

Abstract
Polycyclic polyether natural products have fascinated chemists and biologists alike owing to their useful biological activity, highly complex structure and intriguing biosynthetic mechanisms. Following the original proposal for the polyepoxide origin of lasalocid and isolasalocid1 and the experimental determination of the origins of the oxygen and carbon atoms of both lasalocid and monensin, a unified stereochemical model for the biosynthesis of polyether ionophore antibiotics was proposed2. The model was based on a cascade of nucleophilic ring closures of postulated polyepoxide substrates generated by stereospecific oxidation of all-trans polyene polyketide intermediates2. Shortly thereafter, a related model was proposed for the biogenesis of marine ladder toxins, involving a series of nominally disfavoured anti-Baldwin, endo-tet epoxide-ring-opening reactions3–5. Recently, we identified Lsd19 from the Streptomyces lasaliensis gene cluster as the epoxide hydrolase responsible for the epoxide-opening cyclization of bisepoxyprelasalocid A6 to form lasalocid A7,8. Here we report the X-ray crystal structure of Lsd19 in complex with its substrate and product analogue9 to provide the first atomic structure—to our knowledge—of a natural enzyme capable of catalysing the disfavoured epoxide-opening cyclic ether formation. On the basis of our structural and computational studies, we propose a general mechanism for the enzymatic catalysis of polyether natural product biosynthesis.
Epoxide-opening cascades, including those using the disfavoured anti-Baldwin cyclization (Fig. 1a), have emerged as an important synthetic strategy in the field of organic chemistry10. Whereas little is known about ladder polyether biosynthesis, biosynthesis of ionophore polyethers (Fig. 1b) has been studied extensively11. Although most cyclic ethers in ionophore polyethers are the favoured exo-cyclization products (Fig. 1b, in blue), several compounds, including lasalocid A, contain six-membered rings presumably formed via a disfavoured endo-tet cyclization (Fig. 1b, in red).
Figure 1. Polyether natural products and proposed steps in the cyclic ether formation.

a, An example of a cascade epoxide ring closure implicated in the biosynthesis of ladder polyether. b, Structures of representative ionophore polyethers. c, Plausible mechanism of epoxide-mediated cyclic ether formation leading to the formation of lasalocid A, isolasalocid A and the product analogues.
Lsd19 was co-crystallized with the substrate analogue at pH 4.6, and the crystal structure was determined to 1.59 Å resolution (Supplementary Table 1). Lsd19 consists of two highly homologous domains (Fig. 2a) that belong to the nuclear transport factor 2 (NTF2)-like superfamily12, which includes Δ5-3-ketosteroid isomerases (KSIs)13 and limonene-1,2-epoxide hydrolase (LEH)14. Each domain consists of three α-helices and a cone-like six-stranded β-sheet whose cavity forms a deep substrate-binding pocket. The amino- and carboxy-terminal domains (Lsd19A and Lsd19B, respectively) are linked by a short loop and are oriented in a head-to-tail fashion, resembling the homodimer structure of single-domain NTF2-like proteins13. Lsd19A and Lsd19B have identical backbone conformations; their respective 117 core Cα atoms have a root-mean-squared deviation of just 0.79 Å. There are two significant differences between Lsd19A and Lsd19B. First, Lsd19B has an additional 18-residue-long loop–helix–loop near the entrance to its substrate-binding pocket (Fig. 2a, in green). Second, the active site of Lsd19A contains an uncyclized bisepoxide substrate (Fig. 2b), whereas the active site of Lsd19B contains a doubly cyclized tetrahydrofuran–tetrahydrofuran (THF–THF) product. This difference in active sites is consistent with our previously reported results that indicated that Lsd19A catalyses a single round of cyclization of the bisepoxide substrate, whereas Lsd19B cyclizes the initially formed THF ether/monoepoxide intermediate, predominantly to a tetrahydrofuran–tetrahydropyran (THF–THP) product11.
Figure 2. Crystal structure of Lsd19.
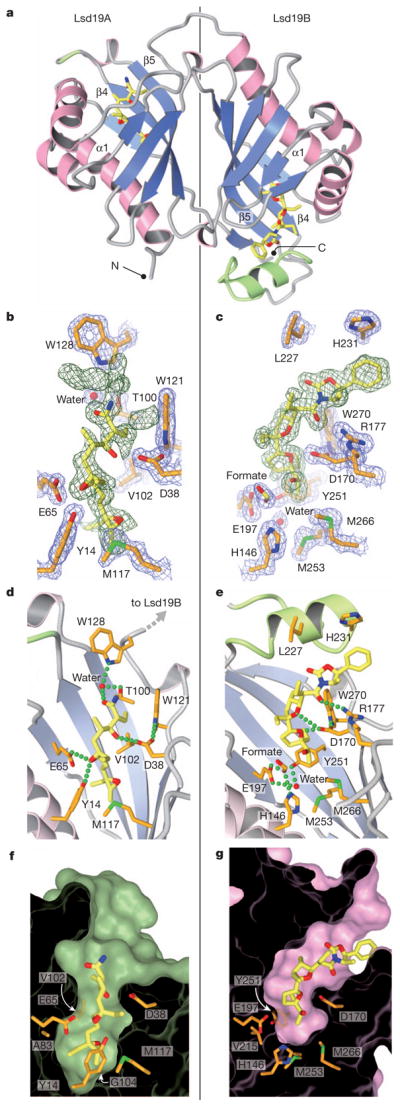
a, The overall fold of Lsd19. The extra loop–helix–loop in Lsd19B and the corresponding insertion site in Lsd19A are shown in green. b, c, Lsd19A (b) and Lsd19B (c) electron density for the ligand (simulated-annealing omit map contoured at 2.0σ, green mesh) and protein side chains (2Fo – Fc map contoured at 1.5σ, blue mesh). Carbon atoms of the bound ligands and protein are shown in yellow and orange, respectively. Oxygen, nitrogen and sulphur atoms are shown in red, blue and green, respectively. Green broken lines represent hydrogen bonds. d, e, Lsd19A (d) and Lsd19B (e) active-site hydrogen-bonding interactions between the ligand and protein side chains. The α1 helix (as labelled in panel a) was removed to clarify the view. f, g, Lsd19A (f) and Lsd19B (g) molecular surface cross-section, showing residues that determine the pocket size and shape.
In Lsd19A, the uncyclized substrate analogue is seen in the active site. This observation is consistent with the fact that Lsd19 is inhibited by the low pH of the crystallization buffer (Supplementary Fig. 1a). This characteristic is shared by many EHs15,16. The electron density for the oxazolidinone portion of the substrate analogue is ill-defined and therefore omitted from the final model (Fig. 2b). As the oxazolidinone is not present in the natural substrate, disorder in this region is not unexpected. In contrast, the portion of the substrate that contains the two epoxide groups that eventually undergo cyclization is clearly defined in the electron density map (Fig. 2b). The terminal 22,23-epoxide sits at the bottom of the pocket and is surrounded by hydrophobic residues, protected from potential non-specific nucleophilic attack. The putative nucleophilic 15-hydroxyl oxygen is hydrogen bonded to Asp 38, whereas the oxygen atom of the electrophilic 18,19-epoxide that undergoes ring opening lies within hydrogen-bonding distance to Tyr 14 and Glu 65. Arg 54 is hydrogen-bonded to the catalytic Asp 38 via a water molecule, presumably promoting deprotonation and favourable positioning of the side-chain carboxylic acid of Asp 38 for ensuing proton abstraction from the 15-hydroxyl group. This active-site configuration is homologous to that of KSI13 and LEH14, which perform similar acid/base catalysis (Fig. 3b). Unlike KSI or LEH, however, Lsd19 must perform conformationally sensitive intramolecular cyclizations on a flexible substrate. Additional interactions between Lsd19A and the 3-methylsalicylate side of the native substrate can help fix the ligand conformation for the ensuing cyclization. Here, a water molecule hydrogen bonded by Thr 100 and Trp 128 forms a hydrogen bond with the carbonyl oxygen of the bound substrate analogue (Fig. 2d). An equivalent oxygen atom is present in the natural substrate, bisepoxyprelasalocid A.
Figure 3. Computational studies of the Lsd19-catalysed epoxide-opening cyclization reactions.

a, Acid- versus base-catalysed cyclization of a model system via 5-exo or 6-endo cyclization, and the respective calculated lowest-energy competing transition structures. B2PLYP/6-311++G(d,p)//B2PLYP/ 6-31G(d) activation free energies (kcal mol−1) and forming/breaking bond distances (in Ångstroms, shown in blue) are given. b, Comparison of active-site geometries in Lsd19B and KSI (PDB accession 1E3V24). c, ‘Theozyme’ calculations, optimizing the competing 5-exo-tet and 6-endo-tet transition structures surrounded by fixed catalytic residues. Relative energies obtained by B2PLYP/6-311++G(d,p)/M06-2X/6-31G(d) (kcal mol−1) and forming/ breaking bond distances are given.
In Lsd19B, an isolasalocid-like THF–THF compound is seen in the active-site pocket instead of the expected lasalocid-like THF–THP reaction product. This unexpected reaction compound is presumably a consequence of the acidic crystallization condition (Supplementary Fig. 1b). We have identified His 146, Asp 170, Glu 197 and Tyr 251 as primary candidates for the catalytic residues in Lsd19B (Fig. 2e). Like in Lsd19A, a hydrogen bond between Asp 170 and His 186 could raise the pKa of His 186. The 13-carbonyl oxygen is hydrogen bonded to Arg 177, providing an additional enzyme–substrate interaction. The binding pocket of Lsd19B is shallower than that of Lsd19A due to the presence of Met 253 at the bottom of the pocket (Fig. 2g). This allows only the singly cyclized monoepoxide to bind in a catalytically relevant orientation, and promotes the critical substrate selectivity in these highly structurally similar domains. Although the shallower binding pocket in Lsd19B affords less substrate-binding energy available for confining the flexible substrate into the catalytically relevant conformation17, the additional loop–helix–loop structure (Fig. 2a, e, in green) seems to compensate for the lost substrate-binding surface.
Analysis of known polyether biosynthetic gene clusters reveals that two copies of polyether epoxide hydrolase (PEH)-encoding genes are present within each cluster: monBI, monBII (monensin)18 and nigBI, nigBII (nigericin)19. For lsd19 (lasalocid)6,7 and nanI (nanchangmycin)20, two copies are encoded as a single gene. The tetronomycin gene cluster21 carries a single copy of PEH gene tmnB, presumably because tetronomycin contains only one epoxide-derived THF ring. Sequence alignment of PEHs indicates that the catalytic residues identified in Lsd19 are highly conserved (Supplementary Fig. 2). This alignment also reveals that PEHs can be divided into two groups: PEH-A (Lsd19A-like) and PEH-B (Lsd19B-like). Only PEH-B has a bulky residue at position 104 that makes the binding pocket shallower, and only PEH-B contains a loop–helix–loop insertion that provides additional ligand-binding surface.
To extend the structural knowledge gained from Lsd19, homology models of other PEHs were constructed. In these models, a direct correlation is observed between the depth of the binding pocket and the length of the substrate chain (Supplementary Fig. 3), leading us to propose a general mode of cyclic ether formation in polyether biosynthesis. With the deeper binding pockets, PEH-As are probably responsible for the formation of internal cyclic ethers. Interestingly, PEH-As that act on longer substrates, such as nigericin, carry extra PEH-A-specific C-terminal residues (Supplementary Fig. 2) positioned at the opening of the binding pocket (Supplementary Fig. 3a), possibly providing additional binding surface for the longer substrates. In contrast, PEH-Bs with shallower binding pockets probably catalyse the formation of terminal cyclic ethers. As seen in Lsd19B, the extra PEH-B-specific loop–helix–loop domain can provide the binding surface for the portion of the substrate protruding out from the shallow substrate-binding pocket.
As the current Lsd19B structure does not address the formation of THP directly, we used computational studies to understand how this enzyme catalyses the disfavoured 6-endo cyclization (Methods and Supplementary Tables 2–4). Quantum mechanical density functional calculations were performed to locate each transition structure for both 5-exo and 6-endo epoxide openings22,23 under acid- and base-catalysed conditions using model substrates, and to calculate the energy difference between the two processes (Fig. 3a and Supplementary Table 2). As expected23, 5-exo-tet cyclization is favoured with both acid and base catalysis. However, the preference for the five-membered ring is reduced under base-catalysed conditions, due to the increased steric hindrance of nucleophilic attack at the more substituted epoxide terminus, as well as the more product-like transition structure with shorter forming O–C bond lengths. The six-membered product is indeed thermodynamically favoured. This provides an indication of how Lsd19B may achieve the six-membered ring formation. We then explored how Lsd19B would influence the regioselectivity of the ring-opening transition state. Docking studies on the Lsd19B structure indicated that Asp 170, Glu 197 and Tyr 251 maintain close contact with the bound ligand. Similarities between the active site organization of Lsd19B and KSI13 (Fig. 3b) further support the idea that Asp 170 acts as a general base, whereas Glu 197 and Tyr 251 act as general acids stabilizing the developing transition structure oxyanion. This arrangement is also remarkably similar to the ‘theozyme’ derived from computations for the anti-Baldwin-cyclization antibody23, which was eventually found to be closely related to the antibody structure24. To test this idea quantitatively, competing 5-exo and 6-endo transition structures were optimized in the presence of Asp 170 and Tyr 251, whose geometry was constrained to the crystal structure coordinates. From this model, values of the activation energy and Gibbs free energy changes for ring closure were computed. These predict a preference for the 6-endo pathway of 2.5 kcal mol−1, enough to achieve nearly 100:1 selectivity (Fig. 3c). The positioning of the catalytic general acid and general base groups causes the 6-endo transition state to be favoured, leading to the thermodyamically more stable THP product.
The structural knowledge gained here has provided a generalized view of the biosynthetic pathway involved in producing polyether natural products using only a pair of epoxide hydrolases. Furthermore, computational studies have provided an explanation for the regiochemical preference for the Lsd19-catalysed cyclization. Active-site pre-organization and general-base catalysis provides enzymatic control to overcome otherwise disfavoured chemical transformations. Similarly, stereo-chemically complex polyether ladder structures can be generated from a simple polyepoxide substrate by Lsd19B-like EHs that can form templating THP unit(s) that facilitate the subsequent cascade of step-wise endo-tet-selective epoxide-opening cyclizations.
METHODS
Lsd19 expression and purification
Production of Lsd19 was performed partly according to the procedure described elsewhere8. Briefly, pCold-based pKW620 carrying the lsd19 gene and the chaperone-encoding vector pG-KJE8 (Takara Bio) were introduced into Escherichia coli BL21(DE3) strain. The culture was grown in Luria Broth medium to OD600 nm of 0.6, and the production of N-terminal His-tagged Lsd19 was induced by 100 μM isopropyl-β-D-galactoside, 0.5 mg ml−1 L-arabinose and 5 ng ml−1 tetracycline. The culture was incubated for another 20 h at 15 °C. Cells were harvested by centrifugation, resuspended in 50 mM sodium phosphate pH 7.4, 300 mM sodium chloride, 10 mM imidazole, 10% (v/v) glycerol and lysed by sonication. After centrifugation at 15,000g for 45 min, the cleared supernatant was mixed with cobalt-agarose beads (Thermo Fisher Scientific). The mixture was incubated for 1 h at 4 °C with moderate shaking. After incubation, the mixture was loaded onto a column and washed with a wash buffer containing 50 mM sodium phosphate pH 7.4, 300 mM sodium chloride, 10 mM imidazole and 10% (v/v) glycerol. Lsd19 was eluted with a wash buffer supplemented with 150 mM imidazole. Fractions containing Lsd19 were pooled and exchanged into a buffer containing 20 mM Tris pH 8.5, 1 mM EDTA and 15 mM β-mercaptoethanol. Lsd19 was further purified by anion exchange chromatography using a HiTrapQ XL column followed by gel filtration on a Superdex200 10/300 GL column (GE Healthcare Life Sciences) in 50 mM potassium phosphate pH 7.0, 1 mM EDTA and 15 mM β-mercaptoethanol. The final protein was >95% pure as judged by polyacrylamide gel electrophoresis, and the yield was approximately 2 mg per litre of culture. The sample was further concentrated to 12 mg ml−1 for storage.
Crystallization, data collection and structure determination
Crystals were obtained by the hanging-drop vapour diffusion method. The bisepoxide substrate analogue was prepared as described8. After combining Lsd19 (6 mg ml−1) with the substrate analogue (100 mM in methanol) at 12.5:1 ratio (v/v) on ice, the sample was mixed with an equal volume of 0.1 M sodium acetate pH 4.6 and 1.6 M sodium formate and equilibrated at 18 °C. Crystals were further optimized using an additive 1,5-diaminopentane dihydrochloride in the presence of 200–700 μl Al’s oil in the well. Before flash freezing, Lsd19 crystals were transferred for about 1 min to a cryoprotectant solution comprised of the crystallization buffer supplemented with 35% glycerol. X-ray diffraction data were collected from a single crystal to a 1.59 Å resolution at beam line 7-1 of Stanford Synchrotron Radiation Lightsource (SSRL; SLAC National Accelerator Laboratory). Data collection was performed on a single crystal at 100 K using a monochromatic X-ray at a wavelength of 0.9795 Å Data were indexed and integrated using XDS25. Structure factor was obtained by CTRUNCATE30. The structure of Lsd19 was determined by molecular replacement with the program MOLREP31 using the Pseudomonas putida biotype B KSI structure (PDB accession 1DMN)37 as a search model. The model was built by Coot32 and ARP/wARP33 and refined by CNS26 and REFMAC5 (CCP4)27. Ramachandran plot of the refined model indicated that all of the residues assumed allowed backbone conformation with 98.2% within the favoured conformation. Other relevant data and refinement statistics are given in Supplementary Table 1.
Computation
Quantum chemical calculations were performed using Gaussian 09, revision A.228. All geometries were fully optimized. Transition structures were identified by a single imaginary harmonic vibrational frequency. Optimizations were performed with default convergence criteria at the B2PLYP/6-31G(d) level of theory using a fine grid for numerical integration. Free energies were computed in the harmonic approximation at 298 K and 1 atm, with unscaled zero-point vibrational energies. Docking Studies were performed with Autodock Vina29.
Single point energy calculations were performed to ensure basis set convergence at the B2PLYP/6-311++G(d,p)//B2PLYP/6-31G(d) level. Calculations were performed at the ‘double-hybrid’ density functional level of theory with the B2PLYP functional34. This method replaces a fraction of the semi-local correlation energy by a non-local correlation energy expression that uses the Kohn–Sham orbitals in second-order perturbation theory and delivers improved energetics over hybrid density functionals. Results obtained at this level were also compared with the hybrid meta-generalized gradient approximation M06-2X density functional. B2PLYP and M06-2X calculations are in accord, both qualitatively and quantitatively, over the preference for 5-exo-tet over 6-endo-tet in the acid- versus base-catalysed cyclization of the model systems. Absolute barriers showed some variations; however, the energy differences between the two cyclization modes were consistent. Changing between double and triple zeta-valence polarized basis sets showed only small effects (Supplementary Table 2).
The effects of solvation were treated with single point energy calculations on the gas-phase geometries using a conductor-like polarizable continuum (CPCM) model35 of water (ρ = 78.36) and dichloromethane (ρ = 8.93), defining the solute cavity by UFF radii. As for gas-phase values, B2PLYP and M06-2X results are in accord over preferred reaction pathway (Supplementary Table 3).
‘Theozyme’ calculations were performed using a truncated model of the active site, constraining the positions of the non-hydrogen atoms in the catalytic residues Glu 197 and Tyr 251 in their crystal structure coordinates. All other atom positions are optimized. Electrostatic effects of the protein interior were described with a CPCM model of low dielectric constant, using diethyl ether as solvent (ρ = 4.24)36. A variety of methods show consistent energy differences between 5-exo and 6-endo pathways (Supplementary Table 4). Absolute energies and Cartesian coordinates for all species in the text are given in the Supplementary Data. Absolute gas-phase energies and free energies (1 atm, 298 K) from B2PLYP/6-31G(d) optimization, B2PLYP/6-311++G(d,p) single point energies, single point energies including CPCM solvation (UAKS solute radii) are all given in kcal mol−1. Imaginary harmonic vibrations are in cm−1.
Supplementary Material
Acknowledgments
This work was supported by the Royal Commission for the Exhibition of 1851 and Fulbright-AstraZeneca Research Fellowship (R.S.P.), the Japan Society for the Promotion of Science (No. LS103) (K.W.), the National Institutes of Health grant GM075962 (K.N.H.), the MEXT research grant on innovative area 22108002 (H.O.), and the National University of Singapore Life Sciences Institute Young Investigator Award (C.-Y.K.). Data collection was performed at the Stanford Synchrotron Radiation Lightsource. We thank D. W. Christianson, D. Hilvert and C. Khosla for critical reading and discussion of the manuscript.
Footnotes
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions A.M. and H.O. prepared the substrate analogue. K.W. cloned and purified Lsd19. X.C. and H.L. purified and crystallized Lsd19. X.C. and I.I.M. collected diffraction data and determined the structure. K.H., X.C. and I.I.M. refined the structure. K.S. provided assistance for crystallography. K.N.H. prepared and analysed models of Lsd19 homologues. R.S.P. and K.N.H. performed the computational study. C.-Y.K. conceived and supervised the project. C.-Y.K. prepared the manuscript with contributions from all co-authors.
Author Information The coordinates and structure factors have been deposited with the Protein Data Bank under accession number 3RGA. Reprints and permissions information is available at www.nature.com/reprints. Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests. Readers are welcome to comment on the online version of this article at www.nature.com/nature.
References
- 1.Westley JW, Blount JF, Evans RH, Jr, Stempel A, Berger J. Biosynthesis of lasalocid II X-ray analysis of a naturally occurring isomer of lasalocid A. J Antibiot (Tokyo) 1974;27:597–604. doi: 10.7164/antibiotics.27.597. [DOI] [PubMed] [Google Scholar]
- 2.Cane DE, Celmer WD, Westley JW. Unified stereochemical model of polyether antibiotic structure and biogenesis. J Am Chem Soc. 1983;105:3594–3600. [Google Scholar]
- 3.Nakanishi K. The chemistry of brevetoxins: a review. Toxicon. 1985;23:473–479. doi: 10.1016/0041-0101(85)90031-5. [DOI] [PubMed] [Google Scholar]
- 4.Baldwin JE. Rules for ring closure. J Chem Soc Chem Comm. 1976:734–736. [Google Scholar]
- 5.Gilmore K, Alabugin IV. Cyclizations of alkynes: revisiting Baldwin’s rules for ring closure. Chem Rev. 2011;111:6513–6556. doi: 10.1021/cr200164y. [DOI] [PubMed] [Google Scholar]
- 6.Shichijo Y, et al. Epoxide hydrolase Lsd19 for polyether formation in the biosynthesis of lasalocid A: direct experimental evidence on polyene-polyepoxide hypothesis in polyether biosynthesis. J Am Chem Soc. 2008;130:12230–12231. doi: 10.1021/ja8040543. [DOI] [PubMed] [Google Scholar]
- 7.Migita A, et al. Identification of a gene cluster of polyether antibiotic lasalocid from Streptomyces lasaliensis. Biosci Biotechnol Biochem. 2009;73:169–176. doi: 10.1271/bbb.80631. [DOI] [PubMed] [Google Scholar]
- 8.Smith L, Hong H, Spencer JB, Leadlay PF. Analysis of specific mutants in the lasalocid gene cluster: evidence for enzymatic catalysis of a disfavoured polyether ring closure. ChemBioChem. 2008;9:2967–2975. doi: 10.1002/cbic.200800585. [DOI] [PubMed] [Google Scholar]
- 9.Minami A, et al. Enzymatic epoxide-opening cascades catalyzed by a pair of epoxide hydrolases in the ionophore polyether biosynthesis. Org Lett. 2011;13:1638–1641. doi: 10.1021/ol200100e. [DOI] [PubMed] [Google Scholar]
- 10.Nakata T. Total synthesisof marine polycyclic ethers. Chem Rev. 2005;105:4314–4347. doi: 10.1021/cr040627q. [DOI] [PubMed] [Google Scholar]
- 11.Westley JW. Antibiotic structure and biosynthesis. J Nat Prod. 1986;49:35–47. [Google Scholar]
- 12.Marchler-Bauer A, et al. CDD: specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 2009;37:D205–D210. doi: 10.1093/nar/gkn845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ha NC, Choi G, Choi KY, Oh BH. Structure and enzymology of Δ5-3-ketosteroid isomerase. Curr Opin Struct Biol. 2001;11:674–678. doi: 10.1016/s0959-440x(01)00268-8. [DOI] [PubMed] [Google Scholar]
- 14.Hopmann KH, Hallberg BM, Himo F. Catalytic mechanism of limonene epoxide hydrolase, a theoretical study. J Am Chem Soc. 2005;127:14339–14347. doi: 10.1021/ja050940p. [DOI] [PubMed] [Google Scholar]
- 15.van der Werf MJ, Overkamp KM, de Bont JA. Limonene-1,2-epoxide hydrolase from Rhodococcus erythropolis DCL14 belongs to a novel class of epoxide hydrolases. J Bacteriol. 1998;180:5052–5057. doi: 10.1128/jb.180.19.5052-5057.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Touhara K, Prestwich GD. Juvenile hormone epoxide hydrolase. Photoaffinity labeling, purification, and characterization from tobacco hornworm eggs. J Biol Chem. 1993;268:19604–19609. [PubMed] [Google Scholar]
- 17.Jencks WP. Binding energy, specificity, and enzymic catalysis: the Circe effect. Adv Enzymol. 1975;43:219–410. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- 18.Leadlay PF, et al. Engineering of complex polyketide biosynthesis—insights from sequencing of the monensin biosynthetic gene cluster. J Ind Microbiol Biotechnol. 2001;27:360–367. doi: 10.1038/sj.jim.7000204. [DOI] [PubMed] [Google Scholar]
- 19.Harvey BM, et al. Insights into polyether biosynthesis from analysis of the nigericin biosynthetic gene cluster in Streptomyces sp DSM4137. Chem Biol. 2007;14:703–714. doi: 10.1016/j.chembiol.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 20.Sun Y, et al. A complete gene cluster from Streptomyces nanchangensis NS3226 encoding biosynthesisof the polyetherionophore nanchangmycin. Chem Biol. 2003;10:431–441. doi: 10.1016/s1074-5521(03)00092-9. [DOI] [PubMed] [Google Scholar]
- 21.Demydchuk Y, et al. Analysis of the tetronomycin gene cluster: insights into the biosynthesis of a polyether tetronate antibiotic. ChemBioChem. 2008;9:1136–1145. doi: 10.1002/cbic.200700715. [DOI] [PubMed] [Google Scholar]
- 22.Gruber K, et al. Structural basis for antibody catalysis of a disfavored ring closure reaction. Biochemistry. 1999;38:7062–7074. doi: 10.1021/bi990210s. [DOI] [PubMed] [Google Scholar]
- 23.Na J, Houk KN, Shevlin CG, Janda KD, Lerner RA. The energetic advantage of 5-exo versus 6-endo epoxide openings: a preference overwhelmed by antibody catalysis. J Am Chem Soc. 1993;115:8453–8454. [Google Scholar]
- 24.Ha NC, Kim MS, Lee W, Choi KY, Oh BH. Detection of large pKa perturbations of an inhibitor and a catalytic group at an enzyme active site, a mechanistic basis for catalytic power of many enzymes. J Biol Chem. 2000;275:41100–41106. doi: 10.1074/jbc.M007561200. [DOI] [PubMed] [Google Scholar]
- 25.Kabsch W. XDS. Acta Crystallogr D. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brunger AT. Version 1.2 of the Crystallography and NMR system. Nature Protocols. 2007;2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 27.Vagin AA, et al. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr D. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- 28.Frisch MJ, et al. Gaussian 09, Revision A.2. Gaussian; 2009. [Google Scholar]
- 29.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Padilla JE, Yeates TO. A statistic for local intensity differences: robustness to anisotropy and pseudo-centering and utility for detecting twinning. Acta Crystallogr D. 2003;59:1124–1130. doi: 10.1107/s0907444903007947. [DOI] [PubMed] [Google Scholar]
- 31.Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr D. 2010;66:22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 32.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nature Protocols. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grimme S. Semiempirical hybrid density functional with perturbative second-order correlation. J Chem Phys. 2006;124:034108. doi: 10.1063/1.2148954. [DOI] [PubMed] [Google Scholar]
- 35.Barone V, Cossi M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J Phys Chem A. 1998;102:1995–2001. [Google Scholar]
- 36.Zhang X, et al. Quantum mechanical design of enzyme active sites. J Org Chem. 2008;73:889–899. doi: 10.1021/jo701974n. [DOI] [PubMed] [Google Scholar]
- 37.Kim DH, et al. Contribution of the hydrogen-bond network involving a tyrosine triad in the active site to the structure and function of a highly proficient ketosteroid isomerase from Pseudomonas putida biotype B. Biochemistry. 2000;39:4581–4589. doi: 10.1021/bi992119u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.