

Abstract
Nonsteroidal anti-inflammatory drugs (NSAIDs) exert their pharmacological activities by inhibiting cyclooxygenase (COX)-1 and COX-2. Previous studies have shown that esters and amides of non-selective inhibitors such as indomethacin are selective against COX-2, which is the therapeutically relevant isoform. Structure-activity analysis indicates that substituted phenyl rings are tolerated as ester components. In the present study, the introduction of inorganic ortho- and meta-carbaborane moieties was explored with the aim to create COX-2 inhibitors and more importantly to investigate the validity of using these boron clusters as drug entities. Interestingly, only the ortho-carbaborane ester was active whereas the meta isomer was not. A similar lack of inhibitory potency was observed when an adamantyl substituent or alkylene spacers at the carbaborane were introduced in the ester functionality.
Keywords: Carborane, Carbaborane, Cyclooxygenase, Indomethacin, Nonsteroidal anti-inflammatory drugs
1. Introduction
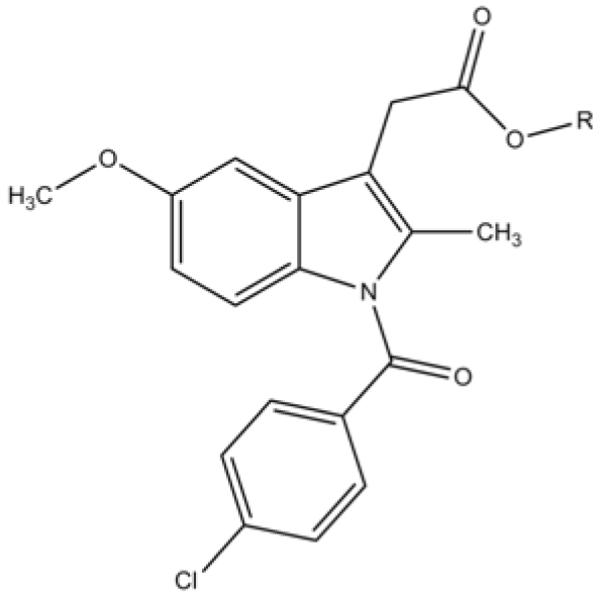
Indomethacin 2-{1-[(4-chlorophenyl)carbonyl]-5-methoxy-2-methyl-1H-indol-3-yl}acetic acid (Fig. 1) is one of the most extensively studied nonsteroidal anti-inflammatory drugs (NSAIDs). It was discovered in 1963 and was developed as an analgesic and anti-inflammatory agent despite the fact that its pharmaceutical target was unknown.1-2
Figure 1.

Indomethacin with acetic acid moiety highlighted.
The detailed mode of action became clear with the discovery of the cyclooxygenase (COX) as target enzyme.3-4 Intense studies uncovered indomethacin as slow, time-dependent COX inhibitor which blocks prostaglandin synthesis.5 It shows a preference for COX-1, the housekeeping COX isozyme, compared to the inducible form of the enzyme, COX-2 (IC50 (COX-1)/IC50 (COX-2) = 0.07).5-6 Inhibition of the latter is responsible for a significant portion of the therapeutic effects of indomethacin, because COX-2 is induced in inflammatory cells, cerebrovascular endothelial cells, and tumor cells and, therefore, is the therapeutically relevant isoform.7-9 The added inhibition of COX-1, particularly in the stomach, contributes to its gastrointestinal side effects.9 Subsequent to the approval and marketing of COX-2-selective inhibitors, cardiovascular side effects emerged with rofecoxib and celecoxib that have been detected with most major classes of selective and non-selective NSAIDs.10-11 This experience illustrates the complexity of developing inhibitors against targets that contribute to a diverse array of physiological responses.
COX-2-selective inhibitors still remain attractive targets because of their reduced gastrointestinal side effects. One strategy for the development of COX-2-selective inhibitors is the conversion of carboxylic acid-containing NSAIDs, such as indomethacin, into neutral ester or amide derivatives.6, 12 This research is therefore aimed to use the inorganic carbaboranes as novel ester entities and to validate the boron cluster isomers as pharmacophores.13-16 Additionally, comparison to the organic analogues phenyl and adamantyl is aimed to rank the utility of the clusters for the use in medicinal chemistry. Carbaboranes are icosahedral cages of ten BH and two CH vertices with the approximate size of a rotating benzene ring or adamantane (Fig. 2).17
Figure 2.

ortho-, meta-, para-Carbaborane between benzene and adamantane.
The CH vertices can be organized either in ortho, meta, or para fashion.17 Both the boron and the carbon atoms can be furnished with various substituents and allow for the integration of the cluster into a myriad of compounds.18 The size of the clusters is virtually the same for all isomers, the intrinsic properties, however, differ.19
In terms of stability and reactivity, meta- and para-carbaborane are very similar. The ortho-isomer stands out as a result of the highest electron deficiency.19 Carbaborane chemistry has already been widely explored.18, 20-29 Thus, various carbaborane-modified alcohols have been prepared and render the synthesis of the target ester compounds easily possible.30-33
2. Results and discussion
2.1. Chemistry
2.1.1. Synthesis of the carbaboranyl alcohols
The carbaboranyl alcohols were prepared from the unsubstituted carbaborane clusters (Scheme 1). The ortho and meta isomers were selected as the initial synthetic targets: the para cluster was omitted because of its similarity to the meta isomer and its high price. The carbaboranes were either directly attached to the indomethacin acid function or separated by CH2 spacers to study the impact of the cluster on the acid group.
Scheme 1.

Reaction scheme to obtain the carbaboranyl alcohols with key reagents drawn. The detailed reagents were used as follows. a) n-BuLi, B(OMe)3; b) CH3COOH/H2O2;32 c) n-BuLi, (CHO)n;34 d) 1.) n-BuLi, TBDMS-Cl, 2.) n-BuLi, C3H6O, 3.) Bu4NF.35
The first step to modify the carbaborane carbon atom is generally the removal of the CH proton using a base. Most commonly n-BuLi is applied. For the ortho isomer the lithium base can be substituted for TBAF in some cases.36
The formation of a boronic ester turned out to be beneficial to obtain the hydroxycarbaboranes (2) easily and in high yields.32 Hydroxycarbaboranes (2) were synthesized in analogy to a procedure described in the literature.32 Purification, however, was slightly modified by replacing chromatography with simple extraction. This was particularly useful for the ortho isomer (2o), which eluated as a broad band on silica. Extraction was possible, because the alcohol could be transferred as an anion into an aqueous base-solution. This aqueous layer could easily be separated from the unreacted starting material, which remained in the organic layer. This procedure allowed us to obtain the hydroxylated product and recover unsubstituted carbaborane, both in high purity. Carbaboranylmethanol (3) was obtained using paraformaldehyde after deprotonation.34, 36 In order to prepare the carbaboranylpropanols (4) the previous protection, normally via silylation, of the second CH group is recommended to suppress the formation of the di-alcohol.35, 37 Introduction of propanol at the unprotected cluster carbon atom could easily be achieved using the ring opening reaction of oxetane.35, 37
2.1.2. Synthesis of the carbaboranyl-indomethacin esters
The synthesis of the carbaboranyl-indomethacin ester (5-7) was carried out using the established method via carboxylic acid activation by N,N’-bis(2-oxo-3-oxazolidinyl)phosphonic chloride (BOP-Cl) (Scheme 2).38
Scheme 2.

Synthesis of the indomethacin-carbaboranyl esters. a) BOP-Cl, NEt3.
Abandonment of aqueous workup was found to facilitate the reaction and additionally increased the yields. The formation of 5o was most difficult. The electron-withdrawing ortho-carbaborane was expected to reduce the nucleophilicity of the alcohol oxygen atom, directly attached to the cluster. This effect decreased the yield of the ester formation. Introduction of a methylene spacer consequently diminished the adverse influence of the cluster and increased the yields. Nevertheless, 5o was prepared in quantities sufficient for growing crystals suitable for X-ray structure analysis (Fig. 3).
Figure 3.

ORTEP of 5o with selected atoms labeled, thermal ellipsoids are drawn at 50% probability.
2.2. COX-inhibition studies
The carbaborane esters were first screened for COX-1 and COX-2 inhibition at 25 μM concentration in a standard assay system that measures the ability of compounds to inhibit the conversion of [14C]-arachidonic acid to [14C]-prostaglandins (Fig. 4).
Figure 4.

Ovine COX-1 and murine COX-2 inhibition studies of compounds 5-7, and 9 at 25 μM concentration.
5o showed by far the best COX-inhibition and inhibited both COX-1 and COX-2 as does indomethacin. A full dose response determination for 5o gave similar IC50 values of 2.6 μM for COX-1 and 4.2 μM for COX-2 (Table 1).
Table 1.
IC50 values of phenyl, carbaboranyl and adamantyl esters in comparison to indomethacin.
| |||
|---|---|---|---|
| R | IC50 (μM) | ||
| COX-1 | COX-2 | ||
| H | (1) | 0.05* | 0.75* |
| o-C2B10H11 | (5o) | 2.60 | 4.20 |
| m-C2B10H11 | (5m) | >25 | >25 |
| C10H15 | (9) | >25 | >25 |
| C6H5 | (10) | >25* | 0.40* |
already reported12
All other compounds revealed only poor COX inhibition in concentrations as high as 25 μM. Compounds 6 and 7 revealed almost the same extent of inhibition. The size of the spacer is therefore of subordinate importance. The same inhibition pattern was also obtained for 5m and the adamantyl ester 9. meta-Carbaborane in general and ortho-carbaborane connected via spacer to the acid function are consequently not suitable to modify indomethacin.
Indomethacin itself revealed IC50 values of 0.05 μM for COX-1 and 0.75 μM for COX-2.6, 12 Esterification with ortho-carbaborane reduced the clear COX-1 selectivity of indomethacin, but also decreased activity.
We evaluated the possibility that 5o inhibits COX enzymes by being hydrolyzed to indomethacin, which we considered unlikely because of the different ratio of COX-1-to-COX-2 inhibition exhibited by 5o. NMR stability measurements in wet methanol revealed that 5o is stable in this solvent for the assay timescale. These two facts indicate that the inhibitory activity of 5o can be attributed to the ester and hydrolysis to indomethacin is, if ever, of minor importance. Comparison of the carbaboranyl esters (5o and 5m) to the corresponding adamantyl (9) and phenyl ester (10) revealed that the ortho isomer behaves more like the phenyl ring whereas the meta isomer is closer to adamantyl in terms of COX inhibition in these special cases. The phenyl ester showed the best COX-2 inhibition while being inactive against COX-1.12
3. Conclusion
A series of indomethacin esters was constructed with the inorganic ortho- and meta-carbaborane clusters. 5o, with the ortho isomer directly attached to the acid function, inhibited COX in the low micromolar range, but with a clearly reduced COX-1 selectivity. All other esters were generally less active. This illustrates the exceptional position of the ortho isomer. Comparison to the carbon analogues showed that the ortho-carbaborane substituent resembles more a phenyl substituent, whereas the meta-carbaborane substituent acted more like an adamantyl substituent with respect to the compounds studied in the COX inhibition assays.
4. Experimental
4.1. Chemistry
4.1.1 General
All reactions were carried out under nitrogen atmosphere by using standard Schlenk techniques. The solvents were purified by a Solvent Purification System SPS-800 Series. 3 and 4 were synthesized according to the literature.34-35 All chemicals were used as purchased. Flash chromatography was carried out on Merck SilicaGel 60 (0.035-0.070 mm). Merck Silica 60 F254 was used for thin-layer chromatography (TLC). The TLC plates were developed with palladium(II) chloride methanol solution. The infrared spectra were recorded on a Perkin-Elmer System 2000 FT-IR spectrometer using a KBr disk. The 1H, 13C, and 11B NMR spectra were recorded on an AVANCE DRX 400 spectrometer (Bruker). The chemical shifts for the 1H, 13C, and 11B NMR spectra are reported in parts per million (ppm) at 400.13, 100.63, and 128.38 M Hz, respectively, with tetramethylsilane as standard for the first two and BF3(OEt2) as external standard for last-named. Proton-coupled 13C NMR spectra were recorded for carbaborane-containing compounds. The number of boron atoms and the 1JBH could not always be determined unambiguously, due to the broad and overlapping signals. The elemental analyses were recorded on a VARIO EL (Heraeus). The melting points were determined in capillaries (GALLENKAMP) and represent uncorrected values. The crystals for molecular structure determination were grown at room temperature. The crystallographic data of 5o were collected on a CCD Oxford Xcalibur S diffractometer (λ(Mo-Kα) = 0.71073 Å) in ω and Φ scan mode. Semi-empirical from equivalents absorption corrections were carried out with SCALE3 ABSPACK and the structures were solved with direct methods.39-40 Structure refinement was carried out with SHELXL-97.41 All non-hydrogen atoms were refined anisotropically, all H atoms were located on difference Fourier maps and refined freely. CCDC 803538 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
4.1.2. 1-Hydroxy-1,2-dicarba-closo-dodecaborane(12) (2o) and 1-Hydroxy-1,7-dicarba-closo-dodecaborane(12) (2m)
The synthesis was carried out as reported.32 The crude product was dissolved in diethyl ether and extracted six times with an equal amount of aqueous NaOH (1 M). The combined aqueous layers were immediately acidified with concentrated HCl to pH = 1. Unreacted carbaborane was recovered from the ether layer after evaporation of the solvent. The product was extracted with four portions of diethyl ether while pH = 1 was maintained. The combined organic layers were dried (MgSO4) and the ether was removed under reduced pressure. Residual traces of solvent were removed via sublimation. The yields resemble those reported.
4.1.3. General procedure to obtain the esters 5-7 and 9
Indomethacin (1 eq.) and BOP-Cl (1 eq.) were suspended in dichloromethane (20 mL/g acid), NEt3 (2 eq.) was slowly added and stirred for 10 min. The corresponding alcohol (1.3 eq., 4 eq. in the case of 8) was added and stirred for at least 12 h. The solvent was evaporated and the solid was purified by column chromatography using different solvent mixtures described as follows. The solvent was removed under reduced pressure to yield the corresponding ester, all as slightly yellow solids.
2-{1-[(4-Chlorophenyl)carbonyl]-5-methoxy-2-methyl-1H-indol-3-yl}acetic acid 1-(1,2-dicarba-closo-dodecaboranyl) ester (5o)
Solvent for purification: Hexanes (80-110 °C)/ethyl acetate 3:1; Yield: 0.20 g (29% from 0.50 g (1)); elemental analysis calcd (%) for C21B10H26O4NCl: C 50.45; H 5.24; found: C 50.53 ; H 5.24 ; mp.: 155-156 °C; ESI MS (+) (CH3COCH3/Na+): m/z: 523.2 (100%, [M+Na]+); 1H NMR (CDCl3, 25 °C, ppm): 7.65 (d, 3JHH = 8 Hz, 2H, CHphenyl), 7.49 (d, 3JHH = 8 Hz, 2H, CHphenyl), 6.85 (d, 3JHH = 8 Hz, 1H, CHindole), 6.84 (d, 4JHH = 2 Hz, 1H, CHindole), 6.70 (dd, 3JHH = 8 Hz, 4JHH = 2 Hz, 1H, CHindole), 4.70 (s, 1H, CclusterH), 3.85 (s, 3H, OCH3), 3.68 (s, 2H, CH2), 3.40 - 1.10 (m, vbr, 10H, C2B10H10), 2.38 (s, 3H, CH3); 11B NMR (CDCl3, 25 °C, ppm): −4.3 (d, 1JBH = 154 Hz, 1B, C2B10H10), −8.9 (d, 1JBH = 141 Hz, 1B, C2B10H10), −11.9 (d, 1JBH = 141 Hz, 6B, C2B10H10), −14.7 (d, 1JBH = 167 Hz, 2B, C2B10H10); 13C NMR (CDCl3, 25 °C, ppm): 168.2 (s, vbr, CO), 166.2 (s, vbr, CO), 156.2 (s, vbr, CindoleOCH3), 139.7 (s, vbr, Cphenyl), 136.6 (s, vbr, Cindole), 133.5 (s, vbr, Cindole), 131.2 (dd, 1JCH = 171 Hz, 2JCH = 7 Hz, CphenylH), 130.7 (s, vbr, Cphenyl), 129.7 (s, vbr, Cindole), 129.2 (dd, 1JCH = 171 Hz, 2JCH = 7 Hz, CphenylH), 115.1 (d, 1JCH = 161 Hz, CindoleH), 112.1 (dd, 1JCH = 161 Hz, 2JCH = 5 Hz, CindoleH), 109.9 (s, vbr, Cindole), 100.7 (dd, 1JCH = 161 Hz, 2JCH = 5 Hz, CindoleH), 93.3 (s, CclusterO), 60.1 (d, 1JCH = 201 Hz, CclusterH), 55.7 (q, 1JCH = 141 Hz, CH3O), 30.6 (t, 1JCH = 131 Hz, CH2), 13.2 (q, 1JCH = 131 Hz, CH3); IR (KBr, cm−1): (w, ν(Ccluster–H), 2931 (w), 2590 (s, ν(B–H)), 1793 (w, ν(C=O)), 1679 (m, ν(C=O)), 1609 (w), 1479 (s), 1457 (w), 1401 (w), 1357 (m), 1326 (s), 1225 (m), 1152 (w), 1091 (s), 1067 (s), 1036 (w), 1015 (w), 925 (w), 834 (w), 755 (w), 729 (w), 690 (w), 561 (w), 481 (w).
Structural data for 5o obtained from n-pentane: C21H26B10ClNO4, Mr = 499.98, triclinic, space group P , a = 779.63(2), b = 1360.07(5), c = 1398.83(5) pm, α = 116.465(4), β = 97.200(3), γ = 99.626(3) °, T = 130 K, V = 1.27512(7) nm3, Z = 2, ρcalcd 1.302 Mg/m3 μ = 0.181 mm−1, 2.83 ≤ Θ ≤ 32.26°, R = 0.0505, wR = 0.1459, GOF = 1.069.
2-{1-[(4-Chlorophenyl)carbonyl]-5-methoxy-2-methyl-1H-indol-3-yl}acetic acid 1-(1,7-dicarba-closo-dodecaboranyl) ester (5m)
Solvent for purification: Hexanes (80-110 °C)/methanol/ethyl acetate 8:2:1. Yield: 0.63 g (90% from 0.50 g (1)); elemental analysis calcd (%) for C21B10H26O4NCl: C 50.45; H 5.24; found: C 49.97; H 5.20; mp.: 113-115 °C; ESI MS (+) (CH3COCH3/Na+): m/z: 523.2 (100%, [M+Na]+), 539.0 (35%, [M+K]+); 1H NMR (CDCl3, 25 °C, ppm): 7.65 (d, 3JHH = 8 Hz, 2H, CHphenyl), 7.47 (d, 3JHH = 8 Hz, 2H, CHphenyl), 6.88 (d, 3JHH = 8 Hz, 1H, CHindole), 6.85 (d, 4JHH = 2 Hz, 1H, CHindole), 6.69 (dd, 3JHH = 8 Hz, 4JHH = 2 Hz, 1H, CHindole), 3.85 (s, 3H, OCH3), 3.61 (s, 2H, CH2), 3.45 - 1.25 (m, vbr, 10H, C2B10H10), 2.84 (s, 1H, CclusterH), 2.35 (s, 3H, CH3); 11B NMR (CDCl3, 25 °C, ppm): −4.6 (d, 1JBH = 154 Hz, 1B, C2B10H10), −11.8 (d, 1JBH = 167 Hz, 2B, C2B10H10), −13.1 (d, 1JBH = 167 Hz, 3B, C2B10H10), −16.0 (d, 1JBH = 167 Hz, 4B, C2B10H10); 13C NMR (CDCl3, 25 °C, ppm): 168.2 (s, vbr, CO), 166.2 (s, vbr, CO), 156.1 (s, vbr, CindoleOCH3), 139.5 (s, vbr, Cphenyl), 136.3 (s, vbr, Cindole), 133.6 (s, vbr, Cindole), 131.2 (dd, 1JCH = 171 Hz, 2JCH = 5 Hz, CphenylH), 130.7 (s, vbr, Cphenyl), 130.0 (s, vbr, Cindole), 129.2 (dd, 1JCH = 171 Hz, 2JCH = 5 Hz, CphenylH), 115.0 (d, 1JCH = 161 Hz, CindoleH), 112.1 (d, vbr, 1JCH = 161 Hz, CindoleH), 110.8 (s, vbr, Cindole), 100.8 (d, vbr, 1JCH = 161 Hz, CindoleH), 96.7 (s, vbr, CclusterO), 55.7 (q, 1JCH = 141 Hz, OCH3), 51.6 (d, 1JCH = 181 Hz, CclusterH), 30.4 (t, 1JCH = 131 Hz, CH2), 13.3 (q, 1JCH = 131 Hz, CH3); IR (KBr, cm−1): (w, ν(Ccluster–H), 2931 (w), 2608 (s, ν(B–H)), 2057 (w), 1785 (m, ν(C=O)), 1682 (s, ν(C=O)), 1623 (s), 1479 (s), 1400 (w), 1361 (m), 1321 (s), 1242 (m), 1224 (m), 1184 (m), 1155 (w), 1112 (s), 1068 (m), 1037 (w), 1014 (m), 927 (w), 832 (w), 803 (w), 755 (w), 732 (w), 690 (w), 561 (w), 482 (w), 418 (w).
2-{1-[(4-Chlorophenyl)carbonyl]-5-methoxy-2-methyl-1H-indol-3-yl}acetic acid 1-(1,2-dicarba-closo-dodecaboranyl)methyl ester (6o)
Solvent for purification: Toluene/ethyl acetate 7:2; Yield: 0.45 g (62% from 0.50 g (1)); elemental analysis calcd (%) for C22B10H28O4NCl: C 51.41, H 5.49; found: C 51.76, H 5.53; mp.: 53-55 °C; ESI MS (+) (CH3COCH3): m/z: 536.0 (100%, [M+Na]+); 1H NMR (CDCl3, 25 °C, ppm): 7.67 (d, 3JHH = 8 Hz, 2H, CHphenyl), 7.49 (d, 3JHH = 8 Hz, 2H, CHphenyl), 6.90 (d, 4JHH = 2 Hz, 1H, CHindole), 6.84 (d, 3JHH = 8 Hz, 1H, CHindole), 6.69 (dd, 3JHH = 8 Hz, 4JHH = 2 Hz, 1H, CHindole), 4.55 (s, 2H, OCH2), 3.84 (s, 3H, OCH3), 3.73 (s, 2H, CH2), 3.53 (s, 1H, CclusterH), 2.95 - 1.25 (m, vbr, 10H, C2B10H10), 2.42 (s, 3H, CH3); 11B NMR (CDCl3, 25 °C, ppm): −2.0 (d, 1JBH = 154 Hz, 1B, C2B10H10), −4.3 (d, 1JBH = 154 Hz, 1B, C2B10H10), −9.3 (d, 1JBH = 154 Hz, 2B, C2B10H10), −11.6 (d, 1JBH = 154 Hz, 2B, C2B10H10), −13.1 (d, JBH = 167 Hz, 4B, C2B10H10); 13C NMR (CDCl3, 25 °C, ppm): 169.4 (s, vbr, CO), 168.2 (s, vbr, CO), 156.2 (s, vbr, CindoleOCH3), 139.6 (s, vbr, Cphenyl), 136.3 (s, vbr, Cindole), 133.5 (s, vbr, Cindole), 131.2 (dd, 1JCH = 161 Hz, 2JCH = 6 Hz, CphenylH), 130.8 (s, vbr, Cphenyl), 130.1 (s, vbr, Cindole), 129.2 (dd, 1JCH = 161 Hz, 2JCH = 6 Hz, CphenylH), 115.2 (d, 1JCH = 161 Hz, CindoleH), 111.8 (dd, 1JCH = 161 Hz, 2JCH = 6 Hz, CindoleH), 111.1 (s, vbr, Cindole), 101.0 (dd, 1JCH = 161 Hz, 2JCH = 6 Hz, CindoleH), 71.5 (s, Ccluster), 64.7 (t, 1JCH = 161 Hz, OCH2), 59.2 (d, 1JCH = 191 Hz, CclusterH), 55.7 (q, 1JCH = 141 Hz, CH3O), 29.9 (t, 1JCH = 131 Hz, CH2), 13.2 (q, 1JCH = 131 Hz, CH3); IR (KBr, cm−1): (w, ν(Ccluster–H)), 2929 (w), 2592 (s, ν(B–H)), 1751 (s, ν(C=O)), 1682 (s, ν(C=O)), 1594 (m), 1479 (s), 1400 (w), 1358 (m), 1321 (s), 1221 (s), 1135 (s), 1089 (m), 1067 (m), 1034 (m), 924 (w), 832 (w), 754 (w), 724 (w), 481 (w).
2-{1-[(4-Chlorophenyl)carbonyl]-5-methoxy-2-methyl-1H-indol-3-yl}acetic acid 1-(1,7-dicarba-closo-dodecaboranyl)methyl ester (6m)
Solvent for purification: Hexanes (80-110 °C)/ethyl acetate 6:1. Yield: 0.15 g (63% from 0.16 g (1)) elemental analysis calcd (%) for C22B10H28O4NCl: C 51.41, H 5.49; found: C 51.90, H 5.54; mp.: 101-102 °C; ESI MS (+) (CH3COCH3): m/z: 536.1 (100%, [M+Na]+), 553.0 (74%, [M+K]+); 1H NMR (CDCl3, 25 °C, ppm): 7.67 (d, 3JHH = 8 Hz, 2H, CHphenyl), 7.47 (d, 3JHH = 8 Hz, 2H, CHphenyl), 6.93 (d, 4JHH = 2 Hz, 1H, CHindole), 6.85 (d, 3JHH = 8 Hz, 1H, CHindole), 6.68 3 (dd, 3JHH = 8 Hz, 4JHH = 2 Hz, 1H, CHindole), 4.30 (s, 2H, OCH2), 3.85 (s, 3H, OCH3), 3.70 (s, 2H, CH2), 3.10 – 0.90 (m, vbr, 10H, C2B10H10), 2.91 (s, 1H, CclusterH), 2.41 (s, 3H, CH3); 11B NMR (CDCl3, 25 °C, ppm): −4.4 (d, 1JBH = 154 Hz, 1B, C2B10H10), −8.7 (d, 1JBH = 154 Hz, 1B, C2B10H10), −10.7 (d, 1JBH = 154 Hz, 2B, C2B10H10), −11.7 (d, 1JBH = 154 Hz, 2B, C2B10H10), −13.4 (d, 1JBH = 154 Hz, 2B, C2B10H10), −15.8 (d, 1JBH = 180 Hz, 2B, C2B10H10); 13C NMR (CDCl3, 25 °C, ppm): 169.3 (s, vbr, CO), 168.3 (s, vbr, CO), 156.1 (s, vbr, CindoleOCH3), 139.4 (s, vbr, Cphenyl), 136.1 (s, vbr, Cindole), 133.8 (s, vbr, Cindole), 131.2 (dd, 1JCH = 171 Hz, 2JCH = 6 Hz, CphenylH), 130.8 (s, vbr, Cphenyl), 130.4 (s, vbr, Cindole), 129.2 (dd, 1JCH = 171 Hz, 2JCH = 6 Hz, CphenylH), 115.0 (d, 1JCH = 161 Hz, CindoleH), 111.8 (dd, 1JCH = 161 Hz, 2JCH = 6 Hz, CindoleH), 111.7 (s, vbr, Cindole), 101.1 (dd, 1JCH = 161 Hz, 2JCH = 6 Hz, CindoleH), 74.9 (s, Ccluster), 64.7 (t, 1JCH = 151 Hz, OCH2), 55.7 (q, 1JCH = 141 Hz, CH3O), 54.9 (d, 1JCH = 181 Hz, CclusterH), 29.9 (t, 1JCH = 131 Hz, CH2), 13.3 (q, 1JCH = 131 Hz, CH3); IR (KBr, cm−1): (w, ν(Ccluster–H)), 2928 (w, ν(C–H)), 2600 (s, ν(B–H)), 2348 (w), 1752 (m, ν(C=O)), 1686 (s, ν(C=O)), 1592 (w), 1479 (s), 1459 (m), 1401 (w), 1358 (m), 1322 (s), 1223 (m), 1138 (s), 1089 (m), 1068 (m), 1036 (w), 1014 (w), 926 (w), 832 (w), 804 (w), 754 (w), 730 (w), 543 (w). 481 (w), 428 (w).
2-{1-[(4-Chlorophenyl)carbonyl]-5-methoxy-2-methyl-1H-indol-3-yl}acetic acid 3-[1-(1,2-dicarba-closo-dodecaboranyl)]propyl ester (7o)
Solvent for purification: Hexanes (80-110 °C)/ethyl acetate 4:1; Yield: 0.18 g (60% from 0.25 g (1)); elemental analysis calcd (%) for C24B10H32O4NCl: C 53.18, H 5.95; found: C 53.73; H 5.97. mp.: 55-57 °C; ESI MS (+) (CH3COCH3): m/z: 565.1 (100%, [M+Na]+); 1H NMR (CDCl3, 25 °C, ppm): 7.67 (d, 3JHH = 8 Hz, 2H, CHphenyl), 7.49 (d, 3JHH = 8 Hz, 2H, CHphenyl), 6.94 (d, 4JHH = 2 Hz, 1H, CHindole), 6.86 (d, 3JHH = 8 Hz, 1H, CHindole), 6.69 (dd, 3JHH = 8 Hz, 4JHH = 2 Hz, 1H, CHindole), 4.07 (t, 2H, 3JHH = 4 Hz, OCH2), 3.83 (s, 3H, OCH3), 3.65 (s, 2H, CH2), 3.25 (s, 1H, CclusterH), 2.90 – 0.90 (m, vbr, 10H, C2B10H10), 2.39 (s, 3H, CH3), 2.00 (m, vbr, 2H, CH2), 1.74 (m, vbr, 2H, CH2); 11B NMR (CDCl3, 25 °C, ppm): −2.4 (d, 1JBH = 154 Hz, 1B, C2B10H10), −5.7 (d, 1JBH = 128 Hz, 1B, C2B10H10), −9.2 (d, 1JBH = 154 Hz, 2B, C2B10H10), −11.9 (d, 1JBH = 154 Hz, 3B, C2B10H10), −13.1 (d, 1JBH = 154 Hz, 3B, C2B10H10); 13C NMR (CDCl3, 25 °C, ppm): 170.5 (s, vbr, CO), 168.3 (s, vbr, CO), 155.9 (s, vbr, CindoleOCH3), 139.5 (s, vbr, Cphenyl), 136.2 (s, vbr, Cindole), 133.6 (s, vbr, Cindole), 131.2 (dd, 1JCH = 171 Hz, 2JCH = 5 Hz, CphenylH), 130.8 (s, vbr, Cphenyl), 130.5 (s, vbr, Cindole), 129.2 (dd, 1JCH = 171 Hz, 2JCH = 5 Hz, CphenylH), 115.0 (d, 1JCH = 161 Hz, CindoleH), 112.2 (s, vbr, Cindole), 110.9 (dd, 1JCH = 161 Hz, 2JCH = 5 Hz, CindoleH), 101.9 (dd, 1JCH = 161 Hz, 2JCH = 5 Hz, CindoleH), 74.1 (s, vbr, Ccluster), 63.0 (t, 1JCH = 151 Hz, OCH2), 61.9 (d, 1JCH = 191 Hz, CclusterH), 55.8 (q, 1JCH = 141 Hz, CH3O), 34.6 (t, 1JCH = 131 Hz, CH2Ccluster), 30.5 (t, 1JCH = 131 Hz, CH2), 28.5 (t, 1JCH = 131 Hz, CH2), 13.3 (q, 1JCH = 131 Hz, CH3); IR (KBr, cm−1): (w, ν(Ccluster–H)), 2957 (w), 2930 (m, ν(C–H)),), 2856 (w), 2836 (w), 2590 (s, ν(B–H)), 2362 (w), 2343 (w), 2066 (w), 1919 (w), 1846 (w), 1737 (s, ν(C=O)), 1684 (s, ν(C=O)), 1592 (m), 1478 (s), 1457 (m), 1438 (m) 1401 (m), 1371 (m),1357 (s), 1322 (s), 1290 (m), 1262 (m), 1223 (s), 1167 (m), 1142 (m), 1090 (m), 1068 (m), 1037 (m), 1015 (m), 957 (w), 926 (m), 835 (m), 803 (w), 755 (m), 723 (w), 689 (w), 669 (w), 602 (w), 562 (w), 548 (w), 482 (w), 433 (w), 416 (w).
2-{1-[(4-Chlorophenyl)carbonyl]-5-methoxy-2-methyl-1H-indol-3-yl}acetic acid 3-[1-(1,7-dicarba-closo-dodecaboranyl)]propyl ester (7m)
Solvent for purification: Hexanes (80-110 °C)/ethyl acetate 3:1. Yield: 0.21 g (73% from 0.19 g (1)); elemental analysis calcd (%) for C24B10H32O4NCl: C 53.18, H 5.95; found: C 53.69; H 5.98; mp.: 53-54 °C; ESI MS (+) (CH3COCH3): m/z: 565.3 (100%, [M+Na]+); 1H NMR (CDCl3, 25 °C, ppm): 7.66 (d, 3JHH = 8 Hz, 2H, CHphenyl), 7.47 (d, 3JHH = 8 Hz, 2H, CHphenyl), 6.93 (d, 4JHH = 2 Hz, 1H, CHindole), 6.85 (d, 3JHH = 8 Hz, 1H, CHindole), 6.67 (dd, 3JHH = 8 Hz, 4JHH = 2 Hz, 1H, CHindole), 4.02 (t, 2H, 3JHH = 4 Hz, OCH2), 3.84 (s, 3H, OCH3), 3.64 (s, 2H, CH2), 3.30 - 1.50 (m, vbr, 10H, C2B10H10), 2.89 (s, 1H, CclusterH), 2.39 (s, 3H, CH3), 1.89 (m, vbr, 2H, CH2), 1.65 (m, vbr, 2H, CH2); 11B NMR (CDCl3, 25 °C, ppm): −4.1 (d, 1JBH = 154 Hz, 1B, C2B10H10), −10.8 (d, 1JBH = 154 Hz, 5B, C2B10H10), −13.6 (d, 1JBH = 167 Hz, 2B, C2B10H10), −15.4 (d, 1JBH = 180 Hz, 2B, C2B10H10); 13C NMR (CDCl3, 25 °C, ppm): 170.6 (s, vbr, CO), 168.3 (s, vbr, CO), 156.0 (s, vbr, CindoleOCH3), 139.3 (s, vbr, Cphenyl), 135.9 (s, vbr, Cindole), 133.9 (s, vbr, Cindole), 131.2 (dd, 1JCH = 171 Hz, 2JCH = 5 Hz, CphenylH), 130.8 (s, vbr, Cphenyl), 130.5 (s, vbr, Cindole), 129.1 (dd, 1JCH = 171 Hz, 2JCH = 5 Hz, CphenylH), 115.0 (d, 1JCH = 161 Hz, CindoleH), 112.4 (s, vbr, Cindole), 111.5 (dd, 1JCH = 161 Hz, 2JCH = 5 Hz, CindoleH), 101.3 (dd, 1JCH = 161 Hz, 2JCH = 5 Hz, CindoleH), 75.1 (s, vbr, Ccluster), 63.7 (t, 1JCH = 141 Hz, OCH2), 55.7 (q, 1JCH = 141 Hz, CH3O), 54.9 (d, 1JCH = 191 Hz, CclusterH), 33.3 (t, 1JCH = 131 Hz, CH2), 30.3 (t, 1JCH = 131 Hz, CH2), 29.0 (t, 1JCH = 131 Hz, CH2), 13.3 (q, 1JCH = 131 Hz, CH3); IR (KBr, cm−1): (w, ν(Ccluster–H)), 2956 (w), 2929 (w, ν(C–H)), 2855 (w), 2835 (w), 2599 (s, ν(B–H)), 2347 (w), 2286 (w), 2063 (w), 1925 (w), 1846 (w), 1736 (s, ν(C=O)), 1685 (s, ν(C=O)), 1592 (m), 1479 (s), 1457 (m), 1438 (w), 1401 (w), 1370 (m), 1357 (m), 1322 (s), 1290 (m), 1262 (m), 1223 (m), 1166 (m), 1142 (m), 1111 (w), 1089 (m), 1068 (m), 1039 (m), 1014 (m), 956 (w), 926 (w), 833 (w), 804 (w), 754 (m), 731 (w), 689 (w), 663 (w), 602 (w), 561 (w), 547 (w), 510 (w), 481 (w), 438 (w), 429 (w), 421 (w), 414 (w).
2-{1-[(4-Chlorophenyl)carbonyl]-5-methoxy-2-methyl-1H-indol-3-yl}acetic acid 1-adamantyl ester (9)
Solvent for purification: Hexanes (80-110 °C)/ethyl acetate 3:1. Yield: 0.76 g (55% from 1 g (1)); elemental analysis calcd (%) for C29H30O4NCl: C 70.79; H 6.15; found: C 70.52, H 6.21; mp.: 81-82 °C; ESI MS (+) (CH3COCH3/Na+): m/z: 492.0 (11%, [M+H]+), 514.0 (100%, [M+Na]+), 530.0 (38%, [M+K]+); 1H NMR (CDCl3, 25 °C, ppm): 7.66 (d, 3JHH = 8 Hz, 2H, CHphenyl), 7.47 (d, 3JHH = 8 Hz, 2H, CHphenyl), 6.97 (d, 1H, 4JHH = 2 Hz, CHindole), 6.89 (d, 3JHH = 8 Hz, 1H, CHindole), 6.66 (dd, 3JHH = 8 Hz, 4JHH = 2 Hz, 1H, CHindole), 3.84 (s, 3H, OCH3), 3.56 (s, 2H, CH2), 2.37 (s, 3H, CH3), 2.15 (s, vbr, 3H, CHadamantane), 2.09 (s, vbr, 6H, CH2adamantane), 1.64 (s, vbr, 6H, CH2adamantane); 13C{1H} NMR (CDCl3, 25 °C, ppm): 169.9 (CO), 168.3 (CO), 156.0 (CindoleOCH3), 139.1 (Cphenyl), 135.7 (Cindole), 134.0 (Cindole), 131.2 (CphenylH), 130.8 (Cphenyl and Cindole), 129.1 (CphenylH), 114.9 (CindoleH), 113.4 (Cindole), 111.6 (CindoleH), 101.4 (CindoleH), 81.2 (Cadamantane), 55.7 (CH3O), 41.3 (CH2 adamantane), 36.1 (CH2 adamantane), 31.9 (CH2), 30.8 (CHadamantane), 13.4 (CH3); IR (KBr, cm−1): (m, ν(C–H)), 2854 (w), 1729 (m, ν(C=O)), 1684 (s, ν(C=O)), 1594 (w), 1478 (m), 1457 (m), 1399 (w), 1357 (m), 1322 (s), 1259 (m), 1223 (m), 1170 (m), 1144 (w), 1089 (m), 1057 (m), 1015 (w), 969 (w), 926 (w), 833 (w), 754 (w), 690 (w), 550 (w), 482 (w).
4.2. COX-Inhibition studies
Concentration-dependent inhibition reactions were performed by pre-incubating the inhibitor and enzyme for 17 min at 25 °C, followed by 3 min at 37 °C prior to the addition of 50 μM [1□14C]-AA for 30 sec at 37 °C. All assays were terminated and analyzed for substrate consumption by thin-layer chromatography (TLC) as previously described.6 All inhibitor concentrations for 50% enzyme activity (IC50) were determined graphically using Prism and were the average of at least two independent determinations.
Acknowledgement
This work was supported by the Studienstiftung des Deutschen Volkes (doctoral grant for M.S.), the Graduate School of Excellence “Building with Molecules and Nano-objects (BuildMoNa)” funded by the Deutsche Forschungsgemeinschaft and by a research grant from the National Institutes of Health (CA89450).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hart FD, Boardman PL. Br. Med. J. 1963;2:965. doi: 10.1136/bmj.2.5363.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kistenmacher TJ, Marsh RE. J. Am. Chem. Soc. 1972;94:1340. doi: 10.1021/ja00759a047. [DOI] [PubMed] [Google Scholar]
- 3.Vane JR. Nat. New Biol. 1971;231:232. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- 4.Ferreira SH, Moncada S, Vane JR. Nat. New Biol. 1971;231:237. doi: 10.1038/newbio231237a0. [DOI] [PubMed] [Google Scholar]
- 5.Prusakiewicz JJ, Felts AS, Mackenzie BS, Marnett LJ. Biochemistry. 2004;43:15439. doi: 10.1021/bi048534q. [DOI] [PubMed] [Google Scholar]
- 6.Kalgutkar AS, Crews BC, Rowlinson SW, Marnett AB, Kozak KR, Remmel RP, Marnett LJ. Proc. Natl. Acad. Sci. USA. 2000;97:925. doi: 10.1073/pnas.97.2.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marnett LJ, Kalgutkar AS. Trends Pharmacol. Sci. 1999;20:465. doi: 10.1016/s0165-6147(99)01385-1. [DOI] [PubMed] [Google Scholar]
- 8.Ghosh N, Chaki R, Mandal V, Mandal SC. Pharmacol. Rep. 2010;62:233. doi: 10.1016/s1734-1140(10)70262-0. [DOI] [PubMed] [Google Scholar]
- 9.Mitchell JA, Warner TD. Nat. Rev. Drug Discov. 2006;5:75. doi: 10.1038/nrd1929. [DOI] [PubMed] [Google Scholar]
- 10.Marnett LJ. Annu. Rev. Pharmacol. Toxicol. 2009;49:265. doi: 10.1146/annurev.pharmtox.011008.145638. [DOI] [PubMed] [Google Scholar]
- 11.Rouzer CA, Marnett LJ. J Lipid Res. 2009;50:29. doi: 10.1194/jlr.R800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kalgutkar AS, Marnett AB, Crews BC, Remmel RP, Marnett LJ. J. Med. Chem. 2000;43:2860. doi: 10.1021/jm000004e. [DOI] [PubMed] [Google Scholar]
- 13.Endo Y, Yoshimi T, Kimura K, Itai A. Bioorg. Med. Chem. Lett. 1999;9:2561. doi: 10.1016/s0960-894x(99)00436-9. [DOI] [PubMed] [Google Scholar]
- 14.Endo Y, Iijima T, Kagechika H, Ohta K, Kawachi E, Shudo K. Chem. Pharm. Bull. (Tokyo) 1999;47:585. doi: 10.1248/cpb.47.585. [DOI] [PubMed] [Google Scholar]
- 15.Endo Y, Iijima T, Yamakoshi Y, Fukasawa H, Miyaura C, Inada M, Kubo A, Itai A. Chem. Biol. 2001;8:341. doi: 10.1016/s1074-5521(01)00016-3. [DOI] [PubMed] [Google Scholar]
- 16.Endo Y, Iijima T, Yamakoshi Y, Yamaguchi M, Fukasawa H, Shudo K. J. Med. Chem. 1999;42:1501. doi: 10.1021/jm9900725. [DOI] [PubMed] [Google Scholar]
- 17.Valliant JF, Guenther KJ, King AS, Morel P, Schaffer P, Sogbein OO, Stephenson KA. Coord. Chem. Rev. 2002;232:173. [Google Scholar]
- 18.Bregadze VI. Chem. Rev. 1992;92:209. [Google Scholar]
- 19.Hermansson K, Wojcik M, Sjöberg S. Inorg. Chem. 1999;38:6039. doi: 10.1021/ic990381l. [DOI] [PubMed] [Google Scholar]
- 20.Armstrong AF, Valliant JF. Dalton Trans. 2007:4240. doi: 10.1039/b709843j. [DOI] [PubMed] [Google Scholar]
- 21.Bregadze VI, Sivaev IB, Glazun SA. Anticancer Agents Med. Chem. 2006;6:75. doi: 10.2174/187152006776119180. [DOI] [PubMed] [Google Scholar]
- 22.Kabalka GW, Yao ML. Anticancer Agents Med. Chem. 2006;6:111. doi: 10.2174/187152006776119144. [DOI] [PubMed] [Google Scholar]
- 23.Kalinin V, Ol’shevskaya V. Russ. Chem. Bull. 2008;57:815. [Google Scholar]
- 24.Sivaev IB, Bregadze VV. Eur. J. Inorg. Chem. 2009;2009:1433. [Google Scholar]
- 25.Goto T, Ohta K, Fujii S, Ohta S, Endo Y. J. Med. Chem. 2010;53:4917. doi: 10.1021/jm100316f. [DOI] [PubMed] [Google Scholar]
- 26.Beer ML, Lemon J, Valliant JF. J. Med. Chem. 2010;53:8012. doi: 10.1021/jm100758j. [DOI] [PubMed] [Google Scholar]
- 27.Scholz M, Steinhagen M, Heiker JT, Beck-Sickinger AG, Hey-Hawkins E. ChemMedChem. 2011;6:89. doi: 10.1002/cmdc.201000368. [DOI] [PubMed] [Google Scholar]
- 28.Bednarska K, Olejniczak AB, Wojtczak BA, Sulowska Z, Lesnikowski ZJ. ChemMedChem. 2010;5:749. doi: 10.1002/cmdc.201000075. [DOI] [PubMed] [Google Scholar]
- 29.Scholz M, Kaluðerovic GN, Kommera H, Paschke R, Will J, Sheldrick WS, Hey-Hawkins E. Eur. J. Med. Chem. 2011;46:1131. doi: 10.1016/j.ejmech.2011.01.030. [DOI] [PubMed] [Google Scholar]
- 30.Cheung M-S, Chan H-S, Xie Z. Organometallics. 2003;23:517. [Google Scholar]
- 31.Drechsel K, Lee CS, Leung EW, Kane RR, Hawthorne MF. Tetrahedron Lett. 1994;35:6217. [Google Scholar]
- 32.Ohta K, Goto T, Yamazaki H, Pichierri F, Endo Y. Inorg. Chem. 2007;46:3966. doi: 10.1021/ic062025q. [DOI] [PubMed] [Google Scholar]
- 33.Zharov I, Saxena A, Michl J, Miller RD. Inorg. Chem. 1997;36:6033. doi: 10.1021/ic970796b. [DOI] [PubMed] [Google Scholar]
- 34.Goto T, Ohta K, Suzuki T, Ohta S, Endo Y. Bioorg. Med. Chem. 2005;13:6414. doi: 10.1016/j.bmc.2005.06.061. [DOI] [PubMed] [Google Scholar]
- 35.Naeslund C, Ghirmai S, Sjöberg S. Tetrahedron. 2005;61:1181. [Google Scholar]
- 36.Nakamura H, Aoyagi K, Yamamoto Y. J. Am. Chem. Soc. 1998;120:1167. [Google Scholar]
- 37.Gomez FA, Hawthorne MF. J. Org. Chem. 1992;57:1384. [Google Scholar]
- 38.Diago-Meseguer J, Palomo-Coll AL, Fernández-Lizarbe JR, Zugaza-Bilbao A. Synthesis. 1980:547. [Google Scholar]
- 39.Altomare A, Cascarano G, Giacovazzo C, Guagliardi A. J. Appl. Crystallogr. 1993;26:343. [Google Scholar]
- 40.SCALE3 ABSPACK: Empirical absorption correction, CrysAlis – Software package. Oxford Diffraction Ltd.; Oxford (UK): 2006. [Google Scholar]
- 41.Sheldrick GM. SHELXL-97, Program for the Refinement of Crystal Structures. University of Göttingen; Göttingen (Germany): 1997. [Google Scholar]