

Abstract
Coagulation factor XI (FXI) plays an important part in both venous and arterial thrombosis, rendering FXIa a potential target for the development of antithrombotic therapy. The kunitz protease inhibitor (KPI) domain of protease nexin-2 (PN2) is a potent, highly specific inhibitor of FXIa, suggesting its possible role in the inhibition of FXI-dependent thrombosis in vivo. Therefore, we examined the effect of PN2KPI on thrombosis in the murine carotid artery and the middle cerebral artery. Intravenous administration of PN2KPI prolonged the clotting time of both human and murine plasma, and PN2KPI inhibited FXIa activity in both human and murine plasma in vitro. The intravenous administration of PN2KPI into WT mice dramatically decreased the progress of FeCl3-induced thrombus formation in the carotid artery. After a similar initial rate of thrombus formation with and without PN2KPI treatment, the propagation of thrombus formation after 10 minutes and the amount of thrombus formed were significantly decreased in mice treated with PN2KPI injection compared with untreated mice. In the middle cerebral artery occlusion model, the volume and fraction of ischemic brain tissue were significantly decreased in PN2KPI-treated compared with untreated mice. Thus, inhibition of FXIa by PN2KPI is a promising approach to antithrombotic therapy.
Introduction
Coagulation factor XI (FXI) has an essential role both in maintaining normal hemostasis and in the pathogenesis of thrombosis.1 Because deficiencies of factor XII (FXII), prekallikrein, and high Mr kininogen are not associated with hemostatic abnormalities but FXI deficiency produces relatively mild, posttraumatic bleeding complications in ∼ 50% of affected individuals,2–4 the more relevant pathway for activation of plasma FXI might be via feedback activation by thrombin or possibly autoactivation by FXIa.5,6 High levels of FXI constitute a risk factor for deep venous thrombosis (DVT)7 and cardiovascular disease in women.8 Although inherited FXI deficiency confers no protection against acute myocardial infarction (AMI),9 a large clinical study showed that the risk of DVT in the 10% of individuals in a population with the highest levels of FXI was > 2-fold greater than that for the remaining 90% of the population, supporting the conclusion that elevated FXI levels constitute an important risk factor for venous thrombosis.7,8,10 There is also a reduced incidence of ischemic stroke in patients with severe FXI deficiency.11 One study followed 600 patients with a first episode of venous thromboembolism and found an increased risk of recurrence, especially among those with increased levels of thrombin-activatable fibrinolysis inhibitor (TAFI) and FXI,12 whereas another study showed that increased levels of TAFI, but not FXI, increased the risk of venous thromboembolism in patients with FVLeiden carrier status.10 These studies are consistent with the results of studies demonstrating increased rabbit jugular vein thrombolysis by FXI Ab neutralization because of diminished indirect activation of TAFI.13 Two studies in mice with targeted FXI gene deletion, either alone14 or in combination with targeted deficiency of protein C,15 showed protection from FeCl3-induced carotid thrombosis14 or from early lethality caused by thrombosis in protein C–deficient animals.15 Another study reported the important role of FXI in the growth of arterial thrombi using a rabbit model of arterial thrombosis,16 whereas another demonstrated the dependence of surface- and tissue factor–initiated thrombus propagation on FXI in primates.17 Finally, the majority of patients with the acute coronary syndrome and coronary artery disease with a history of AMI have increased levels of circulating FXIa that correlate with markers of coagulation and inflammation,18 and patients with severe FXI deficiency have a reduced incidence of DVT.19 Thus, FXI/XIa levels in plasma constitute an important thrombogenic factor, and either FXI or FXIa provide a potential target for antithrombotic therapy.20
Important control mechanisms exist for the regulation of coagulation protease activities, including FXIa. Serine protease inhibitors (referred to as SERPINs) have been shown to be physiologic regulators of FXIa function in plasma, including protease nexin 1, antithrombin III, C1 inhibitor, α-1-protease inhibitor, and α-2-antiplasmin (reviewed in Walsh and Gailani1). Another class of inhibitors, referred to as kunins, have also been identified as important regulators of protease function in plasma. Thus, following platelet activation by physiologic stimulators, protease nexin 2 (PN2) is secreted from α-granules into plasma and inhibits FXIa.21–23 PN2, a kunitz-type inhibitor, is a ∼ 120-kDa isoform of the Alzheimer β-amyloid protein precursor (AβPP) that has been shown to be a highly potent and physiologically relevant inhibitor of FXIa,21,22,24–27 with little or no detectable inhibitory activity against other coagulation enzymes, including thrombin, FVIIa, FIXa, FXa, FXIIa, plasmin, or kallikrein.28 PN2 is a slow, tight binding inhibitor of FXIa with a Ki of 290-450pM.21,24,26 The kunitz protease inhibitor (KPI) domain of PN2 (PN2KPI) is 57 residues in length (Glu289-Ile345 in the 751-amino acid isoform of PN2) and contains the entire FXIa inhibitory function of PN2.25–27 A homologous protein belonging to the same kunitz family, basic (or bovine) pancreatic trypsin inhibitor (BPTI; aprotinin; Trasylol) is also an inhibitor of several human serine proteases including trypsin, plasmin, kallikrein, activated protein C, and FXIa.29,30 Because BPTI has in the past been commonly used clinically to control plasma proteolysis during and after cardiopulmonary bypass surgery, it seems feasible to examine the in vivo inhibitory activity of the KPI domain of PN2 as an antithrombotic agent. Therefore, in the present study, we examined the effect of PN2KPI on thrombus formation in a murine model of FeCl3-induced carotid artery thrombosis and stroke in the mouse middle cerebral artery occlusion (MCAO) model.
Methods
Expression and purification of recombinant KPI (PN2KPI) domain of PN2
The PN2KPI was expressed in Pichia pastoris and purified as reported.31 Briefly, the sequence of the PN2 gene which encodes the KPI domain was inserted into the expression vector PICα, and P pastoris transfected with this expression vector was expanded in BMGY medium before transfer to BMMY medium for PN2KPI expression in the presence of methanol (0.1%). The medium containing expressed PN2KPI was harvested; the protein was precipitated in 100% ammonium sulfate, resuspended in Tris buffer (Tris, 25mM, pH 7.5), and extensively dialyzed against Tris buffer. The PN2KPI was further purified by ion exchange using a Hitrap Q column on AKATA UPC 950 FPLC (Amersham Biosciences), followed by gel filtration using a HiLoad 30, 16/60 column (Amersham). The concentration of purified PN2KPI was determined by bicinchoninic acid (BCA) assay (Pierce). Its FXIa inhibitory effect was characterized using a kinetic assay reported earlier.28,31
Inhibition of plasma FXIa activity by PN2KPI
Two separate assays were used to measure the anticoagulant activity of PN2KPI. The effect of PN2KPI on blood coagulation was examined using the activated partial thromboplastin time (APTT) assay. Normal pooled human plasma, containing 3.2% trisodium citrate, was purchased from George King Biomedical Inc. Mouse blood, obtained by heart puncture, was drawn into syringes rinsed in trisodium citrate and mixed immediately with trisodium citrate to achieve a final concentration of 3.2%. The plasma was separated from whole blood by centrifugation at 1200g for 20 minutes. Twenty-five microliters of mouse plasma was mixed with 75 μL of TBS buffer (Tris, 25mM NaCl, 125mM, BSA 0.1%, pH 7.5) containing various concentrations (0.5-60μM) of KPI and incubated with 25 μL of APTT reagent (Instrumentation Laboratories) at 37°C for 10 minutes, after which 25 μL of 20mM CaCl2 was added, and the clotting time was determined in an Amelung KC4 microcoagulometer (Amelung GmbH).
In the second assay, the inhibitory effect of PN2KPI against either human or mouse plasma FXIa was determined, using a modified APTT assay. Initially, a standard curve between the clotting time and FXI activity was generated as follows. Serial dilutions of mouse or human plasma (12.5 μL) were mixed with an equal volume of human FXI-deficient plasma (obtained from Haematologic Technologies Inc) and 75 μL of TBS buffer. After incubation with 25 μL of APTT reagent for 10 minutes, 25 μL of 20mM CaCl2 was added and the clotting time was determined in an Amelung KC4 microcoagulometer (Amelung GmbH). The FXIa inhibitory activity of PN2KPI on either mouse plasma or human plasma was then measured using the same protocol, except that the TBS buffer contained various concentrations (0-50μM) of PN2KPI. The remaining FXIa activity was determined by plotting the resulting clotting time against the standard curve.
The effect of PN2KPI on blood coagulation administered to mice in vivo was examined in similar fashion as discussed in the previous paragraph. The blood withdrawn from mice treated with PN2KPI was used in both the whole-plasma APTT and in the FXIa activity assay. The clotting time was plotted against the standard curves, respectively, to determine the plasma FXIa concentration.
Animal models and pathologic studies
Carotid artery thrombosis model.
The carotid artery thrombosis model was established in 7- to 11-week-old female C57BL/6 mice following the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the protocol approved by the Temple University Institutional Animal Care and Use Committee. The mice were anesthetized with an intraperitoneal injection of a 1:1 combination of ketamine (100 mg/mL) and xylazine (20 mg/mL) at a dose of 1 mL/kg. Rhodamine (0.2 mL, 0.05%) and PN2KPI (90 μg in 0.2 mL of normal saline) were injected through the tail vein. An incision was made along the midline of the anterior neck and the sternocleidomastoideus of one side was dissected caudally, avoiding cutting the vessels around it, to expose the carotid artery. A slightly curved forceps (Micro Dissecting Forceps, 4 inches, serrated, 70-1010) with its tip wrapped in tape was placed under the carotid artery to detach it from the jugular vein and the vagus nerve. Thrombus formation was initiated by placing filter paper (1 × 1 mm2) soaked with 7.5% FeCl3 solution on the surface of the exposed carotid artery for 3 minutes. After removal of the filter paper, the vessel was rinsed with 0.9% saline solution. The progress of thrombosis was observed with an epiilluminiscence microscope (BX10; Olympus), which was equipped with a digital camera (Cooke 1600; Cooke Corporation). A series of brief videos were recorded using Camware software (Cooke Corporation) at different time points (1, 3, 5, 7,10,15, 20, 30, and 40 minutes) after carotid artery injury. The size of the thrombus was estimated by measurement of the fluorescence intensity of thrombus inside the vessel based on the image acquired from the recorded video with NIH ImageJ software after background correction.
A 2-mm section of the carotid artery within the site of injury was taken as a pathologic sample and was treated with 4% paraformaldehyde solution overnight. The tissue was embedded in optimal cutting temperature compound from Tissue-Tek (Sakura Finetek), frozen with dry ice, and then stored at −80°C until sectioned. The tissue was sliced into 10-μm serial sections and stained with hematoxylin and eosin. The stained tissue sections were visualized with an Olympus BX 51 microscope and photographed using a Nikon DP70 camera.
MCAO model.
The mice were anesthetized as described in “Carotid artery thrombosis model.” The body temperature was maintained at 37 ± 0.5°C using a heating lamp and pad. The intraluminal filament method32 was used to promote MCAO. The external carotid artery (ECA) was ligated distal to the internal carotid artery (ICA)–ECA branch and the distal portion was removed from the ligation point. A blunted 5-0 monofilament nylon suture coated with poly-L-lysine (0.1% in deionized water; Sigma-Aldrich) was introduced through a small incision in the ECA, into the ICA, and then advanced into the circle of Willis, and finally to the origin of the middle cerebral artery (MCA). The nylon suture was then removed after 45 minutes of occlusion, and the ECA was permanently ligated. A laserPro Blood Perfusion Monitor (TSI Inc) was used to monitor regional cerebral blood flow before ischemia, during MCAO, and during reperfusion. The MCAO was considered adequate if regional cerebral blood flow showed a sharp drop to 25% of baseline (pre-ischemia) level. The PN2KPI was administered after the MCA was occluded in a bolus injection at the same dosage used in the carotid thrombosis model. The animals were euthanized with an overdose of pentobarbital (200 mg/kg intraperitoneally) 24 hours after MCAO, and the brains removed. Five 2-mm coronal sections were obtained using a mouse brain matrix (Zivic Instruments). The brain sections were stained with 2% triphenyltetrazolium chloride (TTC; Sigma-Aldrich), and the anterior and caudal faces of each section were scanned using a flatbed color scanner (Microtek Inc). The resulting images were captured as JPEG files and analyzed using ImageJ software. The non-TTC staining area of both sides of each section was measured, and their average was multiplied by the thickness of each section (2 mm) to obtain the infarct volume of that section. The infarct volume of the whole brain was calculated by adding the infarct volume of all 5 sections. To calculate the infarct fraction (%), the volume of the ipsilateral hemisphere was first subtracted from the sum of the infarct volume and contralateral brain volume, and this value was further divided by the contralateral volume as described by Zhang et al,33 Swanson et al,34 and Lin et al.35 The stroke volume and fraction in MCAO model was compared between PN2KPI- and vehicle-treated mice.
Tail bleeding time.
After placing the anesthetized mouse in an animal restrainer, the tail was washed with 70% ethanol and rinsed with prewarmed 0.9% saline. The end of the mouse tail was transected with a scalpel at a position where the diameter of the tail was ∼ 1 mm. The tail was hung over the edge of the table and immersed in 0.9% isotonic saline at 37°C, and the time from the incision to the cessation of the stream of blood was recorded as tail vein bleeding time. If the bleeding did not stop after 10 minutes, the tail was cauterized and the bleeding time was recorded as 600 seconds.36
Statistical analysis
All experimental manipulations and observations were carried out on coded samples/animals by an individual (H.L.) blinded to the experimental group, that is, PN2KPI administration versus control animals. All data are shown as mean values ± SEM and were compared between experimental groups. Differences between the PN2KPI group and the control group were evaluated using a paired 2-tailed t test with statistical significance determined as P < .05.
Results
The expression and purification of PN2KPI
The PN2KPI expressed in P pastoris was highly purified (> 98% pure) after passing through ion exchange and gel filtration columns, as previously reported.28,31 The inhibitory effect against purified FXIa (Ki 0.5-1.4nM) was consistent with that previously reported.31
The inhibition of blood plasma FXIa activity by recombinant KPI
The PN2KPI added to mouse plasma inhibited the APTT assay as shown in Figure 1A. The inhibitory effect of PN2KPI on the APTT was also observed in human plasma, as previously reported.31 The coagulation times in the APTT assay were prolonged when different concentrations (0-50μM) of PN2KPI were added to either human (Figure 1B) or mouse (Figure 1C) plasma. However, the PN2KPI was a more potent and complete inhibitor of coagulation in human than in murine plasma. The IC50 of the PN2KPI inhibitory effect in human plasma (Figure 1B) was ∼ 125nM, whereas in mouse plasma the IC50 was ∼ 2μM (Figure 1C). Plasma samples withdrawn from PN2KPI-treated animals also demonstrated prolonged clotting times in the APTT assay (47.8 ± 0.8 seconds, mean of 5 determinations) and in the FXI activity assay (72.2 ± 1.1 seconds, mean of 5 determinations). When the clotting times obtained in the APTT assay and the FXI activity assay were plotted against their respective standard curves, the estimated plasma concentration of PN2KPI in mouse plasma after PN2KPI treatment was ∼ 1-2μM, and the FXIa activity remaining in the murine plasma at concentrations of PN2KPI > 2μM was ∼ 25%-50% of the activity in untreated murine plasma (see Figure 1C).
Figure 1.
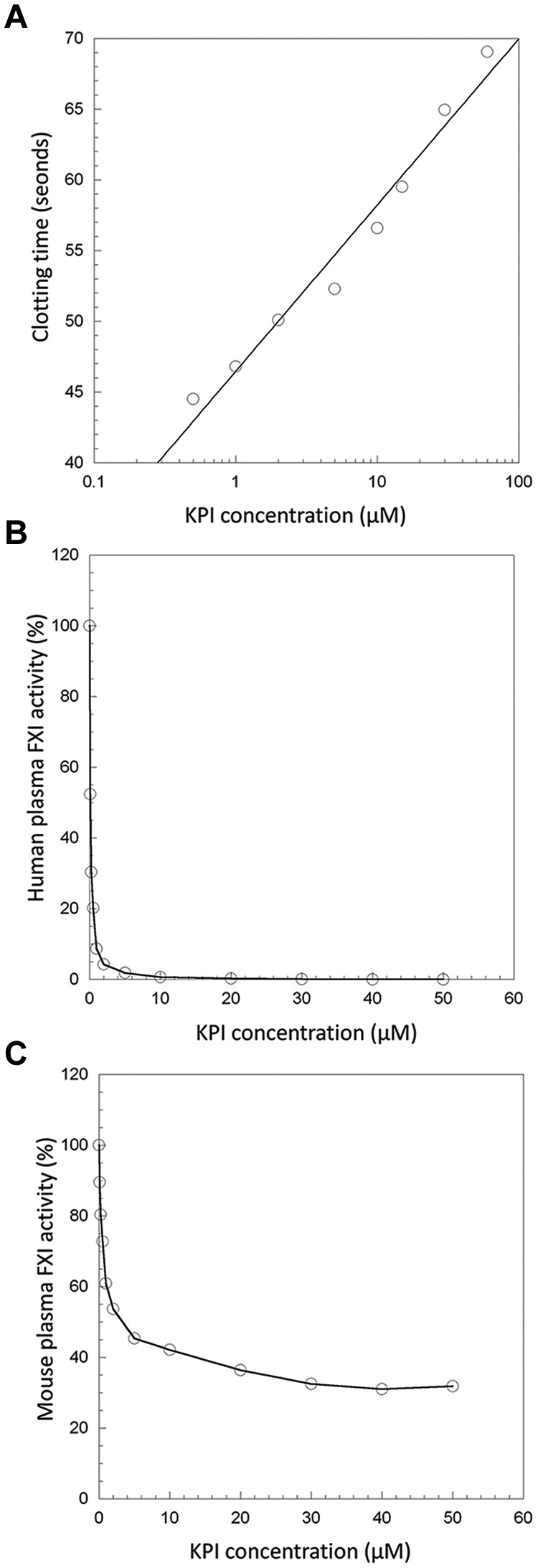
The inhibitory effect of PN2KPI in human and murine plasma. (A) The clotting time of mouse plasma is prolonged with externally added PN2KPI. (B) FXI activity in human plasma is inhibited by externally added PN2KPI and inhibitory activity is saturated at PN2KPI concentrations > 10μM. (C) Mouse FXI activity is ∼ 75% inhibited by human KPI and inhibitory activity is saturated at PN2KPI concentrations > 10μM.
Thrombus formation in the mouse model of FeCl3-induced carotid artery thrombosis
Thrombus formation was initially observed ∼ 3 minutes after FeCl3-induced carotid injury in both PN2KPI-treated and control animals, followed by a rapid growth phase of the thrombus until 10 minutes, during which no significant difference in thrombus size was recorded between the 2 groups of the mice (Figure 2A-B). However, thrombus accumulation continued rapidly in control animals, achieving a maximum plateau at 30-40 minutes after injury. In contrast, thrombus accumulation was curtailed after ∼ 5 minutes postinjury in mice treated with PN2KPI, and in some PN2KPI-treated animals, small portions of formed thrombi were flushed away by the high rate of blood flow. When the growth of thrombus size was plotted against time in the 2 groups of animals (Figure 2C), it was apparent that the initial rate of thrombus growth was similar during the initial 3-minute period after injury in the PN2KPI-treated and control animals, whereas after 5-7 minutes the PN2KPI-treated mice demonstrated a low-level plateau of thrombus accumulation and the control group continued to accumulate thrombus mass that plateaued at 30-40 minutes. The difference in thrombus size between the 2 groups achieved statistical significance after 10 minutes (P < .05). The pathologic sections of the samples of injured carotid artery showed that the sizes of thrombi formed within the lumen of the vessel of PN2KPI-treated mice were much smaller than those obtained from untreated animals, although animals from both groups showed evidence of similar damage caused by FeCl3 (Figure 3).
Figure 2.

Inhibitory effect of PN2KPI on thrombus development in the FeCl3-induced mouse carotid artery thrombosis model. Thrombus formation is recorded by fluorescence microscopy as described in “Carotid artery thrombosis model.” The image of thrombus growth was captured from video clips recorded at different time points. Thrombus growth (A) in a representative untreated mouse and (B) in a PN2KPI-treated mouse was recorded. (C) Thrombus growth, represented by the artificial fluorescence intensity unit (AFU), was measured at various time points (1, 3, 5, 7,10,15, 20, 30, and 40 minutes) in 6 control (□) and 6 PN2KPI-treated (○) mice. Results shown are mean values ± SEM. *Significant differences with P < .05.
Figure 3.

Pathologic sections of carotid artery thrombosis. (A) The histologic section of a normal carotid artery, stained with hematoxylin and eosin; (B-C) FeCl3-treated vessels. Section of (B) a control mouse and (C) from a mouse treated with PN2KPI.
The murine MCAO model
The introduction of a thread into the middle cerebral artery resulted in intravascular thrombosis leading to ischemia of the mouse brain. Thus, inhibition of thrombus formation alleviates the extent of the stroke.37 As shown in Figure 4, the inhibition of FXIa by PN2KPI (Figure 4B), compared with a vehicle-treated mouse (Figure 4A), as shown with TTC staining, reduced the ischemic volume and the fraction of ischemic brain post MCAO. When the volume of ischemic brain in a group (n = 6) of PN2KPI-treated animals was compared with a control group (n = 6) of vehicle-treated animals, a significant reduction in stroke volume and fraction was observed in the mice treated with PN2KPI compared with those from a control group (P < .05, Figure 4C-D).
Figure 4.

Coronal sections of the brains of mice treated either with vehicle or PN2KPI. The ischemic region of brain is indicated by the pale area within the right hemisphere of animals treated with vehicle (A) or PN2KPI (B) 24 hours after right middle cerebral artery occlusion. (C-D) The comparison of the stroke volume and stroke fraction between the 2 groups (n = 6 in each group, P < .05). The bars represent the mean ± SEM of stroke volume values. *Significant differences with P < .05 compared with the control group.
The tail bleeding time
To determine the effect of PN2KPI-treatment on hemostasis, the tail bleeding time was measured in a group of 6 mice treated with PN2KPI, compared with 6 animals treated with vehicle. The bleeding time was similar (P > .05) in PN2KPI-treated mice (2.02 ± 0.73 minutes) compared with those in the control group (2.11 ± 0.49 minutes). Thus, the administration of PN2KPI to mice, sufficient to inhibit both carotid artery thrombosis and ischemic stroke because of MCAO, had no effect on hemostasis.
Discussion
The initiation of blood coagulation in vivo follows the formation of the enzyme complex that occurs when FVII or FVIIa is assembled on the transmembrane protein tissue factor (TF) present in many cell types.38 The complex of TF and FVIIa is almost immediately inhibited when a complex is formed between FXa and the tissue factor pathway inhibitor.39 The loss of the TF effect, that is, the shutdown of the extrinsic pathway, could also be a consequence of thrombus accumulating over the top of the source of TF. Thus, FXa generation results in the immediate inactivation of the enzyme complex that initially forms it (ie, TF-FVIIa). Therefore, the formation of thrombin through the extrinsic pathway is transient and the continuous generation of thrombin by the intrinsic pathway is required to assure normal hemostasis.40 FXI is required for normal functioning of the intrinsic pathway, and an alternative mechanism for the activation of FXI exists that is independent of the contact proteins. FXI can be activated by 4 biologically relevant proteases: FXIIa, FXIa, thrombin, and meizothrombin.5,6 It participates in the contact phase of blood coagulation in the presence of anionic surfaces for optimal surface-mediated in vitro activation by FXIIa.41 However, because deficiencies of FXII, prekallikrein, and high Mr kininogen are not associated with hemostatic abnormalities but deficiency of FXI produces abnormal bleeding complications in at least 50% of affected individuals,3 the more relevant in vivo pathway for FXI activation may be feedback activation by thrombin or, possibly, autoactivation by FXIa.5,6 In addition, platelets are known to secrete inhibitors of FXIa21–23 including PN2, a truncated form of the transmembrane protein AβPP that contains a KPI domain. Although the release of PN2 from activated platelets inhibits plasma FXIa activity in the environment of the hemostatic thrombus, platelet-bound FXIa is protected from inactivation by PN2.26 PN2KPI is a potent inhibitor of FXIa (Ki ∼ 0.65nM) and trypsin (Ki ∼ 0.03nM), whereas Ki values for other plasma coagulation proteases (thrombin, FVIIa, FIXa, FXa, FXIIa, plasmin, and kallikrein, Ki > 185nM) indicate that PN2KPI is highly specific for FXIa among coagulation proteases.28
Recent studies in FXII knockout mice showed that FXII deficiency protects mice from experimentally induced thrombosis without producing bleeding complications.42 The FXI-deficient mouse model was similarly used to demonstrate in various thrombosis models that FXI deficiency also protects against thrombosis without a bleeding phenotype.43 Therefore, FXI/XIa is an ideal target for the development of inhibitors to prevent thrombotic events without an increased risk of bleeding complications.44 Our present studies confirm this hypothesis, and demonstrate that PN2KPI, administered to WT mice to achieve a plasma concentration of ∼ 1-2μM inhibits the propagation of carotid thrombi in a murine FeCl3 carotid injury model. Interestingly, the initial rate of thrombus formation (ie, during the initial 3-7 minutes after injury) was not significantly affected in PN2KPI-treated mice compared with untreated animals. However, the subsequent propagation of thrombi was prevented in PN2KPI-treated animals. This suggests that the initial formation of thrombi occurs in response to the formation of the FVIIa/TF complex after release of TF from injured tissue, and that the FXa generated by FVIIa activation is limited because it is immediately inhibited by tissue factor pathway inhibitor.38 Thus, the extrinsic pathway alone fails to sustain the stability and expansion of thrombosis. The present studies suggest that the intrinsic or consolidation pathway of coagulation, triggered by the generation of FXIa catalyzed by the initial thrombin burst via the FVIIa/TF pathway, is critical to the process of thrombus propagation observed in WT control mice but not in PN2KPI-treated mice.
The MCAO model of ischemic stroke is mediated by both thrombotic and inflammatory mechanisms,37 and deficiency of components of the intrinsic pathway are critically involved in infarct development.45 Both FXII-deficient and FXI-deficient mice are protected from cerebral ischemia: the infarct volumes were 50% less in FXII-deficient compared with wild-type mice at 24 hours after MCAO, and the brain infarct volumes were also markedly diminished in FXI knockout mice.46 By suppression of FXIa activity, recombinant PN2KPI prevents stroke in the MCAO model and significantly reduces the infarct volumes.
Considering its presence in neuronal cells and platelets, it has been suggested that PN2 and other KPI-containing proteins (eg, amyloid precursor-like protein-2 or APLP2) might function as potential cerebral anticoagulants.47,48 Consistent with the results of our present studies, it was found that overexpression of PN2/AβPP in the circulating platelets of transgenic mice resulted in marked inhibition of cerebral thrombosis and larger intracerebral hematomas,47,48 whereas PN2/AβPP deletion in AβPP gene knockout mice resulted in increased cerebral thrombosis and reduced intracerebral hematomas.47 The question raised by these elegant studies relates to the mechanism by which overexpression of PN2/AβPP can inhibit thrombosis and enhance hemorrhage, and deletion of this kunitz inhibitor can promote thrombosis and inhibit hemorrhage. Our previous studies28,31 strongly suggest that the only plasma coagulation enzyme inhibited by physiologic concentrations of PN2KPI achievable in normal human plasma is FXIa. Thus, FXIa is inhibited by PN2KPI with a Ki ∼ 0.5-1.0nM, a concentration considerably higher than the virtually undetectable level of PN2/AβPP in plasma,22 but well below the concentrations (3-5nM) that can be achieved at physiologic concentrations of platelets after secretion from α-granules.22,26 Thus, in the vicinity of a platelet thrombus, the concentration of PN2/AβPP is likely to be sufficient to regulate FXIa activity, but insufficient to have any significant effect on other plasma serine proteases, Ki values (183-5500nM) for which are 36-fold to 1100-fold higher than the inhibitor concentration (3-5nM) achieved after platelet secretion.28
There are several important issues to be addressed in considering PN2KPI as a candidate for antithrombotic therapy in humans. First, as shown in Figure 1, PN2KPI is a more potent and complete inhibitor of FXIa in human plasma than in murine plasma. This could prove to be either a benefit or a liability for human therapy. The fact that the mice retain ∼ 25%-50% FXI levels after PN2KPI administration which protects them from MCAO strongly suggests that partial inhibition of FXIa suffices to protect the animals from cerebrovascular stroke. Second, because PN2KPI is a small protein with (presumably) a short half-life, it would have to be injected at regular intervals which, however, might be prolonged by the construction of fusion proteins.49 Third, the administration of PN2KPI before the induction of thrombosis—for example, in the MCAO model—while it provides proof of principle on the mechanism and effectiveness of the inhibitor, does not represent a real-life situation for human use, in which the PN2KPI would be administered after the appearance of symptoms because of cerebral artery thrombosis. Future studies are planned to examine the effectiveness of PN2KPI administered during middle cerebral artery occlusion and also immediately after restoration of the circulation when reperfusion injury begins. Moreover, studies are planned to investigate the effectiveness of long-term expression of the PN2KPI gene in mice, a strategy that might be used in the prevention of cerebrovascular stroke in patients at high risk for thromboembolism.
The rationale for using PN2KPI as an antithrombotic agent is based in part on the striking structural homology between the human FXIa/KPI complex31 and the bovine trypsin/BPTI complex,50 in addition to the fact that BPTI (as Trasylol) has in the past been frequently used therapeutically in human subjects in the treatment and prevention of hemorrhage, for example, in patients undergoing cardiopulmonary bypass surgery. However, it is no longer used clinically because of numerous adverse reactions, including anaphylaxis. Because PN2KPI is a human protein, it is likely to be well tolerated when administered to humans for thromboprophylaxis or antithrombotic therapy, and unlikely to elicit immunologic or allergic reactions. Thrombosis is a primary cause of mortality and morbidity in the United States and developed world. Each year, 1.1 million Americans experience acute myocardial infarction (AMI). Another million experience a thrombotic cerebrovascular accident, and DVT is a major problem for surgical and other hospitalized patients.51 Thrombi initiated by TF released from disrupted atherosclerotic plaques and formed in the carotid artery constitute a major source of thromboembolism resulting in stroke. Although uncontrolled coagulation leads to massive thrombosis, blood clots are essential to maintain normal hemostasis and prevent blood loss from injury. Excess inhibition of blood coagulation, as occurs frequently with the major anticoagulants, heparin and warfarin, and with the newer thrombin and FXa inhibitors, frequently leads to serious bleeding. In contrast, because PN2KPI is highly effective in preventing experimental thrombosis in mice without discernible excess bleeding complications, it is a candidate to be evaluated for effective and safe antithrombotic therapy in humans.
Acknowledgments
This work was supported by grant HL46213 from the National Institutes of Health.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: W.W. and H.L. contributed equally in the design and performance of the research, in the analysis of data, and contributed to the writing of the manuscript; D.N. prepared the recombinant KPI used in these studies and contributed to the experimental design, analysis of data, and writing of the manuscript; Z.W.R. assisted in the development of the MCA occlusion model and the coding of animals and treatment assignments; R.F.T. contributed to the experimental design, analysis of data, and experimental tools; and P.N.W. contributed to experimental design, analysis of data, and writing of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for D.N. is Foundation for Research on Rare Diseases and Disorders, Chennai, India.
Correspondence: Peter N. Walsh, MD, PhD, Sol Sherry Thrombosis Research Center, Temple University School of Medicine, 3400 North Broad St, Philadelphia, PA 19140; e-mail: pnw@temple.edu.
References
- 1.Walsh PN, Gailani D. Factor XI. In: Colman RW, Marder VJ, Clowes AW, George JN, Goldhaber SZ, editors. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. 5th Ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006. pp. 221–233. [Google Scholar]
- 2.Rosenthal RL, Dreskin OH, Rosenthal N. Plasma thromboplastin antecedent (PTA) deficiency: clinical coagulation, therapeutic and hereditary aspects of a new hemophilia-like disease. Blood. 1955;10(2):120–131. [PubMed] [Google Scholar]
- 3.Ragni MV, Sinha D, Seaman F, Lewis JH, Spero JA, Walsh PN. Comparison of bleeding tendency, factor XI coagulant activity, and factor XI antigen in 25 factor XI-deficient kindreds. Blood. 1985;65(3):719–724. [PubMed] [Google Scholar]
- 4.Rimon A, Schiffman S, Feinstein DI, Rapaport SI. Factor XI activity and factor XI antigen in homozygous and heterozygous factor XI deficiency. Blood. 1976;48(2):165–174. [PubMed] [Google Scholar]
- 5.Naito K, Fujikawa K. Activation of human blood coagulation factor XI independent of factor XII. Factor XI is activated by thrombin and factor XIa in the presence of negatively charged surfaces. J Biol Chem. 1991;266(12):7353–7358. [PubMed] [Google Scholar]
- 6.Gailani D, Broze GJ., Jr Factor XI activation in a revised model of blood coagulation. Science. 1991;253(5022):909–912. doi: 10.1126/science.1652157. [DOI] [PubMed] [Google Scholar]
- 7.Meijers JC, Tekelenburg WL, Bouma BN, Bertina RM, Rosendaal FR. High levels of coagulation factor XI as a risk factor for venous thrombosis. N Engl J Med. 2000;342(10):696–701. doi: 10.1056/NEJM200003093421004. [DOI] [PubMed] [Google Scholar]
- 8.Berliner JI, Rybicki AC, Kaplan RC, Monrad ES, Freeman R, Billett HH. Elevated levels of factor XI are associated with cardiovascular disease in women. Thromb Res. 2002;107(1-2):55–60. doi: 10.1016/s0049-3848(02)00190-1. [DOI] [PubMed] [Google Scholar]
- 9.Salomon O, Steinberg DM, Dardik R, et al. Inherited factor XI deficiency confers no protection against acute myocardial infarction. J Thromb Haemost. 2003;1(4):658–661. doi: 10.1046/j.1538-7836.2003.00195.x. [DOI] [PubMed] [Google Scholar]
- 10.Libourel EJ, Bank I, Meinardi JR, et al. Co-segregation of thrombophilic disorders in factor V Leiden carriers; the contributions of factor VIII, factor XI, thrombin activatable fibrinolysis inhibitor and lipoprotein(a) to the absolute risk of venous thromboembolism. Haematologica. 2002;87(10):1068–1073. [PubMed] [Google Scholar]
- 11.Salomon O, Steinberg DM, Koren-Morag N, Tanne D, Seligsohn U. Reduced incidence of ischemic stroke in patients with severe factor XI deficiency. Blood. 2008;111(8):4113–4117. doi: 10.1182/blood-2007-10-120139. [DOI] [PubMed] [Google Scholar]
- 12.Eichinger S, Schoenauer V, Weltermann A, et al. Thrombin activatable fibrinolysis inhibitor (TAFI) and the risk of recurrent venous thromboembolism. Blood. 2004;103(10):3773–3776. doi: 10.1182/blood-2003-10-3422. [DOI] [PubMed] [Google Scholar]
- 13.Minnema MC, Friederich PW, Levi M, et al. Enhancement of rabbit jugular vein thrombolysis by neutralization of factor XI. In vivo evidence for a role of factor XI as an anti-fibrinolytic factor. J Clin Invest. 1998;101(1):10–14. doi: 10.1172/JCI781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosen ED, Gailani D, Castellino FJ. FXI is essential for thrombus formation following FeCl3-induced injury of the carotid artery in the mouse. Thromb Haemost. 2002;87(4):774–776. [PubMed] [Google Scholar]
- 15.Chan JC, Ganopolsky JG, Cornelissen I, et al. The characterization of mice with a targeted combined deficiency of protein c and factor XI. Am J Pathol. 2001;158(2):469–479. doi: 10.1016/S0002-9440(10)63989-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamashita A, Nishihira K, Kitazawa T, et al. Factor XI contributes to thrombus propagation on injured neointima of the rabbit iliac artery. J Thromb Haemost. 2006;4(7):1496–1501. doi: 10.1111/j.1538-7836.2006.01973.x. [DOI] [PubMed] [Google Scholar]
- 17.Gruber A, Hanson SR. Factor XI-dependence of surface- and tissue factor-initiated thrombus propagation in primates. Blood. 2003;102(3):953–955. doi: 10.1182/blood-2003-01-0324. [DOI] [PubMed] [Google Scholar]
- 18.Butenas S, Undas A, Gissel MT, Szuldrzynski K, Zmudka K, Mann KG. Factor XIa and tissue factor activity in patients with coronary artery disease. Thromb Haemost. 2008;99(1):142–149. doi: 10.1160/TH07-08-0499. [DOI] [PubMed] [Google Scholar]
- 19.Salomon O, Steinberg DM, Zucker M, Varon D, Zivelin A, Seligsohn U. Patients with severe factor XI deficiency have a reduced incidence of deep-vein thrombosis. Thromb Haemost. 2011;105(2):269–273. doi: 10.1160/TH10-05-0307. [DOI] [PubMed] [Google Scholar]
- 20.Deng H, Bannister TD, Jin L, et al. Synthesis, SAR exploration, and x-ray crystal structures of factor XIa inhibitors containing an alpha-ketothiazole arginine. Bioorg Med Chem Lett. 2006;16(11):3049–3054. doi: 10.1016/j.bmcl.2006.02.052. [DOI] [PubMed] [Google Scholar]
- 21.Smith RP, Higuchi DA, Broze GJ., Jr Platelet coagulation factor XIa-inhibitor, a form of Alzheimer amyloid precursor protein. Science. 1990;248(4959):1126–1128. doi: 10.1126/science.2111585. [DOI] [PubMed] [Google Scholar]
- 22.Van Nostrand WE, Schmaier AH, Farrow JS, Cunningham DD. Protease nexin-II (amyloid beta-protein precursor): a platelet alpha-granule protein. Science. 1990;248(4956):745–748. doi: 10.1126/science.2110384. [DOI] [PubMed] [Google Scholar]
- 23.Bush AI, Martins RN, Rumble B, et al. The amyloid precursor protein of Alzheimer's disease is released by human platelets. J Biol Chem. 1990;265(26):15977–15983. [PubMed] [Google Scholar]
- 24.Van Nostrand WE, Wagner SL, Farrow JS, Cunningham DD. Immunopurification and protease inhibitory properties of protease nexin-2/amyloid beta-protein precursor. J Biol Chem. 1990;265(17):9591–9594. [PubMed] [Google Scholar]
- 25.Zhang Y, Scandura JM, Van Nostrand WE, Walsh PN. The mechanism by which heparin promotes the inhibition of coagulation factor XIa by protease nexin-2. J Biol Chem. 1997;272(42):26139–26144. doi: 10.1074/jbc.272.42.26139. [DOI] [PubMed] [Google Scholar]
- 26.Scandura JM, Zhang Y, Van Nostrand WE, Walsh PN. Progress curve analysis of the kinetics with which blood coagulation factor XIa is inhibited by protease nexin-2. Biochemistry. 1997;36(2):412–420. doi: 10.1021/bi9612576. [DOI] [PubMed] [Google Scholar]
- 27.Badellino KO, Walsh PN. Protease nexin II interactions with coagulation factor XIa are contained within the Kunitz protease inhibitor domain of protease nexin II and the factor XIa catalytic domain. Biochemistry. 2000;39(16):4769–4777. doi: 10.1021/bi9925468. [DOI] [PubMed] [Google Scholar]
- 28.Navaneetham D, Sinha D, Walsh PN. Mechanisms and specificity of factor XIa and trypsin inhibition by protease nexin 2 and basic pancreatic trypsin inhibitor. J Biochem. 2010;148(4):467–479. doi: 10.1093/jb/mvq080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grzesiak A, Krokoszynska I, Krowarsch D, Buczek O, Dadlez M, Otlewski J. Inhibition of six serine proteinases of the human coagulation system by mutants of bovine pancreatic trypsin inhibitor. J Biol Chem. 2000;275(43):33346–33352. doi: 10.1074/jbc.M006085200. [DOI] [PubMed] [Google Scholar]
- 30.Stassen JM, Lambeir AM, Matthyssens G, et al. Characterisation of a novel series of aprotinin-derived anticoagulants. I. In vitro and pharmacological properties. Thromb Haemost. 1995;74(2):646–654. [PubMed] [Google Scholar]
- 31.Navaneetham D, Jin L, Pandey P, et al. Structural and mutational analyses of the molecular interactions between the catalytic domain of factor XIa and the Kunitz protease inhibitor domain of protease nexin 2. J Biol Chem. 2005;280(43):36165–36175. doi: 10.1074/jbc.M504990200. [DOI] [PubMed] [Google Scholar]
- 32.Hata R, Mies G, Wiessner C, et al. A reproducible model of middle cerebral artery occlusion in mice: hemodynamic, biochemical, and magnetic resonance imaging. J Cereb Blood Flow Metab. 1998;18(4):367–375. doi: 10.1097/00004647-199804000-00004. [DOI] [PubMed] [Google Scholar]
- 33.Zhang M, Adler MW, Abood ME, Ganea D, Jallo J, Tuma RF. CB2 receptor activation attenuates microcirculatory dysfunction during cerebral ischemic/reperfusion injury. Microvasc Res. 2009;78(1):86–94. doi: 10.1016/j.mvr.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10(2):290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- 35.Lin KN, Liu RS, Yeh TP, Wang SJ, Liu HC. Posterior ischemia during an attack of transient global amnesia. Stroke. 1993;24(7):1093–1095. doi: 10.1161/01.str.24.7.1093. [DOI] [PubMed] [Google Scholar]
- 36.Denis C, Methia N, Frenette PS, et al. A mouse model of severe von Willebrand disease: defects in hemostasis and thrombosis. Proc Natl Acad Sci U S A. 1998;95(16):9524–9529. doi: 10.1073/pnas.95.16.9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nieswandt B, Kleinschnitz C, Stoll G. Ischaemic stroke: a thrombo-inflammatory disease?. J Physiol. 2011;589(Pt 17):4115–4123. doi: 10.1113/jphysiol.2011.212886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steinberg M, Nemerson Y. Activation of factor X. In: Colman RW, Hirsh J, Marder VJ, Salzman EW, editors. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. 2nd Ed. Philadelphia, PA: J. B. Lippincott; 1987. pp. 112–119. [Google Scholar]
- 39.Broze GJ, Jr, Warren LA, Novotny WF, Higuchi DA, Girard JJ, Miletich JP. The lipoprotein-associated coagulation inhibitor that inhibits the factor VII-tissue factor complex also inhibits factor Xa: insight into its possible mechanism of activation. Blood. 1988;71(2):335–343. [PubMed] [Google Scholar]
- 40.Broze GJ, Jr, Girard TJ, Novotny WF. Regulation of coagulation by a multivalent Kunitz-type inhibitor. Biochemistry. 1990;29(33):7539–7546. doi: 10.1021/bi00485a001. [DOI] [PubMed] [Google Scholar]
- 41.Ratnoff OD, Davie EW, Mallett DL. Studies on the action of Hageman factor: evidence that activated Hageman factor in turn activates plasma thromboplastin antecedent. J Clin Invest. 1961;40:803–819. doi: 10.1172/JCI104314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Renné T, Pozgajová M, Grüner S, et al. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med. 2005;202(2):271–281. doi: 10.1084/jem.20050664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang X, Cheng Q, Xu L, et al. Effects of factor IX or factor XI deficiency on ferric chloride-induced carotid artery occlusion in mice. J Thromb Haemost. 2005;3(4):695–702. doi: 10.1111/j.1538-7836.2005.01236.x. [DOI] [PubMed] [Google Scholar]
- 44.Löwenberg EC, Meijers JC, Monia BP, Levi M. Coagulation factor XI as a novel target for antithrombotic treatment. J Thromb Haemost. 2010;8(11):2349–2357. doi: 10.1111/j.1538-7836.2010.04031.x. [DOI] [PubMed] [Google Scholar]
- 45.Stoll G, Kleinschnitz C, Nieswandt B. Molecular mechanisms of thrombus formation in ischemic stroke: novel insights and targets for treatment. Blood. 2008;112(9):3555–3562. doi: 10.1182/blood-2008-04-144758. [DOI] [PubMed] [Google Scholar]
- 46.Kleinschnitz C, Stoll G, Bendszus M, et al. Targeting coagulation factor XII provides protection from pathological thrombosis in cerebral ischemia without interfering with hemostasis. J Exp Med. 2006;203(3):513–518. doi: 10.1084/jem.20052458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu F, Davis J, Miao J, et al. Protease nexin-2/amyloid beta-protein precursor limits cerebral thrombosis. Proc Natl Acad Sci U S A. 2005;102(50):18135–18140. doi: 10.1073/pnas.0507798102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu F, Previti ML, Van Nostrand WE. Increased severity of hemorrhage in transgenic mice expressing cerebral protease nexin-2/amyloid beta-protein precursor. Stroke. 2007;38(9):2598–2601. doi: 10.1161/STROKEAHA.106.480103. [DOI] [PubMed] [Google Scholar]
- 49.Osborn BL, Olsen HS, Nardelli B, et al. Pharmacokinetic and pharmacodynamic studies of a human serum albumin-interferon-alpha fusion protein in cynomolgus monkeys. J Pharmacol Exp Ther. 2002;303(2):540–548. doi: 10.1124/jpet.102.037002. [DOI] [PubMed] [Google Scholar]
- 50.Hanson WM, Domek GJ, Horvath MP, Goldenberg DP. Rigidification of a flexible protease inhibitor variant upon binding to trypsin. J Mol Biol. 2007;366(1):230–243. doi: 10.1016/j.jmb.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.American Heart Association. Heart Disease and Stroke Statistics 2010 Update. [Accessed 2010]. http://circ.ahajournals.org/cgi/content/full/121/7/e46.