fibrin-stabilizing factor. Am J Physiol. 1977;233:H346–H349. doi: 10.1152/ajpheart.1977.233.3.H346. [DOI] [PubMed] [Google Scholar]
- 42.Lynch GW, Slayter HS, Miller BE, McDonagh J. Characterization of thrombospondin as a substrate for factor XIII transglutaminase. J Biol Chem. 1987;262:1772–1778. [PubMed] [Google Scholar]
- 43.Hvas AM, Sorensen HT, Norengaard L, Christiansen K, Ingerslev J, Sorensen B. Tranexamic acid combined with recombinant factor VIII increases clot resistance to accelerated fibrinolysis in severe hemophilia A. J Thromb Haemost. 2007;5:2408–2414. doi: 10.1111/j.1538-7836.2007.02755.x. [DOI] [PubMed] [Google Scholar]