

Abstract
The availability of the complete genome sequences has facilitated access to essential information to identify proteins. The determination of Arabidopsis genome sequence has had a great impact to annotate data. The genome sequencing of Sorghum bicolor has been only recently completed and hither to the global response to abiotic stresses in this important crop remains largely unexplored. We used 2-D gel electrophoresis based proteomic approach refined with MALDI-TOF to analyze drought-stress response proteins in sorghum. Major changes in protein complement of sorghum were observed in hydroponic cultures at 96 hours under drought stress. Six most highly expressed proteins were excised for functional identification. Here, we developed a method to obtain functional distances between GO terms and analyzed distance values to allocate shortest path (SP) in GO hierarchy. The shortest paths for expressed proteins were noted for most informative common ancestor (MICA) terms, viz. binding, catalytic activity and primary metabolic process. We observed the expressed proteins belonged to the functional group of signal transduction mechanisms, carbohydrate transport and metabolism. These identified functions of proteins suggest a different mechanism of drought-stress tolerant in sorghum. The novel approach applied in this study may have great importance in further identifying proteins involved in abiotic and biotic stress conditions in crops.
Keywords: Proteomic analysis, 2D Gel electrophoresis, Gene Ontology, Drought-stress, Sorghum bicolor
Background
The food productivity decreases due to the effect of various abiotic stresses. Drought and salinity are the major abiotic environment stresses, which negatively affect growth and productivity of plants. Minimizing these losses and to improve and manage food productivity by developing stress tolerant crops is a major challenge globally. More than one abiotic stress can occur simultaneously; one abiotic stress decreases plant's ability to resist a second stress. Plant's responses to salt and drought stress have much in common [1]. The primary effect of drought is to generate osmotic stress, whereas salinity also induces osmotic stress by its effect on the ionic homeostasis in the plant cell. Reduction in the ability of plants to take up water under salinity quickly causes reduction in growth rate, along with a group of other metabolic changes that are identical with drought stress. Photosynthesis is among the primary process that are affected by drought and salinity which cause reduced synthesis of photosynthetic pigments [1]. These changes in the amount of photosynthetic pigments are closely associated to plant biomass yield. Besides photosynthesis, reduced water potential also disrupts membranes along with other essential physiological and biochemical processes such as respiration, ion uptake, carbohydrates and nutrient metabolism [2]. Reduced water limits CO2 fixation and reduces NADP+ regeneration in calvin cycle. These adverse conditions increase the formation of reactive oxygen species (ROS) such as H2O2 (hydrogen peroxide), O2– (superoxide) and OH (hydroxyl) radicals, through enhanced leakage of electrons to molecular oxygen. ROS is involved in the stress signal transduction pathway as a secondary messenger, however excessive ROS production causes oxidative stress, which damages plant's photosynthetic pigments, membrane lipids, proteins and nucleic acids. To keep the levels of active oxygen species under control, plants have antioxidant defense systems to protect cells from oxidative damage [3].
Sorghum bicolor is a stress-tolerant cereal crop species with considerable economic importance in drought and salinity affected arid and semiarid regions of the world. The crop is grown for food, feed and biofuels. The genome sequencing of S. bicolor has been recently completed [4]. Although, currently a large amount of data are available on gene expression in response to salt, drought and other stresses, the global response to these abiotic stresses in sorghum remains largely unexplored. The increasing availability of genomic sequences of several plants in combination with high-throughput bioinformatics tools and databases provide new opportunities for understanding role of proteins. The stress defence proteins network has been recently explained in Arabidopsis and rice using multi-parallel gene expression analysis techniques [5]. Currently, several computational approaches for functional identification of proteins such as sequence similarity, phylogenetic profiles, protein-protein interaction (PPI) and gene expression are available [6]. Protein-protein interaction method is based on the assumption that interacting proteins usually share same function [7]. In this paper, we report Gene Ontology term's functional distances analysis method to identify six expressed drought responsive proteins in sorghum. Gene Ontology (GO) term's semantic similarity provides functional relationship between proteins. The semantic similarity between two proteins is usually calculated based on the GO terms similarity [8]. To identify semantic similarity between GO terms, several methods have been reported earlier, viz. Resnik's, Lin'S and JiangConrath's [9]. Gene Ontology is a structured and controlled vocabulary, which characterizes the functional annotation of proteins. Gene Ontology is composed of three independent ontologies: biological process (BP), molecular function (MF), and cellular component (CC). In directed acyclic graph (DAG) the GO terms are structured as ‘isa’ and ‘part-of’ relationships in GO database [10].
The Gramene plant genome database [11] (Release 34b) includes a wealth of information of proteins from Arabidopsis, rice and other plant species. However, only little information in sorghum has been proposed till date. In the present study, 2-DE based proteomic technique with MALDI-TOF was used to separate and identify six highly expressed proteins in sorghum. The drought (no water supply) until 96 hours to investigate cellular responses was imposed in 7 d old sorghum plants. To identify drought responsive six expressed proteins, we proposed a new method to identify proteins using functional distances between GO terms.
Methodology
Plant material:
Seeds of Sorghum bicolor (L.) genotype csv-17 were used as plant material and grown hydroponically in Hoagland's solution. After 7 days of germination, the drought stress (no water supply) was imposed on germinated seeds and leaves.
Two-dimensional (2-DE) gel electrophoresis:
The method Mechin et al. [12] was followed for the extraction of proteins from the plant sample. Protein samples were purified using 2D-cleanup kit (Bio-Rad) and the protein pellet was finally resuspended in sample rehydration buffer (8M urea, 2% CHAPS, 15mM DTT and 0.5% IPG buffer pH 4–7). Protein spots were visualized by staining with coomassie brilliant blue G-250. Gel images were captured by GS800 densitometer (Bio-Rad, USA).
Protein spots identification by mass spectrometry:
The gel spots were washed with proteomic grade de-ionized water and proteins were identified by using MALDI-TOF mass spectrometer (Ultraflex III, Bruker Daltonics, Germany). The expressed proteins were analyzed using mascot sequence matching software (http://www.matrixscience.com) using MSDB in the taxonomy group of green plants. While performing mascot the search parameters were: maximum of one missed cleavage by trypsin, fixed modification of oxidation, charged state of +1, peptide mass tolerance of 50 ppm, and fragment mass tolerance of ±1.0 Da.
Data identification and network illustration:
A total of six expressed proteins in sorghum were identified to be common with from closely related in Oryza sativa proteins by homology comparison. Homology search was carried out using blastp at Uniprot database (http://www.uniprot.org/blast/) with the following parameters: Database-UniprotKB; Threshold-0.1; Matrix-blosum-62. The molecular weights of expressed proteins were determined using the Compute pI/Mw search tool at expasy server (http://web.expasy.org/compute_pi). The COGnitor program (http://www.ncbi.nlm.nih.gov/COG/old/xognitor) was utilized for functional annotation of proteins. Protein families were obtained using InterProScan search tool (http://www.ebi.ac.uk/Tools/pfa/iprscan) against hidden markov model (HMM) Pfam database [13]. The GO-terms were obtained using Gene Ontology database that cross linked at Uniprot database. We used GO hierarchies of Arabidosis thaliana to obtain GO directed acyclic graphs (DAG). This graph was explained using cytoscape 2.8.1 version [14].
Functional distance algorithm:
We proposed an algorithm to obtain functional distances using sum of distance values between GO terms from root to its associated terms in GO hierarchy. The ProteInOn tool (http://xldb.di.fc.ul.pt/tools/proteinon/) was utilized to calculate the semantic similarities with JiangConrath's measurement without ignore inferred from electronic annotation (IEA). The shortest path was confirmed by analysis of distance values in their corresponding paths. The smallest distance value of analyzed path was referred as shortest path. The shortest path algorithm is given as follows: D1 = 100 - SS (t1, t 2) → (1) d = arctan = PathA[D1 + Dn] × π → (2) 2 Where, D1 is the functional distance between GO term's (t1, t2) and SS is semantic similarity. The pathA is the path of GO ontologies that connected to the root term. The total distance (d) to identify shortest path was calculated by summing of functional distance values between GO terms.
Discussion
Sorghum seedlings were grown hydroponically and drought stress imposed on 7 d old germinated seeds. The drought stress induced chlorosis and reduced growth of leaves. The MALDITOF/ MS analysis identified expression of six drought inducible proteins as compared to corresponding control with water in sorghum seedlings (Figure 1).
Figure 1.
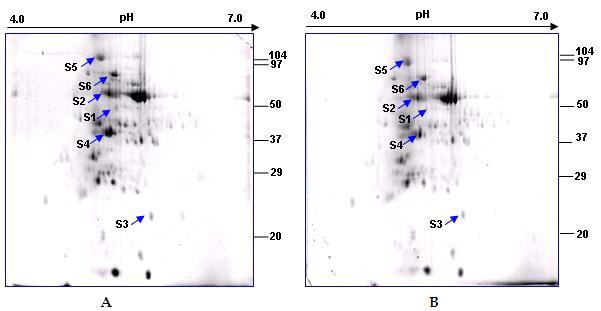
2-DE gel showing spots differentially expressed (>1.5- fold difference) in leaves from Sorghum bicolor plants grown in drought stress (no water supply) (A) as compared to the control; (B) the identified spots are marked with arrows.
The whole genome sequencing of Sorghum bicolor has been recently completed [4] and till date no comparable information on drought inducible proteins from this crop is available. Hence, the identified homologues in sorghum were described as hypothetical proteins. Here, very effective bioinformatics approaches were applied to put functional context of expressed drought responsive proteins Table 1 (see supplementary material). The COGnitor program based results revealed that differentially expressed proteins belonged to functional categories of signal transduction mechanisms (spot S1; S2) and, carbohydrate transport and metabolism (spot S5). Among the expressed proteins, cupin1 (spot S3), glycoside hydrolase (spot S4) and raffinose synthase (spot S6) did not match with any COG record. Hence, the families of all six expressed proteins were identified Table 1 (see supplementary material).
In this study, we have developed an effective method to identify proteins using functional distances between hierarchical structured GO terms. The shortest path was identified on the bases of the analysis of distance values of their corresponding paths. In the results the smallest distance value has been referred as shortest path Table 2 (see supplementary material). While finding shortest paths for assigned GO terms of six expressed proteins, the most informative common ancestor (MICA) terms were noted. The identified MICA terms were facilitated to discover functional annotation of drought stress responsive proteins. The shortest path method developed in this study was applied on hierarchical structured GO terms in molecular function, biological process and cellular component. The GO terms (n1-n8) were associated with root term molecular function (GO: 0003674) (Figure 2). We identified shortest paths for these terms using sum of term's semantic similarity. The distance score 13.4 was identified for (n1) term by the following calculation: arctan=1.3+3.6+7.9+0.6; distance (d) = arctan×π/2. This value has been referred as shortest path for (n1) GO-term. The same calculation was performed to find shortest path for other GO-terms that associated with the root term, molecular function Table 2 (see supplementary material). While finding the shortest paths, MICA [(M1) “catalytic activity” (GO: 0003824)] was noted for terms [(n1) hydrolase activity, hydrolyzing O-glycosyl compounds (GO: 0004553) (spot S4)], [(n2) galactinol-sucrose galactosyltransferase activity (GO: 0047274) (spot S6)], [(n3) protein serine/threonine kinase activity (GO: 0004674) (spot S1; S2)]. Several functional studies have revealed that signaling pathways leading to biotic and abiotic stress resistance are mediated by calcium-dependent protein kinases (CDPKs) [15]. Previously, the expression of glycoside hydrolase proteins under drought stress has been reported in Arabidopsis and rice [16]. These proteins are involved in the metabolism of various carbohydrate containing compounds and play other major functional roles in the plants.
Figure 2.

GO hierarchy network of highly expressed proteins under drought stress. In the graph M1, M2 and M3 are MICA terms and (n1-n11) are GO terms of expressed proteins.
The second MICA [(M2) “binding” (GO: 0005488)] was noted for terms [(n4) manganese ion binding (GO: 0030145) (spot S3)], [(n5) cation binding (GO: 0043169) (spot S4], [(n6) calcium ion binding (GO: 0005509) (spot S1)] and [(n7) ATP binding (GO: 0005524) (spot S1; S2]. The MICA term “binding” and its associated terms are involved in numerous functions including in electron carriers, maintenance of charge balance, and enzyme activation. These binding processes play important role in minimizing the adverse effects of abiotic stresses in crop plants [17]. Thus it may be concluded that spots (S1, S2, S3, S4, and S6) were strongly associated with proteins that are involved in binding and catalytic activity process.
The GO hierarchy of root term biological process (GO: 0008150) was associated with GO-terms [(n9) carbohydrate metabolic process (GO: 0005975) (spots S4, S6)] and [(n10) trehalose biosynthetic process (GO: 0005992) (spot S5)] (Figure 2). To identify shortest path from root to its associated GO-terms, we have proposed a method based on algorithm described in methodology. Consequently, we noted the MICA [(M3) “primary metabolic process” (GO: 0044238)] was noted for its GO-terms. The GO-terms (n9) and (n10) share a same hierarchy path (Figure 2). Previously it has been reported that the several carbohydrates viz., sucrose, glucose, fructose and starch accumulate under salt stress and play a major role in osmotic adjustment, carbon storage and radical scavenging [18]. Earlier studies have evaluated that proline, spermine, sucrose, mannitol, and fructans increase during drought stress [19]. In the cellular component root term (GO: 0005575) did not have any significant MICA for its associated GO term [(n11) apoplast (GO: 0048046)]. The apoplast is involved in numerous processes of plants, such as development, nutrition, signalling, detoxification and defense [20].
Conclusion
It can be concluded that the approach applied in this study can be used to identify proteins. We described isolation and characterization of six drought stress responsive expressed in sorghum using 2DE-MS proteomic approaches. We developed a new method to obtain functional distances between GO terms and the analysis of distance values to allocate the shortest path (SP) in GO hierarchy. Further, we observed that identified proteins belonged to functional categories of signal transduction mechanisms. These identified proteins may possibly lead to a distinct mechanism of drought-stress adaptation in sorghum. The approach applied in this study may have great importance in further identifying proteins involved in abiotic and biotic stress conditions in crops.
Supplementary material
Acknowledgments
The authors gratefully acknowledge liberal use of the facilities of the DBT supported Centre of Bioinformatics at Banasthali University, India.
Footnotes
Citation:Sharma et al, Bioinformation 8(13): 595-599 (2012)
References
- 1.R Munns, et al. Plant Cell Environ. 2002;25:239. doi: 10.1046/j.0016-8025.2001.00808.x. [DOI] [PubMed] [Google Scholar]
- 2.CA Jaleel, et al. C R Biol. 2008;331:272. [Google Scholar]
- 3.R Mittler. Trends Plant Sci. 2002;7:405. doi: 10.1016/s1360-1385(02)02312-9. [DOI] [PubMed] [Google Scholar]
- 4.AH Paterson, et al. Nature. 2009;457:551. [Google Scholar]
- 5.S Amme, et al. J Exp Bot. 2006;57:1537. doi: 10.1093/jxb/erj129. [DOI] [PubMed] [Google Scholar]
- 6.M Pellegrini. Curr Opin Chem Biol. 2001;5:46. doi: 10.1016/s1367-5931(00)00165-4. [DOI] [PubMed] [Google Scholar]
- 7.U Karaoz, et al. Proc Natl Acad Sci USA. 2004;101:2888. [Google Scholar]
- 8.S Jain, GD Bader. BMC Bioinformatics. 2010;11:562. doi: 10.1186/1471-2105-11-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.G Yu, et al. Bioinformatics. 2010;26:976. doi: 10.1093/bioinformatics/btq064. [DOI] [PubMed] [Google Scholar]
- 10.MA Harris, et al. Nucleic Acids Res. 2004;32:D258. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.P Jaiswal. Methods Mol Biol. 2011;678:247. doi: 10.1007/978-1-60761-682-5_18. [DOI] [PubMed] [Google Scholar]
- 12.V Mechin, et al. Methods Mol Biol. 2007;355:1. doi: 10.1385/1-59745-227-0:1. [DOI] [PubMed] [Google Scholar]
- 13.A Bateman, et al. Nucleic Acids Res. 2004;32:D138. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.P Shannon, et al. Genome Res. 2003;13:2498. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.AA Ludwig, et al. J Exp Bot. 2004;55:181. doi: 10.1093/jxb/erh008. [DOI] [PubMed] [Google Scholar]
- 16.Z Minic. Planta. 2008;227:723. doi: 10.1007/s00425-007-0668-y. [DOI] [PubMed] [Google Scholar]
- 17.S Mahajan, N Tuteja. Arch Biochem Biophys. 2005;444:139. doi: 10.1016/j.abb.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 18.YG Yin, et al. J Exp Bot. 2010;61:563. doi: 10.1093/jxb/erp333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.J Krasensky, C Jonak. J Exp Bot. 2012;63:1593. doi: 10.1093/jxb/err460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.V Dani, et al. Proteomics. 2005;5:737. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.