

Abstract
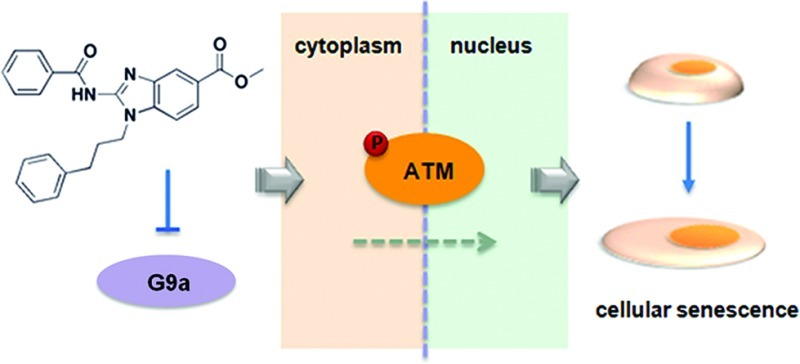
Post-translational modifications of histones alter chromatin structure and play key roles in gene expression and specification of cell states. Small molecules that target chromatin-modifying enzymes selectively are useful as probes and have promise as therapeutics, although very few are currently available. G9a (also named euchromatin histone methyltransferase 2 (EHMT2)) catalyzes methylation of lysine 9 on histone H3 (H3K9), a modification linked to aberrant silencing of tumor-suppressor genes, among others. Here, we report the discovery of a novel histone methyltransferase inhibitor, BRD4770. This compound reduced cellular levels of di- and trimethylated H3K9 without inducing apoptosis, induced senescence, and inhibited both anchorage-dependent and -independent proliferation in the pancreatic cancer cell line PANC-1. ATM-pathway activation, caused by either genetic or small-molecule inhibition of G9a, may mediate BRD4770-induced cell senescence. BRD4770 may be a useful tool to study G9a and its role in senescence and cancer cell biology.
Histone methyltransferases (HMTs) and demethylases (HDMs) dynamically alter the methylation state of histone proteins. Somatic mutation and amplification of HMTs are frequently observed in human cancers, and at least 22 out of 50 arginine and lysine HMTs encoded in the human genome have been associated with cancer or other diseases in humans or mice.1 Methylation of lysine 9 on histone H3 (H3K9) is associated with transcriptional silencing, and this mark is often found in the promoter regions of aberrantly silenced tumor-suppressor genes in cancer cells.2 Euchromatin histone methyltransferase 1 (EHMT1), also known as GLP or KMT1D, forms a heteromeric complex with G9a (also called EHMT2 or KMT1C) to yield H3K9 methyltransferase activity in euchromatin.3 Knockdown of G9a significantly reduces di- and trimethylation of H3K9 in cell culture and in mice.4,5
Few selective small-molecule inhibitors of chromatin-modifying enzymes exist. Current methyltransferase inhibitors fall into two categories: H3 peptide substrate-competitive inhibitors and S-adenosylmethionine (SAM) cofactor-competitive inhibitors (Figure 1a). The substrate-competitive compound BIX-01294 was identified as a selective G9a inhibitor by high-throughput screening.6 Despite its relative selectivity, BIX-01294 shows toxicity apparently not linked to its HMT inhibitory activity. A structural analogue, UNC0638, was recently reported to have increased potency and reduced cell toxicity.7 Cofactor-competitive inhibitors include BIX-01338, discovered in the same screen as BIX-01294, and the natural product chaetocin.8 Both compounds are non-selective, with similar IC50 values against G9a and the HMT SUV39H1. BIX-01338 neither modulates cellular H3K9 methylation status nor inhibits cancer cell growth.6 Inspired by the isoform selectivity exhibited by certain inhibitors of kinases and histone deacetylases (Figure 1a), we were interested in developing new SAM-competitive inhibitors selective toward subsets of HMTs or even a single HMT. Such compounds should be useful tools for the study of methyltransferases.
Figure 1.
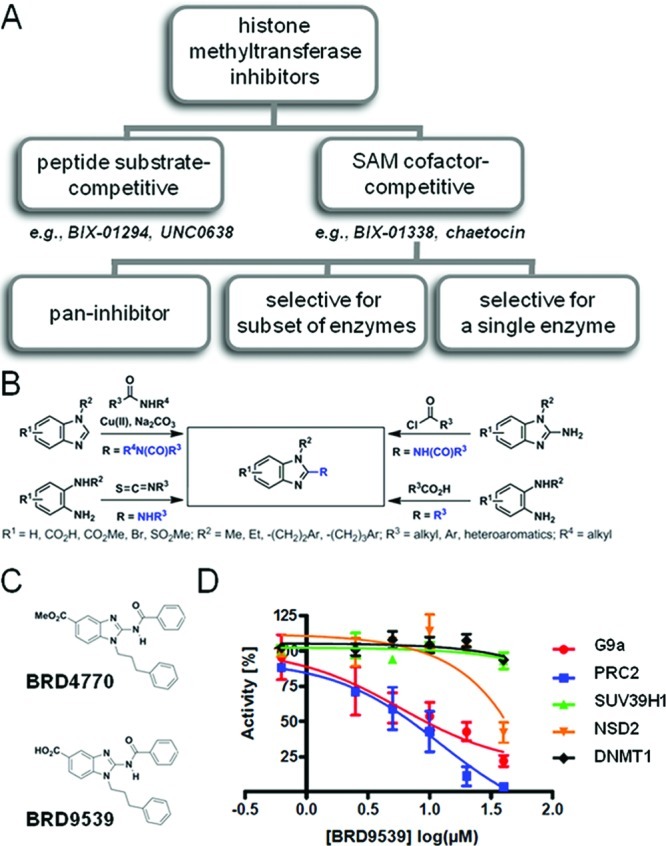
Small-molecule inhibitors of G9a. (A) Classification of existing histone methyltransferases inhibitors (HMTi). Representative substrate- and cofactor-competitive HMTi’s are shown. (B) Synthetic chemistry scheme yielding 117 benzimidazoles as potential SAM mimetics for competitive inhibition of HMTs. (C) Chemical structures of BRD4770 and BRD9539. (D) Biochemical activities of G9a, PRC2, SUV39H1, NSD2, and DNMT1 following treatment with indicated concentrations of BRD9539. Data represent the mean and standard error of four independent reactions.
Here, we describe the discovery of BRD4770, a SAM mimetic and analogue of BIX-01338 that selectively inhibits a subset of HMTs, including G9a, in biochemical assays and in cells. Similar to knockdown of G9a,4,9,10 BRD4770 induced a senescent phenotype in a pancreatic cancer cell line. BRD4770 also inhibited both anchorage-dependent and -independent cell proliferation and induced G2/M cell-cycle arrest. The protein kinases ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related protein (ATR) are thought to be important in DNA damage-induced senescence.11 We show that BRD4770 activates the ATM pathway without inducing DNA damage, while the ATR pathway is not affected. BRD4770 is a novel probe for studying G9a and its role in cellular senescence.
Although an S-adenosylmethionine (SAM)-competitive inhibitor of G9a, BIX-01338, has been reported, it inhibits several methyltransferases, including SUV39H1, and lacks cellular activity.6 To improve selectivity and achieve G9a inhibition in cells, we synthesized a focused library of 117 2-substituted benzimidazoles as potential SAM mimetics (Figure 1b). These compounds were tested for biochemical inhibition of G9a; 41 showed >45% inhibition at 50 μM (Supporting Figure S1a). Knockdown of G9a resulted in an enlarged and flattened cell morphology, and visual examination revealed that eight compounds induced similar morphological changes. We also measured cellular ATP levels, as a surrogate of cell viability, in HeLa cells treated with this compound collection. After a 2-day treatment, 17 compounds decreased ATP levels in a dose-dependent manner (Supporting Figure S1a). Overall, five compounds were active in all three assays (Supporting Figure S1b). We focused on BRD4770, which induced the strongest morphological change in cells, and BRD9539, the potential active form of BRD4770 in cells (Figure 1c).
We next examined the biochemical activity of BRD9539 against five methyltransferases: G9a, SUV39H1, PRC2, NSD2, and DNMT1. BRD9539 inhibited G9a activity in a dose-dependent manner, with an IC50 of 6.3 μM (Figure 1d). The inhibitory effect of BRD9539 decreased with increasing SAM concentrations (Supporting Figure S2), but detailed kinetic analysis is required to determine whether BRD9539, like its analogue BIX-01338, is SAM-competitive. Existing SAM-competitive inhibitors, such as BIX-01338 and chaetocin, inhibit G9a but also inhibit SUV39H1.8,12 BRD9539 also inhibited PRC2 activity with a similar IC50. No inhibition of SUV39H1 and DNMT1 was observed up to 40 μM. Partial inhibition of NSD2 was observed only at 40 μM. BRD9539 is a more potent biochemical inhibitor than its methyl-ester analogue BRD4770, with 20% remaining G9a activity compared to 45% of BRD4770 at screening concentration. However, BRD9539 had no activity in cell-based assays, presumably due to impaired cell permeability compared to that of BRD4770. We assume that BRD4770 is converted to BRD9539 in cells since methyl esters of biologically active carboxylic acids are rapidly hydrolyzed in cells and often used as cell-active “pro-drugs” of the less cell-permeable carboxylic acids. In addition, the activities of 16 other chromatin-modifying enzymes and 100 kinases involved in cell-cycle regulation and cancer cell biology were tested for activity in the presence of 5 or 10 μM BRD9539; no activity was seen in any of these assays (Supporting Table S1 and S2).
To evaluate BRD4770 activity in cells, we analyzed the methylation state at various histone lysines by mass spectrometry. A significant decrease in di- and trimethylation levels of H3K9, along with an increase in monomethylation level, was observed after 24-h treatment with 10 μM BRD4770. In comparison, BIX-01294, a substrate-competitive inhibitor of G9a, also decreased H3K9 di- and trimethylation, but with an increase of unmodified H3K9, possibly due to a different mechanism of action (Figure 2a). Methylation at other positions, such as H3K27 and H3K36, appeared unaffected. We also assessed H3K9 methylation by Western blot. Treatment of 5 μM BRD4770 for 24 h decreased H3K9 trimethylation level by 23% in PANC-1 cells (Figure 2b). Although we observed a slight decrease in H3K36 trimethylation, the trimethylation levels at other positions, such as H3K4, H3K27, and H3K79, remained largely unchanged (Figure 2b). The EC50 of BRD4770 in PANC-1 cells, determined by Western blotting for trimethylated H3K9, was approximately 5 μM (Figure 2c), which was significantly higher compared to existing substrate-competitive inhibitors (Supporting Figure S3). These data are consistent with inhibition of G9a in cells following BRD4770 treatment. While our biochemical experiments suggested that BRD9539 also inhibits PRC2 activity, we have not observed evidence of PRC2 inhibition in cells with BRD4770 in our experiments to date.
Figure 2.

Cellular effects of BRD4770 on histone methylation. (A) Mass-spectrometric analysis of methylation levels of histone H3 lysines 9, 27, and 36 in cells treated with DMSO (black bars), 10 μM BRD4770 (gray bars), or 10 μM BIX-01294 (white bars) for 24 h. (B) Western blot analysis of trimethylation levels of histone H3 lysines 4, 9, 27, 36, and 79 in PANC-1 cells after 24-h treatment with the indicated concentrations of BRD4770. Tubulin was used as an internal loading control. (C) Western blot analysis of relative trimethylation levels of histone H3 lysine 9 in PANC-1 cells after 24-h treatment with the indicated concentrations of BRD4770. Data represent the mean and standard error of four independent replicates. Caspase-3/7 activity in PANC-1 cells was measured after 24 h (dashed line) or 72 h (solid line) treatment with (D) BRD4770, (E) BIX-01294, or (F) UNC0638. Results were normalized by cellular ATP levels, and data represent the mean and standard error of six independent replicates.
The selective substrate-competitive inhibitor BIX-01294 inhibits GLP and G9a6 but is toxic to cells, which may be caused by off-target effects in cells. Thus, we sought to determine whether BRD4770 induced apoptosis by measuring caspase-3/7 activity. Even after 72 h treatment, BRD4770 did not increase caspase activity in PANC-1 cells at any concentration (Figure 2d). In contrast, BIX-01294 strongly induced caspase activity after only 24 h (Figure 2e). UNC0638, an analogue of BIX-01294 with improved potency and lower cell toxicity, also induced caspase activity after 72 h, but only at a much higher concentration compared to its cellular EC50 (Figure 2f). Thus, the apoptotic effect appears to be unrelated to G9a inhibition. These results suggest that BRD4770 may be a useful probe of G9a activity in cells, without apparent toxicity at its effective concentration.
Knockdown of G9a inhibits cell growth and induces cellular senescence in PC3 prostate cancer cells.4 To assess the effects of inhibiting G9a in pancreatic cancer cells, we compared knockdown of G9a (Supporting Figure S4) to treatment with BRD4770 in PANC-1 cells. Both genetic and small-molecule inhibition of G9a resulted in enlarged and flattened cell morphology, with increased senescence-associated β-galactosidase staining (Figure 3a). Anchorage-dependent and -independent cell growth were monitored in PANC-1 cells by nuclear staining and growth in soft agarose, respectively. BRD4770 treatment reduced the number of cells after 72 h (Figure 3b). Compound-treated cells showed reduced histone H3 phosphorylation at serine 10 (Supporting Figure S5), suggesting reduced proliferation. Colony formation in soft agarose was also significantly reduced following BRD4770 treatment (Figure 3c,d). Using fluorescence-activated cell sorting, we found that treatment with BRD4770 increased the cell population in G2/M and decreased the fraction of G0/G1 cells (Figure 3e).
Figure 3.

Comparison of genetic and small-molecule inhibition of G9a in PANC-1 cells. (A) Senescence-associated β-galactosidase expression in PANC-1 cells following knockdown of G9a with two independent siRNA constructs or treatment with BRD4770 for 5 days. Scale bars = 50 μm. (B) Nuclear count in PANC-1 cells treated for 72 h with BRD4770, fixed, and stained with Hoechst dye. Total nuclei per well were counted by automated microscopy (see Methods). Data represent the mean and standard error of 16 independent replicates. * indicates p < 0.003, ** indicates p < 0.001 (t test). (C) Brightfield images of PANC-1 cells after 3-day treatment with BRD4770, followed by 10-day culture in soft agarose. Scale bar = 50 μm. (D) Quantification of PANC-1 cell growth in soft agarose by DNA measurement (see Methods). * indicates p < 0.001 (t test). (E) Evaluation of cell cycle in PANC-1 cells treated for 3 days with DMSO (left panel) or BRD4770 (right panel). Inset, calculated percentage of cells in each phase.
These data led us to wonder whether the compound induces cell-cycle arrest. ATM and ATR are important regulators of cell-cycle arrest caused by DNA damage, including senescence.13,14 To investigate the mechanism further underlying cell-growth inhibition induced by BRD4770, we examined the effect of BRD4770 treatment on ATM and ATR pathway activation. Since ATM and ATR are regulated by autophosphorylation, we assessed their phosphorylation levels by immunofluorescent staining. Treatment with BRD4770 led to increases in phosphorylated ATM and nuclear translocation of phosphorylated ATM in PANC-1 cells (Figure 4a). We did not observe similar changes in ATR (Figure 4b). Consistent with activation of ATM but not ATR, BRD4770 treatment increased phosphorylation of Chk2 and decreased cdc25C levels (downstream targets of the ATM pathway) but did not increase phosphorylation of Chk1 (a downstream target of ATR) (Figure 4c). Knockdown of G9a, and to a lesser extent GLP, yielded similar results (Figure 4d).
Figure 4.

Effects of G9a inhibition on the ATM and ATR pathways. Immunofluorescent analysis of phosphorylation and nuclear translocation of (A) ATM and (B) ATR in PANC-1 cells treated with the indicated concentrations of BRD4770 for 72 h. Scale bars = 50 μm. (C) Western blots for levels of phosphorylated Chk1 (Ser345), phosphorylated Chk2 (Thr68), and total cdc25C protein expression in PANC-1 cells treated with the indicated concentrations of BRD4770 for 72 h. Tubulin was used as an internal loading control. (D) Assessment of levels of same proteins in PANC-1 cells in which GLP and G9a were knocked down by siRNA, individually and in combination, for 72 h.
Although ATM pathway activation is usually linked to DNA damage, especially that caused by double-stranded breaks, it can also be induced in the absence of DNA damage.15,16 To determine whether BRD4770 causes ATM activation by inducing DNA damage, we stained for phospho-H2AX, which is rapidly phosphorylated and localized to sites of DNA damage in response to double-stranded breaks.17 No increase in phospho-H2AX staining was observed by flow cytometry or by fluorescence microscopy (Supporting Figure S6a). Similarly, no increase in DNA damage was observed in individual cells following BRD4770 treatment by comet assay, measuring the tail moment length (Supporting Figure S6b). Our data suggest BRD4770 causes ATM activation in the absence of DNA damage.
Changes in chromatin structure have been implicated in ATM activation and cellular senescence, but the precise mechanism remains uncharacterized.18 For example, treatment with HDAC inhibitors can trigger cellular senescence by inducing ATM phosphorylation.18−20 Here we show that treatment with an HMT inhibitor causes similar phenotypes. It is unclear if changing histone methylation is sufficient to induce ATM pathway activation and senescence, or whether additional changes in chromatin structure, such as telomere structure, DNA methylation, and histone acetylation, are induced by BRD4770 as a secondary effect and contribute to the overall phenotype. For example, BRD4770 also induces increased levels of lysine acetylation in cells (Supporting Figure S7) without inhibiting histone deacetylases (Supporting Table S1).
Cellular senescence may result from a variety of stresses, mainly mediated by two tumor-suppressor pathways involving p53 and p16-pRB.11,21 However, the senescent phenotype resulting from genetic or chemical inhibition of G9a in PANC-1 cells may be independent of these two pathways. PANC-1, a human pancreatic ductal carcinoma cell line that resists apoptotic cell death, harbors four of the most common mutations in pancreatic cancer, including homozygous deletion of CDKN2A (which encodes p16), and alterations in TP53 (deletion of one allele and mutation at codon 273 within the DNA-binding domain of the other allele).22 BRD4770 activates the ATM pathway and induces senescence in this genetic context. Overall, our data suggest that BRD4770 is a novel small-molecule probe of G9a activity and its role in senescence of cancer cells.
Methods
Cell Culture and Compound Treatment
PANC-1 and HeLa cells (ATCC) were cultured in DMEM medium containing 10% (v/v) FBS and 100 U mL–1 penicillin-streptomycin. BRD4770, BRD9539, BRD2502, BRD9398, BRD1490, and BI-37 were synthesized in our lab. Chemical characterization of BRD4770 and BRD9539 is included in Supporting Information. BIX-01294, UNC0638, and chaetocin were purchased from Sigma-Aldrich.
Enzymatic Assays
Biochemical activity of G9a was measured as described.6 Biochemical assays for SUV39H1 and DNMT1 activity were from BPS Bioscience. Chaetocin was used as a positive control in G9a and SUV39H1 assays. PRC2 and NSD2 activity were measured using dissociation-enhanced lanthanide fluorescent immunoassay (DELFIA) in white 384-well streptavidin-coated plates (PerkinElmer). For detailed assay procedure please see Supporting Information.
Mass-Spectrometric Analysis of Histone Methylation
HeLa cells were seeded in 10 cm dishes and treated with BRD4770 or DMSO for 24 h. Histones were extracted according to the manufacturer’s protocol (Abcam). Histone bands were isolated from SDS-PAGE gel and treated with double propionylation and trypsin. Samples were analyzed on an Orbitrap mass spectrometer as described.6
Gene Silencing
Small-interfering RNAs against GLP (s36392) and G9a (s21469, s21470) were obtained from Applied Biosystems. siRNAs (25 nM) were transfected into PANC-1 cells using Lipofectamine 2000 (Invitrogen), with medium changed 24 h later. Transfected cells were cultured for 72 h, and RNA and protein were collected for real-time PCR and Western blot analysis, respectively.
Western Blotting
Cells were lysed in RIPA buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% (w/v) NP-40, 1% (w/v) sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg mL–1 leupeptin, protease inhibitor and phosphatase inhibitor). Total protein was separated by 4–12% SDS-PAGE and transferred to a PVDF membrane (iBlot system, Invitrogen). Blots were developed using chemiluminescence detection (SuperSignal, Thermo Fisher Scientific Image Station 4000MM Pro, Kodak). Images were quantified using ImageJ image analysis software. Antibody concentrations are included as Supporting Information.
Immunofluorescence and Microscopy
PANC-1 cells were seeded in black optical 96-well plates and treated with compounds for 72 h. Cells were fixed using 4% (v/v) paraformaldehyde for 20 min, permeabilized for 20 min with 0.1% (v/v) Triton X-100, blocked with PBS containing 2% (w/v) BSA at 4 °C overnight, and incubated at 4 °C overnight with either p-ATM (S1981) mouse mAb (10H11.E12, Cell Signaling) or p-ATR (S428) rabbit Ab (Cell Signaling) at 1:250 dilution. Cy3-labeled secondary antibodies (Jackson ImmunoResearch) were added with Hoechst 33342 (Invitrogen) and observed with an Axiovert 200 M fluorescence microscope (Zeiss) at 400X magnification. Cell number analysis was performed on an ImageXpress Micro automated microscope (Molecular Devices) using a 4X objective with laser-based focusing. Image analysis was performed using the Cell Count module in the MetaXpress software application (Molecular Devices).
Senescence Analysis
PANC-1 cells were seeded in 12-well plates, treated with compounds and siRNAs, and stained with Senescence β-Galactosidase Staining Kit (Cell Signaling), with detection of blue color by light microscopy.
Anchorage-Independent Soft Agarose Assay
PANC-1 cells were seeded and treated with BRD4770 in 6-well plates for 72 h. Cells were trypsinized and tested for soft agar colony formation using CytoSelect 96-Well Cell Transformation Assay (Cell Biolabs), using the CyQuant GR dye to measure total cellular nucleic acid levels. Fluorescence was detected with an Analyst HT plate reader (LJL Biosystems) using a 485/520 nm filter set.
Flow Cytometry
Treated cells were washed in PBS, fixed in ice-cold 70% (v/v) ethanol for 30 min at 4 °C, followed by two washes with PBS. Next, 100 μg mL–1 RNase was added and incubated at 37 °C for 15 min, 50 μg mL–1 propidium iodide was added, and cell cycle was analyzed with a BD LSR II flow cytometer (BD Biosciences).
Acknowledgments
We thank L. Briggs for technical assistance, J. Jaffe and S. Carr for advice and assistance with proteomic analysis of histone methylation, and A. Stern for helpful suggestions. This work was supported by the National Institute of General Medicine Sciences (GM38627 to S.L.S.). S.L.S. is a Howard Hughes Medical Institute Investigator.
Supporting Information Available
This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Author Present Address
⊥ Department of Chemistry, Duke University, Durham, NC 27708.
Author Present Address
# CeMM - Research Centre for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria.
Author Contributions
¶ These authors contributed equally to this work.
Supplementary Material
References
- Albert M.; Helin K. (2010) Histone methyltransferases in cancer. Semin. Cell Dev. Biol. 21, 209–220. [DOI] [PubMed] [Google Scholar]
- Yoo C. B.; Jones P. A. (2006) Epigenetic therapy of cancer: past, present and future. Nat. Rev. Drug Discovery 5, 37–50. [DOI] [PubMed] [Google Scholar]
- Tachibana M.; Ueda J.; Fukuda M.; Takeda N.; Ohta T.; Iwanari H.; Sakihama T.; Kodama T.; Hamakubo T.; Shinkai Y. (2005) Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 19, 815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y.; Shen L.; Ahmed S.; Boumber Y.; Sekido Y.; Haddad B. R.; Issa J. P. (2008) Downregulation of histone H3 lysine 9 methyltransferase G9a induces centrosome disruption and chromosome instability in cancer cells. PLoS One 3, e2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M.; Sugimoto K.; Nozaki M.; Ueda J.; Ohta T.; Ohki M.; Fukuda M.; Takeda N.; Niida H.; Kato H.; Shinkai Y. (2002) G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 16, 1779–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubicek S.; O’Sullivan R. J.; August E. M.; Hickey E. R.; Zhang Q.; Teodoro M. L.; Rea S.; Mechtler K.; Kowalski J. A.; Homon C. A.; Kelly T. A.; Jenuwein T. (2007) Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol. Cell 25, 473–481. [DOI] [PubMed] [Google Scholar]
- Vedadi M.; Barsyte-Lovejoy D.; Liu F.; Rival-Gervier S.; Allali-Hassani A.; Labrie V.; Wigle T. J.; Dimaggio P. A.; Wasney G. A.; Siarheyeva A.; Dong A.; Tempel W.; Wang S. C.; Chen X.; Chau I.; Mangano T. J.; Huang X. P.; Simpson C. D.; Pattenden S. G.; Norris J. L.; Kireev D. B.; Tripathy A.; Edwards A.; Roth B. L.; Janzen W. P.; Garcia B. A.; Petronis A.; Ellis J.; Brown P. J.; Frye S. V.; Arrowsmith C. H.; Jin J. (2011) A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat. Chem. Biol. 7, 566–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiner D.; Bonaldi T.; Eskeland R.; Roemer E.; Imhof A. (2005) Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3–9. Nat. Chem. Biol. 1, 143–145. [DOI] [PubMed] [Google Scholar]
- Huang J.; Dorsey J.; Chuikov S.; Perez-Burgos L.; Zhang X.; Jenuwein T.; Reinberg D.; Berger S. L. (2010) G9a and Glp methylate lysine 373 in the tumor suppressor p53. J. Biol. Chem. 285, 9636–9641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M. W.; Hua K. T.; Kao H. J.; Chi C. C.; Wei L. H.; Johansson G.; Shiah S. G.; Chen P. S.; Jeng Y. M.; Cheng T. Y.; Lai T. C.; Chang J. S.; Jan Y. H.; Chien M. H.; Yang C. J.; Huang M. S.; Hsiao M.; Kuo M. L. (2010) H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res. 70, 7830–7840. [DOI] [PubMed] [Google Scholar]
- Campisi J.; d’Adda di Fagagna F. (2007) Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8, 729–740. [DOI] [PubMed] [Google Scholar]
- Kristensen L. S.; Nielsen H. M.; Hansen L. L. (2009) Epigenetics and cancer treatment. Eur. J. Pharmacol. 625, 131–142. [DOI] [PubMed] [Google Scholar]
- von Zglinicki T.; Saretzki G.; Ladhoff J.; d’Adda di Fagagna F.; Jackson S. P. (2005) Human cell senescence as a DNA damage response. Mech. Ageing Dev. 126, 111–117. [DOI] [PubMed] [Google Scholar]
- Rotman G.; Shiloh Y. (1999) ATM: a mediator of multiple responses to genotoxic stress. Oncogene 18, 6135–6144. [DOI] [PubMed] [Google Scholar]
- Bonilla C. Y.; Melo J. A.; Toczyski D. P. (2008) Colocalization of sensors is sufficient to activate the DNA damage checkpoint in the absence of damage. Mol. Cell 30, 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soutoglou E.; Misteli T. (2008) Activation of the cellular DNA damage response in the absence of DNA lesions. Science 320, 1507–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou E. P.; Pilch D. R.; Orr A. H.; Ivanova V. S.; Bonner W. M. (1998) DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273, 5858–5868. [DOI] [PubMed] [Google Scholar]
- Bakkenist C. J.; Kastan M. B. (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499–506. [DOI] [PubMed] [Google Scholar]
- Lee J. S. (2007) Activation of ATM-dependent DNA damage signal pathway by a histone deacetylase inhibitor, trichostatin A. Cancer Res. Treat. 39, 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pospelova T. V.; Demidenko Z. N.; Bukreeva E. I.; Pospelov V. A.; Gudkov A. V.; Blagosklonny M. V. (2009) Pseudo-DNA damage response in senescent cells. Cell Cycle 8, 4112–4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado M.; Blasco M. A.; Serrano M. (2007) Cellular senescence in cancer and aging. Cell 130, 223–233. [DOI] [PubMed] [Google Scholar]
- Sipos B.; Moser S.; Kalthoff H.; Torok V.; Lohr M.; Kloppel G. (2003) A comprehensive characterization of pancreatic ductal carcinoma cell lines: towards the establishment of an in vitro research platform. Virchows Arch. 442, 444–452. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.