

Abstract
Accumulation of misfolded α-synuclein in Lewy bodies and Lewy neurites is the pathological hallmark of Parkinson’s Disease (PD). To identify ligands having high binding potency toward aggregated α-synuclein, we synthesized a series of phenothiazine derivatives and assessed their binding affinity to recombinant α-synuclein fibrils using a fluorescent thioflavin T competition assay. Among 16 new analogues, the in vitro data suggest that compound 11b has high affinity to α-synuclein fibrils (Ki = 32.10 ± 1.25 nM) and compounds 11d, 16a and 16b have moderate affinity to α-synuclein fibrils (Ki ≈ 50 to 100 nM). Further optimization of the structure of these analogues may yield compounds with high affinity and selectivity for aggregated α-synuclein.
Keywords: Parkinson’s disease, Lewy bodies, α-Synuclein, Phenothiazine, Fluorescence
1. Introduction
Parkinson’s Disease (PD) is the second most common neurodegenerative disease after Alzheimer’s Disease (AD). Its main clinical features include rest tremor, bradykinesia, rigidity and loss of postural reflexes.1 The motor symptoms of PD are caused by degeneration of dopaminergic neurons in the substantia nigra, which is accompanied by Lewy bodies (LBs) and Lewy neuritis (LNs). In addition to motor symptoms, individuals with PD also have a high risk of dementia2 and postmortem analysis has demonstrated a correlation between dementia and LBs in cortical neurons.3–6 To date, the correlation between the density of LBs/LNs in substantia nigra and the severity of these diseases is not clear. 7 Mechanisms responsible for initiation and progression of pathogenic processes in PD are still not entirely understood. Aggregated α-synuclein is the major component of LBs and LNs.8–10 α-Synuclein, a presynaptic terminal protein containing 140 amino acids,11 plays an important function in the central nervous system (CNS) by regulating synaptic vesicle recycling and synthesis, vesicular storage and release of neurotransmitter.12–15 One analysis approach suggests that aggregated α-synuclein outside LBs and LNs is 10-fold higher than the amount of aggregated α-synuclein inside LBs and LNs.16 The formation of α-synuclein aggregates may result in synaptic dysfunction and neuronal cell death.17–20 Quantifying α-synuclein aggregation in vivo will be very useful to improve early diagnosis of PD and to monitor disease progression. Furthermore, the ability to quantify aggregated α-synuclein in vivo may be useful to monitor therapeutic efficacy in early clinical studies of disease-modifying treatments. Therefore, small molecules that have suitable pharmaceutical properties as fibrillar α-synuclein ligands and that can be labeled with Position Emission Tomography (PET) radionuclides such as C-11 and F-18, will have great opportunity to serve as PET probes for quantifying α-synuclein aggregation in the brain. In addition, inhibition of the progress of α-synuclein protein aggregation may be a potential strategy for treating PD and its associated diseases. Thus, investigators have attempted to identify highly potent ligands for α-synuclein fibrils.21–24 To achieve the goal of developing highly potent α-synuclein ligands, we focused on exploring the derivatives of phenothiazine. Herein, we report our initial work on the synthesis of new analogues of phenothiazine and the analysis of their binding affinity toward α-synuclein fibrils. Our current work was inspired by (1) to date, no small molecular ligand for α-synuclein fibrils has been reported to have the capability to prevent α-synuclein accumulation in vivo; (2) no suitable PET tracer has been reported that can be used to assess aggregated α-synuclein accumulation in the brain in vivo; (3) phenothiazine structure has been proposed as a promising pharmacophore for the therapy of neurodegenerative diseases25.26 by protecting the dopaminergic neurons against oxidative stress; the representative structures including phenothiazine (1), N-acylaminophenothiazine (2) and N-alkylphenothiazine (3) (Figure 1). Furthermore, it was reported that phenothiazines inhibit α-synuclein filament assembly with IC50 values in the low micromolar range. 21
Figure 1.

Three chemical structures reported previously for neuronal protection
2. Results and discussion
2.1. Chemistry
Based on our structure-activity relationship analysis, we optimized the structure of phenothiazine and synthesized a series of new analogues as shown in Schemes 1–3.
Scheme 1.

Synthesis of compound 6
Reagents and conditions:
a) 1-bromo-4-methoxybenzene, Cul, L-proline, K2CO3, DMSO, 90°C; b) S, I2, 1,2-dichlorobenzene, 150°C
Scheme 3.

Synthesis of N-substituted analogues 12, 13a–c, 14a–b, 15, 16a–c.
Reagents and conditions:
a) NaH, CH3l, DMF, 0°C, overnight; b) H2, Pd-C (10%), 90 psi, ambient temperature, overnight, c) CH3l, Na2CO3, MeCN, ambient temperature, overnight; d) CH3COCl, DCM, ambient temperature, overnight; e) BBr3/DCM (1M), DCM, −78°C, overnight. f) 1-bromo-2-fluroethane, 3-bromopropyne or 3-bromo-1-iodopropene, Na2CO3, MeCN; g) HCl (3 M), H2O, reflux, 5h;
Using copper iodide as catalyst, in the presence of L-proline and potassium carbonate, compound 4 was reacted with 1-bromo-4-methoxybenzene in DMSO to afford bis(4-methoxyphenyl)amine (5) following Ullman reaction (Scheme 1). The target compound 3, 7-dimethoxy-10H-phenothiazine (6) was obtained by cyclization of intermediate 5 with sulfur at 150 °C using dichlorobenzene as solvent and iodine as the catalyst.
The synthesis of compounds 11a–e was achieved as shown in Scheme 2. Hydrolysis of compound 8a in the presence of potassium hydroxide afforded 2-amino-5-methoxybenzothiol, which was coupled with either 4-chloro-3-nitrobenzonitrile (7a) or 1-chloro-2, 4-dinitrobenzene (7b) to obtain the substituted diphenylsulfide intermediate 9a or 9b under mild acidic condition. Acetic anhydride was used to protect the amino group in 9a and 9b to afford acetamides 10a and 10b which were cyclized via Smile rearrangement27 under the strong basic conditions to afford the target compounds 11a and 11b. Following the similar synthetic procedure followed for 9a and 9b, compound 9c was obtained starting with 7b and 2-amino-benzothiol (8b) in the presence of sodium hydroxide. Compound 9c was acylated with acetic anhydride to obtain 10c. Compound 10c was treated with N-bromosuccinimide (NBS) in dimethylformamide (DMF) or iodine monochloride (ICl) in acetic acid to afford 10d or 10e respectively. Cyclization of 10c, 10d and 10e followed the similar procedure described for 11a to obtain target compounds 11c, 11d and 11e.
Scheme 2.

Synthesis of compounds 11a–e
Reagents and conditions:
a) i: KOH/H2O, reflux, overnight; ii: AcOH, EtOH/H2O, ambient temperature, 2h; b) NaOH, EtOH/H2O, ambient temperature, 2h; c) pyridine, Ac2O, ambient temperature, 3h; d) NBS, DMF, 100°C, overnight; e) ICl, AcOH, reflux, 3h; f) KOH, Acetone/EtOH, reflux, 2h;
To test how the proton on the nitrogen of the middle ring affects the binding potency of analogues to α-synuclein fibrils, a methyl group or an acetyl group was substituted on the nitrogen. Analogues 12, 13a–c, 14a–b, 15 and 16a–c were synthesized as shown in Scheme 3. Compound 12 was directly obtained by the N-methylation of 11b using methyl iodide in DMF in the presence of sodium hydride. Compound 13a was generated by reducing compound 12 via hydrogenation using hydrogen atmosphere in the presence of 10% palladium on activated carbon in ethanol. Further N-methylation of 13a using one or two equivalents of methyl iodide in acetonitrile yielded compounds 13b and 13c. Compounds 14a and 14b were obtained via N-acetylation of compound 11b or 11c using acetyl chloride. Removing the methyl group from 14a with boron tribromide afforded the corresponding phenol analogue 15. O-Alkylation of compound 15 using 1-bromo-2-fluoroethane, 3-bromopropyne or 3-bromo-1-iodopropene, followed by hydrolysis in the presence of 3N HCl aqueous solution gave the compounds 16a, 16b and 16c. To obtain enough quantity of target compounds for in vitro studies, some reactions discussed above were repeated and scaled-up. However, in the current work, we did not optimize the reaction conditions and determine the scalability of reactions. Compound 1 was purchased from Sigma-Aldrich Co., and compounds 2 and 3 were synthesized following the literature reported procedures 28, 29 and converted them to corresponding oxalate salts.
2.2. Thioflavin T fluorescence assay for α-synuclein fibrils
At present, no existing standard protocol can be used to measure the binding affinity of compounds for α-synuclein fibrils. Determining the binding affinities of compounds toward proteins with radioligands has higher accuracy and sensitivity than with fluorescent probes. Nevertheless, until today, no radioligand was reported to be suitable for determining binding affinities of compounds toward aggregated α-synuclein. Moreover, thioflavin S has been used to determine the binding affinity of compounds toward β-amyloid, tau and α-synuclein aggregates.30–33 In the current work, we used fluorescent methodology to determine the relative binding potency of compounds toward α-synuclein fibrils and to identify potential ligands that can be radiolabeled with I-125 or H-3 to further characterize their binding properties.
Thioflavin T (ThT) is a fluorescent dye with an identified chemical structure. It is weakly fluorescent in the presence of monomeric α-synuclein or in an α-synuclein fibril-free system. In the presence of α-synuclein fibrils, ThT fluorescence intensity increases by several orders of magnitude at the emission wavelength maximum (λem = 483 nm).31 To determine the binding affinity of compounds toward α-synuclein fibrils, first, the ThT fluorescent emission spectrum was confirmed to be consistent with previously reported data (Figure 2, left); second, we incubated ThT with α-synuclein fibrils and measured ThT’s maximum fluorescent emission wavelength (λem = 485 nm) under its excitation wavelength (λex = 440 nm). No increase in fluorescent emission was observed when ThT was incubated in the presence of monomeric α-synuclein fibrils or in α-synuclein fibril free buffer. Furthermore, the ratio of ThT’s fluorescence intensity in the presence of α-synuclein fibrils to ThT’s fluorescence intensity in either monomeric α-synuclein fibrils or α-synuclein fibril free buffer is about 30-fold. Therefore, the fluorescence intensity of ThT in either monomeric α-synuclein or α-synuclein free buffer is negligible and unlikely to interfere with the measurement of α-synuclein fibril binding affinity for compounds.
Figure 2.

left: fluorescence emission spectra of ThT in the corresponding buffer alone (blue), monomer (red) and fibrils (green) at λex = 440 nm; right: saturation curve of ThT (3 μM) for α-synuclein fibrils (1.5 μM) in Tris buffer (30 mM, pH = 7.4) at different incubation times: 30 min (circle), 60 min (square), 90 min (triangle) at room temperature. The Kd for ThioT binding to fibrils was 948 nM and the Bmax was 5672 afu.
To determine the binding affinity for new compounds toward α-synuclein fibril, we first evaluated the saturation binding curve of ThT fluorescence intensity in the presence of α-synuclein fibrils, from which 60 min was chosen as the optimized incubation time (Figure 2, right). In our system, we obtained the Kd value of 948 ± 271 nM for ThT binding to α-synuclein fibrils, which is different from other reported values, 15 μM and 588 ± 2 nM.31, 32 Differences in Kd values may be explained by differences in α-synuclein fibril preparations and incubation conditions.32, 33 Importantly, we observed consistent α-synuclein fibril binding affinity when using different batches of fibrils in our experimental protocol.
2.3. In vitro evaluation of binding affinities of phenothiazine compounds to α-synuclein fibrils
Compounds were incubated with α-synuclein fibrils to determine whether any fluorescence emission was present at λem = 485 nm, which would have permitted binding affinity (Kd) values to be directly assessed using the same procedure as ThT. All of the analogues reported here had no significant fluorescence, and their binding affinity for α-synuclein fibrils was determined using an indirect ThT competition assay, in which the competition of ThT binding to α-synuclein fibrils was determined at various compound concentrations. This approach enabled IC50 and Ki values of the new compounds to be determined and compared. Results of the competitive binding assays for each compound are shown in supplementary Figure 1 and supplementary Table 1. The corresponding Ki values are listed in Table 1. Although fluorescence quenching can potentially interfere with measurement of competitive binding, the data for the individual compounds closely fit a competitive binding model. Absorbance spectra were measured at the IC50 concentration for each compound. Absorbance was less than 0.001 in the range of 400–500 nm for all of the compounds including 11b as shown in supporting information Figure 3, indicating that absorbance at the excitation or emission wavelengths did not interfere with the fluorescence assay.
Table 1.
Binding affinity (Ki± SD, nM)a of synthesized compounds determined by ThT competition assay
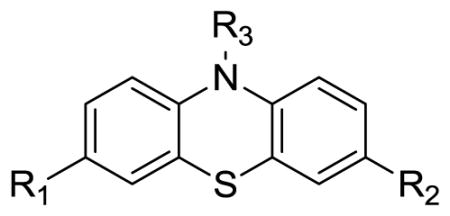
| |||||
|---|---|---|---|---|---|
| Compound # | R1 | R2 | R3 | Ki (nM) | LOG Pb |
| 1 | H | H | H | > 500 | 4.15 |
| 2 | H | H | N2C5OH11 | > 500 | 2.29 |
| 3 | H | H | N3C7H16 | >500 | 2.85 |
| 6 | OCH3 | OCH3 | H | 121.8 ± 5.1 | 3.98 |
| 11a | OCH3 | CN | H | 346 ± 37 | 3.50 |
| 11b | OCH3 | NO2 | H | 32.1 ± 1.3 | 3.79 |
| 11c | H | NO2 | H | 116.5± 1.9 | 3.88 |
| 11d | Br | NO2 | H | 75.3 ± 8.4 | 4.65 |
| 11e | I | NO2 | H | 106.0 ± 9.3 | 4.91 |
| 12 | OCH3 | NO2 | CH3 | > 500 | 4.14 |
| 13a | OCH3 | NH2 | CH3 | > 500 | 3.12 |
| 13b | OCH3 | NHCH3 | CH3 | > 500 | 3.78 |
| 13c | OCH3 | N(CH3)2 | CH3 | > 500 | 4.52 |
| 14a | OCH3 | NO2 | COCH3 | > 500 | 1.86 |
| 14b | H | NO2 | COCH3 | > 500 | 2.76 |
| 15 | OH | NO2 | COCH3 | > 500 | 1.93 |
| 16a | OCH2CH2F | NO2 | H | 49.0 ± 4.9 | 4.02 |
| 16b | OCH2CH=CHI | NO2 | H | 57.9 ± 2.7 | 5.72 |
| 16c | OCH2C≡ CH | NO2 | H | > 500 | 3.83 |
Ki values (mean ± SEM) were determined in at least three experiments and were calculated using Kd of 948 nM for ThT binding to α-synuclein fibrils as shown in Figure 2
Calculated value at pH 7.4 with ACD/Lab, version 7.0 (Advanced Chemistry Development, Inc., Canada)
Based on in vitro data generated by the protocol described in the experimental section, it was found that the dimethoxy substituted phenothiazine analogue 6 had a Ki value of 121.8 ± 5.1 nM. When a cyano group was used to replace one of the methoxy groups in 6, the binding affinity (Ki value) for 11a was decreased 2.8-fold to 346 ± 37 nM; contrarily, when a nitro group was used to replace one methoxy group in 6, the binding affinity (Ki value) for 11b was increased about 3.8-fold to reach 32.1 ± 1.3 nM. Compound 11b is the most potent compound in our current work. In addition, the calculated log P value using ACD/log D program is 3.79 which suggests the lipophilicity of 11b is a little higher than the desired range (range from 1 to 3), but it is still acceptable with a high possibility of crossing the blood-brain-barrier.
To test the possibility of improving affinity of compounds binding to the α-synuclein fibrils, compounds 11c–e were synthesized by replacing the methoxy group in the structure of compound 11b using H, Br and I respectively. Compared to compound 11b (Ki = 32.1 ± 1.3 nM), the binding affinities of all three compounds toward α-synuclein fibrils, 11c, 11d and 11e were decreased; the Ki values were decreased to 116.5 ± 1.9, 75.3 ± 8.4, 106.0 ± 9.3 nM for 11c, 11d and 11e respectively. The order of binding potency to α-synuclein fibrils is -OCH3 > -Br > -I > -H.
To confirm if the free proton on the nitrogen of the middle ring is necessary for having high α-synuclein fibril binding potency, compounds 12, 13a–b, 14a–b, 15 were made; the competitive binding data demonstrated that none of these compounds displayed higher α-synuclein fibril binding affinity (Ki > 500 nM) compared to compounds 6 and 11a–e that have a free proton on the nitrogen of the middle ring.
Since compound 11b displayed relatively high affinity (32.10 ± 1.25 nM), the methoxy was replaced by fluoroethoxy, (3-iodoallyl)oxy and prop-2-yn-1-yloxy to synthesize compounds 16a–c. The in vitro data indicated that both compounds 16a and 16b had relatively high affinities with Ki value of 49.0 ± 4.9 nM and 57.9 ± 2.7 nM respectively, which are comparable to the affinity of compound 11b (32.1 ± 1.3 nM. Log P = 3.79). However, compound 16c displayed significantly lower affinity (Ki > 500 nM). The structures of compound 16a and 16b contain fluorine or iodine atom, providing positions for labeling with F-18 or I-125.
We also measured the binding affinities for three previously described phenothiazine analogues shown in Figure 1. Compound 1 (phenothiazine) had low affinity (Ki > 500 nM) indicating that the substitutions described in this study are important to confer binding affinity to the phenothiazine pharmacophore structure. Compound 2 and 3 also had low affinity (Ki > 500 nM), consistent with the low affinities observed for other analogues with substitutions on the nitrogen of the middle ring. To further evaluate the specificity of the ThT competition assay, we tested eticlopride hydrochloride, a known selective ligand to the dopamine D2 receptor, and observed no significant competition for ThT in the assay (supporting information). Finally, we evaluated the possibility that the observed changes in ThT fluorescence in the binding assays could result from fibril dissociation rather than competition for ThT binding to α-synuclein fibrils. We used SYPRO Ruby staining to quantify α-synuclein fibrils in SDS-PAGE gels following centrifugation of fibrils at 100,000 × g and conversion to monomer by boiling in SDS sample buffer. We observed no change in the mass of α-synuclein fibrils during incubation with each of compounds 11b, 16a, or 16b at a concentration of 3 μM. Since each compound produces greater than 90% inhibition of ThT fluorescence at a concentration of 3 μM, the lack of fibril dissociation indicates that changes in fluorescence reflect competition for ThT binding.
Collectively, based on our structure-activity relationship studies, we found that: (1) there is a high possibility of identifying highly potent ligands for imaging α-synuclein fibril aggregates by optimizing the structure of phenothiazine; (2) the presence of an electron withdrawing nitro group in one of the aromatic rings favors the α-synuclein fibril binding affinity; (3) the free proton of the amino group in the middle ring is essential for maintaining high binding affinity of compounds toward α-synuclein fibrils; otherwise, the binding affinities of compounds will drop dramatically. Compounds having moderate binding affinity for α-synuclein fibrils (Ki < 60 nM) will be assessed for their in vitro binding affinity toward Aβ1-40/42, tau proteins or other neurotransmitters, receptors, transporters, enzymes, and ion channels to determine their binding specificity for α-synuclein fibrils in future studies. Based on previous studies in the development of PET/SPECT ligands for imaging Aβ amyloid in vivo, we anticipate that compounds with binding affinities less than 10 nM for α-synuclein fibrils will need to be obtained.36, 37 Further optimizing the phenothiazine analogues to identify ligands that have high α-synuclein potency (Ki < 10 nM) and specificity, suitable lipophilicity (Log P ranges from 1 to 3) and other biological properties is necessary to achieve the goal of identifying a candidate PET/SPECT ligand for imaging α-synuclein fibril aggregation in the brain.
3. Conclusion
In this work, we successfully optimized the structure of phenothiazine for binding α-synuclein fibrils by synthesizing a series of new analogues and assessing their α-synuclein fibril binding affinity in a ThT competition assay. Based on the in vitro binding affinity screening data, several lead compounds, 11b, 11d, 16a and 16b were identified with high potency for α-synuclein fibrils with Ki < 100 nM. Particularly, compounds 11b, 16a and 16b have Ki values as 32.10 ± 1.25 nM; 48.96 ± 4.94 nM and 57.94 ± 2.67 nM respectively. After further validating the binding specificity of these three compounds toward α-synuclein fibrils, the radioactive [11C]11b, [18F]16a will be synthesized for further study to test the feasibility of assessing α-synuclein fibril aggregation in vivo; [125I]16b could be used as radioactive ligand to establish radioactive methodology to screen the binding affinity of other compounds toward the α-synuclein fibrils. In addition, the in vitro data reported here will provide very useful SAR information to guide further design and synthesis of new analogues to achieve the goal of identifying highly potent small molecules that have high affinity and selectivity for α-synuclein fibrils.
4. Experimental
All reagents and chemicals were purchased from Sigma-Aldrich Corporation (Milwaukee, WI) or VWR international, Inc. (Earthy city, MO) and used without further purification unless otherwise stated. The solvent “hexane” means “n-hexane” unless otherwise stated. The air and water sensitive reactions were carried out under nitrogen. The melting points of all the intermediates and final compounds were determined on Hake-Buchler melting point apparatus and are uncorrected. 1H NMR spectra were recorded on Varian-300MHz and 13C NMR spectra were recorded on Varian-400MHz which were maintained by the Chemistry Department of Washington University in St. Louis. Spectra are referenced to the deuterium lock frequency of the spectrometer. The chemical shifts (in ppm) of residual solvents were found to be at 7.26 for CHCl3 and at 2.50 for DMSO. The following abbreviations were used to describe peak patterns when appropriate: br s = broad singlet, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet. Elemental analysis or HPLC methods (> 95%) were used to determine the purity of the target compounds.
Plate reader & Software was used for Fluorescence Scan: TECAN infinite M100 Plate Reader, i-control 1.7 TECAN software was used to run plate reader. Plate Reader & Software used for Binding Assay: Biotek Synergy 2 Plate Reader, Gen 5 software was used to run plate reader, Fluorescence Filters: Excitation 440/30, Emission 485/20. Optical Setting Top 50% and Sensitivity = 60. Absorbance scans were performed in quartz cuvettes in a Beckman Coulter DU 800 spectrophotometer.
4.1. Chemistry
Bis(4-methoxyphenyl)amine (5)
4-Aminoanisole (300 mg, 2.5 mmol), 4-bromoanisole (360 mg, 2 mmol), CuI (75 mg, 0.4 mmol), L-proline (95 mg, 0.8 mmol) and K2CO3 (1.1 g, 8 mmol) were placed in a 50 mL flask and the DMSO (10 mL) was added. The reaction mixture was stirred and heated at 100 °C for 2 d. The reaction mixture was quenched by adding water (50 mL) and extracted with ethyl acetate. The organic phase was dried over anhydrous Na2SO4 and concentrated. The crude product was purified on a silica gel column using ethyl acetate/hexane (1/4, v/v) to yield white solid (0.15 g, 33%). 1H NMR (CDCl3): δ 3.76 (s, 6H), 5.29 (br s, 1H), 6.81 (d, J = 9.0 Hz, 4H), 6.93 (d, J = 9.0 Hz, 4H). mp 92.4 – 95.0 °C.
3, 7-Dimethoxy-10H-phenothiazine (6)
Compound 5 (150 mg, 0.655 mmol), sulfur (91 mg, 2.3 mmol) and I2 (29 mg, 0.1 mmol) were added into 1, 2-dichlorobenzene (10 mL). The reaction mixture was heated at 150 °C for 12 h. the reaction mixture was cooled down to room temperature and purified on a silica gel column using ethyl acetate/hexane (1/4, v/v) as mobile phase to yield yellow solid (50 mg, 29 %).1H NMR (CDCl3): δ 3.64 (s, 6H), 6.57–6.61 (m, 6H), 8.14 (br s, 1H). 13C NMR (DMSO-d6): δ 55.8, 111.9, 113.6, 115.2, 117.4, 136.7, 154.7. Anal. calcd for C14H13NO2S: C, 64.84; H, 5.05; N, 5.40. Found: C, 64.57; H, 5.15; N, 5.21. mp 194.2 – 196.1 °C.
4-((2-Amino-5-methoxyphenyl)thio)-3-nitrobenzonitrile (9a)
Compound 8a (500 mg, 2.8 mmol) was suspended in 10 mL of aqueous KOH (50%) solution and refluxed overnight. The reaction solution was cooled down to room temperature and added dropwise to the solution of 7a (510 mg, 2.8 mmol) in ethanol (20 mL)/AcOH (50 mL) in ice-water bath. The reaction mixture was stirred for additional 3 h. The precipitate was filtrated and washed with water/ethanol (1/1, v/v) to afford red solid. (510 mg, 60%). 1H NMR (CDCl3): δ 3.77 (s. 3H), 3.99 (br s, 2H), 6.85 (d, J = 8.4 Hz, 1H), 6.98 (m, 3H), 7.58 (d, J = 8.7 Hz, 1H), 8.56 (s, 1H). mp 163.6 – 165.1 °C.
2-((2, 4-Dinitrophenyl)thio)-5-methoxyaniline (9b)
Synthetic procedure described for 9a was followed starting with 8a (5 g, 28 mmol) and 7b (6.7 g, 28 mmol), yellow solid (7.3 g, 81%). 1H NMR(CDCl3):δ 3.76 (s. 3H), 4.00 (br s, 2H), 6.85 (d, J = 8.4 Hz, 1H), 6.96–7.06 (m, 3H), 8.18 (d, J = 9.0 Hz, 1H), 9.13 (s, 1H). mp 169.4 – 171.7 °C.
2-((2, 4-Dinitrophenyl)thio)aniline (9c)
Compound 7b (10 g, 49 mmol) in ethanol was added dropwise into the solution of 8b (6.8 g, 54 mmol), NaOH (2.16 g, 54 mmol) in ethanol (50 mL). The reaction mixture was stirred at ambient temperature for 2 h. the precipitate was filtered and washed by ethanol to obtain yellow solid (11.4 g, 88 %). 1H NMR (CDCl3): δ 4.28 (br s, 2H), 6.87 (m, 2H), 7.03 (d, J = 9.0 Hz, 1H), 7.42 (m, 2H), 8.17 (d, J = 9.0 Hz, 1H), 9.12 (s, 1H). mp 150.7 – 151.8 °C.
N-(2-((4-cyano-2-nitrophenyl)thio)-4-methoxy-phenyl)acetamide (10a)
Acetic anhydride (10 mL) and pyridine (2 mL) were added into a flask containing compound 9a (0.5 g, 1.66 mmol). The solution was stirred for 3 h at ambient temperature and quenched by ice-cold water. The precipitate was filtrated and washed with water to afford yellow solid (0.46 g, 81%) 1H NMR (CDCl3): δ 2.91 (s. 3H), 3.82 (s, 3H), 6.89 (d, J = 8.4 Hz, 1H), 7.08 (s, 1H), 7.15 (d, J = 9.3 Hz, 1H), 7.60 (d, J = 8.7 Hz, 1H), 7.66 (br s, 1H). 8.32 (d, J = 9.0 Hz, 1H). 8.58 (s, 1H). mp 195.0 – 198.9 °C.
N-(2-((2, 4-dinitrophenyl)thio)-5-methoxyphenyl)-acetamide (10b)
Synthetic procedure described for 10a was followed starting with 9b (0.3 g, 0.93 mmol) to afford yellow solid (0.31 g, 95%). 1H NMR (CDCl3): δ 2.05 (s, 3H), 3.81 (s, 3H), 6.95 (d, J = 9.0 Hz, 1H), 7.09 (s, 1H), 7.16 (d, J = 9.0 Hz,1H), 7.65(br s, 1H), 8.19 (d, J = 9.0 Hz,1H), 8.33 (d, J = 8.7 Hz, 1H), 9.14 (s, 1H). mp 124.6 – 125.6 °C.
N-(2-((2, 4-dinitrophenyl)thio)phenyl)acetamide (10c)
Synthetic procedure described for 10a was followed starting with 9b (1.1 g, 3.8 mmol) to afford yellow solid (1.25 g, 99%). 1H NMR (CDCl3): δ 2.10 (s, 3H), 6.88 (d, J = 8.7 Hz,1H), 7.26 (t, J = 6.3 Hz,1H), 7.59 (m, 2H), 7.94 (br s, 1H), 8.19 (d, J = 9.0 Hz,1H), 8.54 (d, J = 8.4 Hz,1H), 9.14 (s, 1H). mp 182.7 – 184.0 °C.
N-(5-bromo-2-((2, 4dinitrophenyl)thio)phenyl)acetamide (10d)
Compound 10c (2.0 g, 6 mmol) and NBS (4.0 g, 24 mmol) was dissolved in DMF (5 mL). The reaction mixture was heated at 100 °C overnight. The reaction solution was quenched in water (100 mL). The precipitate was filtered and purified on silica gel column chromatography using ethyl acetate/hexane(1/2, v/v) as mobile phase to get yellow solid (2.3 g, 90%). 1H NMR (CDCl3): δ 2.11 (s, 3H), 6.91 (d, J = 9.0 Hz, 1H), 7.73 (m, 2H), 7.91 (br s, 1H), 8.25 (d, J = 9.0 Hz, 1H), 8.51 (d, J = 9.3 Hz, 1H), 9.16 (s, 1H). mp 193.7 – 195.7 °C.
N-(2-((2, 4-dinitrophenyl)thio)-5-iodophenyl)-acetamide (10e)
Compound 10c (0.5 g, 1.5 mmol) was dissolved in acetic acid (20 mL). ICl (1 M) (10 mL) was added into above solution under nitrogen atmosphere. The mixture was refluxed for 3 d and quenched in water (200 mL). After filtration, the residue was purified on silica gel column chromatography using ethyl acetate/hexane (1/2, v/v) as mobile phase to yield yellow solid (220 mg, 32%). 1H NMR (CDCl3): δ 2.08(s, 3H), 6.90 (d, J = 9.3Hz, 1H), 7.87 (m, 2H), 7.99 (br s, 1H), 8.23 (d, J = 8.7Hz, 1H), 8.31 (d, J = 8.4Hz, 1H), 9.11 (s, 1H). mp 224.1 – 226.0 °C.
General procedure A
Potassium hydroxide (98 mg, 1.74 mmol)) was added into the solution of compound 10a (300 mg, 0.87 mmol) in acetone (20 mL) in portion under reflux. The reaction solution was refluxed for 2 h and quenched in ice-cold water. The precipitate was filtrated and recrystalized in acetone/water (3/2, v/v) to achieve the corresponding product 11a. Compounds 11b–11e were prepared by the same procedure.
7-Methoxy-10H-phenothiazine-3-carbonitrile (11a)
Yellow solid (100 mg, 45%). 1H NMR (DMSO-d6): δ 3.67 (s, 3H), 6.62 (m, 4H), 7.34 (m, 2H), 9.02 (br s, 1H). 13C NMR (DMSO-d6): δ 55.8, 102.7, 112.1, 113.7, 114.3, 116.1, 117.0, 117.0, 119.5, 129.9, 132.6, 133.4, 146.7, 155.8. Anal. calcd for C14H10N2OS: C, 66.12; H, 3.96; N, 11.02. Found: C, 66.13; H, 3.86; N, 11.03. mp 198.0 – 198.9 °C.
3-Methoxy-7-nitro-10H-phenothiazine (11b)
Synthetic procedure described for 11a was followed starting with 10b (100 mg, 280 mmol). Violet solid (61 mg, 79%) 1H NMR (DMSO-d6): δ 3.66 (s, 3H), 6.58–6.64 (m, 4H), 7.71 (s, 1H), 7.83 (d, J = 9.0 Hz, 1H), 9.39 (br s, 1H). 13C NMR (DMSO-d6): δ 55.8, 112.0, 113.3, 113.8, 116.2, 116.6, 116.8, 122.1, 125.2, 132.2, 140.8, 148.4, 156.3. Anal. calcd for C13H10N2O3S: C, 56.92; H, 3.67; N, 10.21. Found: C, 56.64; H, 3.54; N, 10.07. mp 168.8 – 170.1 °C.
3-Nitro-10H-phenothiazine (11c)
Synthetic procedure described for 11a was followed starting with 10c (300 mg, 0.9 mmol). Violet product (160 mg, 73%). 1H NMR (DMSO-d6): δ 6.69 (m, 2H), 6.84 (t, J = 7.2 Hz, 1H), 6.93 (d, J = 7.2 Hz, 1H), 7.02 (t, J = 7.2 Hz, 1H), 7.73 (s, 1H), 7.85 (d, J = 8.7 Hz, 1H), 9.51 (br s, 1H). 13C NMR (DMSO-d6): δ 113.8, 115.7, 115.8, 117.3, 122.0, 124.1, 125.0, 126.7, 128.5, 139.2, 141.4, 148.2. Anal. calcd for C12H8N2O2S: C, 59.00; H, 3.30; N, 11.47. Found: C, 59.02; H, 3.26; N, 11.33. mp 219.1 – 219.6 °C.
3-Bromo-7-nitro-10H-phenothiazine (11d)
Synthetic procedure described for 11a was followed starting with 10d (240 mg, 0.6 mmol) to afford violet solid (87 mg, 45%). 1H NMR (DMSO-d6): δ 6.60 (d, J = 8.4 Hz, 1H), 6.67 (d, J = 8.4 Hz, 1H), 7.17 (m, 2H), 7.73(s, 1H), 7.85(d, J = 9.0 Hz, 1H), 9.59 (br s, 1H). 13C NMR (DMSO-d6): δ 114.0, 114.8, 116.7, 117.2, 118.4, 122.1, 125.2, 128.6, 131.0, 138.7, 141.7, 147.6. Anal. calcd for C12H7BrN2O2S: C,44.60; H, 2.18; N, 8.67. Found: C, 44.54; H, 2.26; N, 8.56. mp > 250 °C.
3-Iodo-7-nitro-10H-phenothiazine (11e)
Synthetic procedure described for 11a was followed starting with 10e (100 mg, 0.22 mmol) to afford violet solid (290 mg, 36%). 1H NMR (DMSO-d6): δ6.45 (d, J = 8.7Hz, 1H), 6.64 (d, J = 9.3Hz, 1H), 7.22 (s, 1H), 7.30 (d, J = 9.3Hz, 1H), 7.69 (s, 1H), 7.82 (d, J = 8.7Hz, 1H), 9.54 (br s, 1H). 13C NMR (DMSO-d6): δ 86.0, 114.0, 116.9, 117.6, 118.4, 122.1, 125.1, 134.0, 136.9, 139.1, 141.6, 147.6. HRMS (ESI) m/z calcd for C12H7IN2O2S [M•] 369.9276. Found: 369.9269. HPLC purity: 97%. mp > 250 °C.
3-Methoxy-10-methyl-7-nitro-10H-phenothiazine (12)
Sodium hydride (60 %) (29 mg, 0.73 mmol) was added into the solution of compound 11b (100 mg, 0.36 mmol) in DMF (10 mL) at 0 °C. the reaction mixture was stirred under nitrogen atmosphere for 30 min and warmed to room temperature. CH3I (103 mg, 0.73 mmol) in DMF (2 mL) was added to the above solution and stirred for additional 2 h. The reaction mixture was quenched by adding water (100 mL) and extracted with ethyl acetate. The combined organic extracts were dried over anhydrous Na2SO4 and concentrated on rotary evaporator. The residue was purified on the silica gel column chromatography using ethyl acetate/hexane (1/2, v/v) as mobile phase to afford red solid (90 mg, 87%). 1H NMR (DMSO-d6): δ 3.28 (s, 3H), 3.63 (s, 3H), 6.71–6.76 (m, 2H), 6.87–6.94 (m, 2H), 7.84 (s, 1H), 7.96 (d, J = 9.3 Hz, 1H). 13C NMR (DMSO-d6): δ 36.3, 55.9, 112.9, 113.7, 114.0, 116.9, 122.1, 122.3, 124.7, 136.7, 141.7, 151.8, 156.5. Anal. calcd for C14H12N2O3S: C, 58.32; H, 4.20; N, 9.72. Found: C, 58.11; H, 4.11; N, 9.75. mp 174.7 – 175.5 °C.
7-Methoxy-10-methyl-10H-phenothiazin-3-amine (13a)
Compound 12 (500 mg, 1.7 mmol) and 10% Pd on activated carbon (20 mg) were suspended in ethanol (15 mL). Under H2 (90 psi), the reaction mixture was stirred at ambient temperature overnight. The reaction solution was filtrated and concentrated in vacuum. The product was purified on the silica gel column chromatography using ethyl acetate/hexane (1/2, v/v) as mobile phase to afford yellow solid (350 mg, 80%). 1H NMR (DMSO-d6): δ 3.15 (s, 3H), 3.69 (s, 3H), 4.79 (br s, 2H), 6.42–6.45 (m, 2H), 6.64 (d, J = 9.3 Hz, 1H), 6.73–6.80 (m, 3H). 13C NMR (DMSO-d6): δ 35.3, 55.8, 112.8, 112.9, 112.9, 113.4, 114.6, 115.2, 122.8, 123.9, 136.1, 140.4, 144.4, 154.6. Anal. calcd for C14H14N2OS: C, 65.09; H, 5.46; N, 10.84. Found: C, 65.29; H, 5.53; N, 10.59. mp 141.6 – 142.9 °C.
7-Methoxy-N, 10-dimethyl-10H-phenothiazin-3-amine (13b) and 7-methoxy-N, N, 10-trimethyl-10H-phenothiazin-3-amine (13c)
Methyl iodide (280 mg, 2 mmol) in acetonitrile (2 mL) was added into the solution of compound 13a (200 mg, 0.77 mmol) in acetonitrile (10 mL) with Na2CO3 (210 mg, 2 mmol). The reaction mixture was sealed under nitrogen atmosphere and stirred at 80 °C overnight. The reaction mixture was cooled to room temperature and partitioned between ethyl acetate and aqueous phase. The organic extract was purified by silica gel column chromatography using ethyl acetate/hexane (1/3, v/v) as mobile phase to yield two yellow solids, compound 13b (32 mg, 15%) and compound 13c (57 mg, 26%). 13b: 1H NMR(DMSO-d6): δ 2.58 (s, 3H), 3.66 (s, 3H), 3.73 (m, 4H), 6.39 (m, 2H), 6.74 (m, 4H). 13C NMR (DMSO-d6): δ 30.6, 35.3, 55.8, 110.5, 111.1, 112.8, 112.9, 114.7, 115.2, 123.1, 123.9, 136.0, 140.4, 146.1, 154.7. Anal. calcd for C15H16N2OS: C, 66.15; H, 5.92; N, 10.29. Found: C, 66.36; H, 6.09; N, 10.24. mp 170.9 – 171.7 °C. 13c: 1H NMR (DMSO-d6): δ 2.80 (s, 6H), 3.70 (s, 3H), 3.76 (m, 3H), 6.62 (m, 2H), 6.80 (m, 4H). 13C NMR (DMSO-d6): δ 35.3, 41.1, 55.8, 111.9, 112.4, 112.8, 113.0, 114.8, 115.1, 123.1, 123.8, 136.7, 140.2, 147.0, 154.8. Anal. calcd for C16H18N2OS: C, 67.10; H, 6.33; N, 9.78. Found: C, 67.36; H, 6.25; N, 9.79. mp 185.0 – 186.5 °C.
1-(3-Methoxy-7-nitro-10H-phenothiazin-10-yl)ethanone (14a)
Acetyl chloride (850 mg, 11 mmol) was added into the solution of compound 11b (1 g, 3.6 mmol) in dichloromethane (20 mL). The reaction mixture was stirred overnight at ambient temperature. The solvent and excessive acetyl chloride was removed in vacuum. The residue was dissolved into ethyl acetate and washed by water and saturated NaCl solution. The organic extract was dried by anhydrous Na2SO4 and purified on silica gel column chromatography using ethyl acetate/hexane (1/2, v/v) as mobile phase to yield yellow solid (0.4 g, 89%).1H NMR (CDCl3): δ 2.23 (s, 3H), 3.83 (s, 3H), 6.90 (d, J = 9.0 Hz, 1H), 6.98 (s, 1H), 7.32 (d, J = 8.7 Hz, 1H), 7.72 (d, J = 8.7 Hz, 1H), 8.18 (d, J = 8.7 Hz, 1H), 8.29 (s, 1H). 13C NMR (CDCl3): δ 22.9, 55.7, 112.7, 114.0, 122.0, 122.9, 127.4, 127.9, 130.7, 133.2, 134.3, 144.7, 145.6, 158.5, 169.2. Anal. calcd for C15H12N2O4S: C, 56.95; H, 3.82; N, 8.86. Found: C, 56.72; H, 3.89; N, 8.70. mp 155.9 – 156.8 °C.
1-(3-Nitro-10H-phenothiazin-10-yl) ethanone (14b)
Synthetic procedure described for 14a was followed starting with 11c (150 mg, 0.6 mmol) to afford yellow solid (110 mg, 62%). 1H NMR (DMSO-d6):δ 2.18 (s, 3H), 7.36 (t, J = 7.2 Hz, 1H), 7.45 (t, J = 7.2 Hz, 1H), 7.61 (d, J = 7.8 Hz, 1H), 7.69 (d, J = 7.8 Hz, 1H), 7.86 (d, J = 7.8 Hz, 1H), 8.24 (d, J = 8.4 Hz, 1H), 8.42 (s, 1H). 13C NMR (DMSO-d6): δ 23.1, 122.7, 123.3, 127.7, 127.9, 128.3, 128.5, 128.6, 131.2, 134.3, 138.1, 144.5, 145.8, 168.7. Anal. calcd for C14H10N2O3S: C, 58.73; H, 3.52; N, 9.78. Found: C, 58.60; H, 3.61; N, 9.62. mp 144.0 – 145.8 °C.
1-(3-Hydroxy-7-nitro-10H-phenothiazin-10-yl)ethanone (15)
The solution of BBr3 (1 M) in dichloromethane (1 mL) was added dropwise into the solution of compound 11a (100 mg, 0.32 mmol) in dichloromethane (10 mL) at −78 °C. the reaction solution was stirred overnight at ambient temperature. The solvent was removed in vacuum. The residue was partitioned between ethyl acetate and water. The organic extract was dried by anhydrous Na2SO4 and purified on silica gel column chromatography using ethyl acetate/hexane (1/2, v/v) as mobile phase to yield yellow solid (79 mg, 81%). 1H NMR (DMSO-d6): δ 2.15 (s, 3H), 6.82 (d, J = 9.0 Hz, 1H), 6.93 (s, 1H), 7.47 (d, J = 9.0 Hz, 1H), 7.82 (d, J = 9.0 Hz, 1H), 8.22 (d, J = 9.0 Hz, 1H), 8.39 (s, 1H), 10.00 (br s, 1H). 13C NMR (DMSO-d6): δ 23.0, 114.3, 115.3, 122.6, 123.2, 128.4, 128.5, 129.3, 132.3, 134.2, 145.1, 145.6, 156.7, 169.1. HRMS (ESI) m/z calcd for C14H10N2O4S [M+1] 303.0440. Found: 303.0435. HPLC purity: 98%. mp 202.3 – 205.1 °C.
3-(2-Fluoroethoxy)-7-nitro-10H-phenothiazine (16a)
Compound 15 (120 mg, 0.4 mmol) was dissolved in anhydrous DMF (10 mL), NaH (24 mg, 0.6 mmol) was added under 0 °C and stirred for 30 min. 1-Bromo-2-fluoroethane (150 mg, 0.6 mmol) was added and stirred overnight at room temperature. The reaction mixture was quenched with water (100 mL) and extracted with ethyl acetate. After removal of solvent, the residue was suspended in aqueous HCl solution (3 M) in methanol/water (1:1) and refluxed for 5 h. the reaction mixture was quenched in water and extracted with ethyl acetate, the organic extract was dried by anhydrous Na2SO4 and purified on silica gel column chromatography using ethyl acetate/hexane (1.2, v/v) as mobile phase to get violet solid (40 mg, 33%). 1H NMR (DMSO-d6): δ 4.09 (t, J = 3.6 Hz, 1H), 4.19 (t, J = 3.6 Hz, 1H), 4.60 (t, J = 3.6 Hz, 1H), 4.76 (t, J = 3.6 Hz, 1H), 6.64 (m, 4H), 7.72 (s, 1H), 7.83 (d, J = 9.3 Hz, 1H), 9.41 (br s, 1H). 13C NMR (DMSO-d6): δ 67.8, 68.0, 81.6, 83.3, 112.8, 113.4, 114.6, 116.3, 116.6, 116.9, 122.1, 125.2, 132.6, 140.9, 148.4, 155.1. Anal. calcd for C15H10N2O3S: C, 60.39; H, 3.38; N, 9.39. Found: C, 60.15; H, 3.50; N, 9.15. mp 189.3 – 191.4 °C.
(E)-3-(3-iodoallyloxy)-7-nitro-10H-phenothiazine (16b)
Prop-2-yn-1-ol (0.7 g, 12 mmol), Pd(Ph3P)2Cl2 (42 mg, 0.06 mmol) and HSnBu3 (3 g, 10 mmol) was dissolved in anhydrous tetrahydrofuran and stirred at ambient temperature for 1 h. The solvent was removed in vacuum. The residue was purified on the silica gel column using ethyl acetate/hexane (1/9, v/v) as mobile phase to yield colourless liquid, (E)-3-(tributylstannyl)prop-2-en-1-ol (1.2 g, 34%) as intermediate.
Iodine solution in chloroform (0.1 M) was added into the solution of (E)-3-(tributylstannyl)prop-2-en-1-ol (200 mg, 0.58 mmol) in CHCl3 (10 mL) and stirred at ambient temperature for 2 h. The reaction mixture was quenched by aqueous Na2S2O5 (5%) and extracted with ethyl acetate. The organic extract was concentrated and purified on silica gel column using ethyl acetate/hexane (1/3, v/v) as mobile phase to yield colorless liquid ((E)-3-iodoprop-2-en-1-ol) 85 mg, 79%. 1H NMR (CDCl3): δ2.04 (t, J = 5.7 Hz, 1H), 4.08 (t, J = 5.7 Hz, 2H), 6.39 (d, J = 14.4 Hz, 1H), 6.68 (d, J = 14.4 Hz, 1H).
Triphenylphosphine (133 mg, 0.51 mmol) was added into the solution of (E)-3-iodoprop-2-en-1-ol in CH2Cl2 (10 mL) at 0 °C and stirred for 1 h, following by the addition of CBr4 (186 mg, 0.56 mmol) and stirred for additional 2 h at ambient temperature. The solvent was removed in vacuum. The residue was purified by silica gel column chromatography using ethyl acetate/hexane (1/9, v/v) as mobile phase to get colorless liquid, (E)-3-bromo-1-iodoprop-1-ene (40 mg, 35%). 1H NMR (CDCl3): δ3.87 (d, J = 7.8 Hz, 2H), 6.54 (d, J = 14.4 Hz, 1H), 6.71 (d, J = 14.4 Hz, 1H).
Compound 15 (50 mg, 0.17 mmol), (E)-3-bromo-1-iodoprop-1-ene (50 mg, 0.2 mmol) and Na2CO3 (200 mg, 0.2 mmol) was mixed in DMF (10 mL) and stirred for 3 h at ambient temperature. The reaction mixture was quenched in water (50 mL) and extracted with ethyl acetate. The organic extract was concentrated in vacuum. The residue was suspended in HCl (3 M)/MeOH (1:1, v/v) and refluxed for 5 h. the precipitate was filtrated to get violet solid.16b (25 mg, 36%).1H NMR (DMSO-d6): δ 4.41 (s, 2H), 6.61–6.71 (m, 6H), 7.71(s, 1H), 7.83 (d, J = 9.0Hz, 1H), 9.41 (br s, 1H). 13C NMR (DMSO-d6): δ 69.9, 82.6, 113.0, 113.4, 114.7, 116.2, 116.6, 116.9, 122.1, 125.2, 132.6, 140.8, 141.2, 148.4, 154.7. Anal. calcd for C15H11IN2O3S: C, 42.27; H, 2.60; N, 6.57. Found: C, 42.46; H, 2.72; N, 6.38. mp > 250 °C.
3-Nitro-7-(prop-2-yn-1-yloxy)-10H-phenothiazine (16c)
Synthetic procedure described for 16b was followed starting with 15 (70 mg, 0.23 mmol) and 3-bromopropyne (38 mg, 0.25 mmol) to afford violet solid (55 mg, 86%). 1H NMR (DMSO-d6): δ 3.58 (s, 1H), 4.71 (s, 2H), 6.62–6.67 (s, 4H), 7.73 (s, 1H), 7.84 (d, J = 9.0Hz, 1H), 9.43 (br s, 1H). 13C NMR (DMSO-d6): δ 56.2, 78.7, 79.5, 113.2, 113.5, 115.0, 116.2, 116.5, 116.8, 122.1, 125.2, 133.1, 148.4, 154.1. Anal. calcd for C14H10N2O3S: C, 54.89; H, 3.62; N, 9.15. Found: C, 54.87; H, 3.54; N, 8.92. mp 226.4 – 227.0 °C.
4.2. Thioflavin T fluorescence assay for α-synuclein fibrils
4.2.1. Production of purified recombinant α-synuclein protein
α-Synuclein recombinant protein was produced in E. Coli. 38, 39 BL21(DE3)RIL E. Coli were transformed with a pRK172 bacterial expression plasmid containing the human α-synuclein coding sequence. Freshly transformed BL21 colonies were inoculated into 2 L baffled flasks containing 250 mL sterilized TB (1.2% bactotryptone, 2.4% yeast extract, 0.4% glycerol, 0.17 M KH2PO4, 0.72 M K2HPO4) with 50 μg/ml ampicillin, and incubated overnight at 37 °C with shaking. Overnight cultures were pelleted by centrifugation at 3,900 × g for 10 min at 25 °C. Bacterial pellets were resuspended in 20 mL osmotic shock buffer (30 mM Tris-HCl, 2 mM EDTA, 40% Sucrose, pH 7.2) by gentle vortexing and incubated at room temperature for 10 minutes. The cell suspension was then centrifuged at 8,000 × g for 10 min at 25 °C and the pellet was resuspended in 22.5 mL cold H2O before adding 9.4 μL 2 M MgCl2 to each tube. The suspension was incubated on ice for 3 min prior to centrifugation at 20,000 x g for 15 min at 4 °C. The supernatant was transferred to a fresh tube, streptomyocin was added to a final concentration of 10 mg/mL and then centrifuged at 20,000 x g for 15 min at 4 °C. The supernatant from this step was collected and dithiothreitol (DTT) and Tris-HCl were added to final concentrations of 1 mM and 20 mM respectively, before boiling for 10 min to precipitate heat-sensitive proteins, which were pelleted at 20,000 x g for 15 minutes at 4 °C. The supernatant was collected and filtered through a 0.45 μm surfactant free cellulose acetate filter (Corning) before loading onto a 1 mL DEAE Sepharose column equilibrated in 20 mM Tris-HCl pH 8, 1 mM EDTA, and 1 mM DTT. The DEAE column was washed with 20 mM Tris-HCl pH 8, 1 mM EDTA, 1 mM DTT before eluting α-synuclein protein in 20 mM Tris-HCl, pH 8, buffer with 1 mM EDTA, 1 mM DTT and 0.3 M NaCl. The purified α-synuclein protein was dialyzed overnight in 10 mM Tris-HCl, pH 7.6, 50 mM NaCl, 1 mM DTT. Preparations contained greater than 95% α-synuclein protein as determined by SDS-PAGE and BCA assay with a typical yield of 30 mg protein per 250 ml culture.
4.2.2. Fibril Preparation
The purified, recombinant α-synuclein monomer (2 mg/mL) was incubated in Tris-HCl (20 mM), NaCl (100 mM) at 37 °C while shaking at 1000 rpm in an Eppendorf Thermomixer in a 37 °C temperature-controlled room for 72 h. To determine the concentration of fibrils, fibril reaction mixture (100 μL) was centrifuged at 18,000 ×g for 10 min to separate fibrils from monomer. The concentration of α-synuclein monomer in the supernatant was determined in a bicinchoninic acid (BCA) protein assay along with a bovine serum albumin (BSA) standard curve. The measured decrease in monomer concentration was used to determine the concentration of fibrils in the 72 h fibril reaction mixture.
To prepare fibrils for binding assays, the fibril mixture prepared above was centrifuged at 18,000 × g for 10 min. The supernatant was discarded and the fibril pellet was resuspended in Tris-HCl buffer (30 mM, pH = 7.4) to achieve the desired concentration of fibrils for use in the assay.
4.2.3. Determination of maximum excitation/emission wavelength of ThT-α-synuclein fibrils
The ThT solution (6.0 μM) in Tris-HCl buffer (30 mM, pH = 7.4, 40 μL) was added into each of three wells containing α-synuclein fibrils suspension (3.0 μM) in Tris-HCl buffer (30 mM, pH = 7.4, 40 μL) in a 96 well plate for fluorescence detection. The mixture was incubated at room temperature for 1 h with gentle shaking. The reaction plate was scanned by the excitation wavelength range from 430 to 465 nm. The maximum excitation wavelength (λex) was determined according to the fluorescent intensity-excitation wavelength curve. At λex, the emission wavelength was scanned to get maximum emission wavelength (λem). λex and λem for the free ThT and ThT-monomeric α-synuclein was determined by the same procedure described above.
4.2.4. Measurement of the dissociation constant of ThT-α-synuclein fibrils (Kd)
ThT solutions of various concentration from 10 nM to 40 μM in Tris-HCl buffer (30 mM, pH = 7.4, 40 μL) were added into the 96 well plate containing α-synuclein fibrils (3.0 μM) in Tris-HCl buffer (30 mM, pH = 7.4, 40 μL). The mixture was incubated at room temperature for 1h with shaking. The fluorescence intensity for each well was measured at λex and λem, using a Biotek fluorescence plate reader with a 440/30 excitation filter and 485/20 emission filter. The ThT-α-synuclein fibrils saturation binding curve and Kd value were analyzed using Prism 5 software (Graphpad).
4.2.5. Measurement of compounds’ affinity to α-synuclein fibrils (Ki)
20 μL ThT solution (12.0 μM) in Tris-HCl buffer (30 mM, pH = 7.4,) was added into a 96 well plate containing 20 μL α-synuclein fibrils (6.0 μM) in Tris-HCl buffer (30 mM, pH = 7.4,) plus 40 μL compound at different concentration in Tris-HCl buffer (30 mM, pH = 7.4,) plus 10% DMSO. The final concentration of ThT in the assay was 3 μM, approximately 3 times the Kd, which predicts approximately 75% occupancy of the ThT binding sites. The mixture was incubated at room temperature for 1 h with shaking. The fluorescence intensity for each well was measured at λex and λem. IC50 values for each compound were determined by fitting the data to the equation Y=Bottom + (Top-Bottom)/(1 + 10(X-LogIC50)) using nonlinear regression by Kaleidagraph software, where Top and Bottom are the Y values for the top and bottom plateaus of the binding curve. We derived the Ki values from the IC50 values using the Cheng-Prusoff equation,40 Ki=IC50/(1+[ThT]/Kd).
4.2.6. Measurement of α-synuclein fibril dissociation in the presence of phenothiazine compounds
Phenothiazine compounds were added to reactions containing α-synuclein fibrils (1.5 μM) in 30 mM Tris-HCl buffer pH7.4 with 3 μM ThT solution in a total volume of 800 μl and incubated at room temperature for 1 hour. Separate reactions contained 1) no phenothiazine compound, 2) 3 μM compound 11b, 3) 3 μM compound 16a, and 4) 3 μM compound 16b. Following the incubation period the reactions (n=3 for control and each compound) were centrifuged at 100,000 × g for 20 min at 4° C to pellet α-synuclein fibrils. The supernatants were removed and the fibrils in the pellet were resuspended in SDS-PAGE sample buffer (recipe), sonicated in a (sonicator model) sonicator at room temperature, and then incubated at 95 °C for 10 min to solubilize the fibrils by dissociation to monomeric α-synuclein. 15 μL of solubilized fibrils from each reaction were loaded onto an SDS-PAGE (Bio-Rad Criterion) gel and electrophoresed for 1 hour at 175 V. Protein was quantified by staining with SYPRO Ruby protein gel stain (Invitrogen) and then imaged with a Kodak Imager Station 440 using UV excitation. A standard curve was included on the gel to ensure that the reactions samples contained levels of α-synuclein within the linear range of SYPRO Ruby detection.
Supplementary Material
Acknowledgments
The authors like to thank Dr. Robert H. Mach for his advice on the project. Financial support for these studies was provided by the National Institutes of Health under Grants NS061025 (Z.T.), NS075527 (Z.T.)MH092797 (Z.T.) and Barnes-Jewish Hospital Foundation/Washington University Institute of Clinical & Translational Sciences (P,K). Mass spectra were acquired at the NIH NCRR Biomedical Mass Spectrometry Resource at Washington University (NIH 2P41RR000954).
Abbreviations
- PD
Parkinson’s Disease
- AD
Alzheimer’s disease
- LBs
Lewy bodies
- LNs
Lewy neuritis
- CNS
central nervous system
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- ThT
Thioflavin T
- AFU
arbitrary fluorescence units
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jankovic J. J Neurol Neurosurg Psychiatry. 2008;79:368. doi: 10.1136/jnnp.2007.131045. [DOI] [PubMed] [Google Scholar]
- 2.Hely MA, Reid WG, Adena MA, Halliday GM, Morris JG. Mov Disord. 2008;23:837. doi: 10.1002/mds.21956. [DOI] [PubMed] [Google Scholar]
- 3.Tsuboi Y, Uchikado H, Dickson DW. Parkinsonism and Related Disorders. 2007;13:S221. doi: 10.1016/S1353-8020(08)70005-1. [DOI] [PubMed] [Google Scholar]
- 4.Braak H, Rüb U, Jansen Steur EN, Del Tredici K, de Vos RA. Neurology. 2005;64:1404. doi: 10.1212/01.WNL.0000158422.41380.82. [DOI] [PubMed] [Google Scholar]
- 5.Halliday G, Hely M, Reid W, Morris J. Acta Neuropathol. 2008;115:409. doi: 10.1007/s00401-008-0344-8. [DOI] [PubMed] [Google Scholar]
- 6.Hurtig HI, Trojanowski JQ, Galvin J, Ewbank D, Schmidt ML, Lee VM, Clark CM, Glosser G, Stern MB, Gollomp SM, Arnold SE. Neurology. 2000;54:1916. doi: 10.1212/wnl.54.10.1916. [DOI] [PubMed] [Google Scholar]
- 7.Jellinger KA. Acta Neuropathol. 2008;116:1. doi: 10.1007/s00401-008-0406-y. [DOI] [PubMed] [Google Scholar]
- 8.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Nature. 1997;388:839. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 9.Baba M, Nakajo S, Tu P, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T. Am J Pathol. 1998;152:879. [PMC free article] [PubMed] [Google Scholar]
- 10.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. Proc Natl Acad Sci USA. 1998;95:6469. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maroteaux L, Campanelli JT, Scheller RH. J Neurosci. 1988;8:2804. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng F, Vivacqua G, Yu SJ. Chem Neuroan. 2011;42:242. doi: 10.1016/j.jchemneu.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 13.Cabin DE, Shimazu K, Murphy D, Cole NB, Gottschalk W, Mcllwain KL, Orrison B, Chen A, Ellis CE, Paylor R, Lu B, Nussbaum RLJ. Neurosci. 2002;22:8797. doi: 10.1523/JNEUROSCI.22-20-08797.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murphy DD, Rueter SM, Trojanowski JQ, Lee VMYJ. Neurosci. 2000;20:3214. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu S, Ninan I, Antonova I, Battaglia F, Trinchese F, Narasanna A, Kolodilov N, Dauer W, Hawkins R, Arancio O. EMBO J. 2004;23:4506. doi: 10.1038/sj.emboj.7600451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schulz-Schaeffer WJ. Acta Neuropathol. 2010;120:131. doi: 10.1007/s00401-010-0711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kazantsev AG, Kolchinsky AM. Arch Neurol. 2008;65:1577. doi: 10.1001/archneur.65.12.1577. [DOI] [PubMed] [Google Scholar]
- 18.Bate C, Gentlemen S, Williams A. Mol Neurodegeneration. 2010;5:55. doi: 10.1186/1750-1326-5-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee VM, Trojanoski JQ. Neuron. 2006;52:33. doi: 10.1016/j.neuron.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 20.Kramer ML, Schulz-Schaeffer WJJ. Neurosci. 2007;27:1405. doi: 10.1523/JNEUROSCI.4564-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masuda M, Suzuki N, Taniguchi S, Oikawa T, Nonaka T, Iwatsubo T, Hisanaga S, Goedert M, Hasegawa M. Biochemistry. 2006;45:6085. doi: 10.1021/bi0600749. [DOI] [PubMed] [Google Scholar]
- 22.Fodero-Tavoletti MT, Mulligan RS, Okamura N, Furumoto S, Rowe CC, Kudo Y, Masters CL, Cappai R, Yanai K, Villemagne VL. European J Pharm. 2009;617:54. doi: 10.1016/j.ejphar.2009.06.042. [DOI] [PubMed] [Google Scholar]
- 23.Lindquist SL, Outeiro T, Labaudiniere R, Fleming J, Bulawa CE, Weigel C, Liang F, Gupta S, Ripka A. U.S. Patent appl. No 60/787,113. 2010
- 24.Lansbury PT, Justman CJ, Shinobu L. U.S. Patent 60/953,379. 2009
- 25.Hajieva P, Mocko JB, Moosmann B, Behl CJ. Neurochem. 2009;110:118. doi: 10.1111/j.1471-4159.2009.06114.x. [DOI] [PubMed] [Google Scholar]
- 26.González-Muñoz GC, Arce MP, López B, Pérez C, Villarroya M, López MG, García AG, Conde S, Rodríguez-Franco MI. European J Med Chem. 2010;45:6152. doi: 10.1016/j.ejmech.2010.09.039. [DOI] [PubMed] [Google Scholar]
- 27.Marivingt-Mounir C, Mettey Y, Vierfond JJ. Heterocyclic Chem. 1998;35:843. [Google Scholar]
- 28.Corral C, Lissavetzky J, Madronero R. Eur J Med Chem. 1978;13:389. [Google Scholar]
- 29.Gonzalez-Munoz GC, Arce MP, Lopez B, Perez C, Romero A, del Barrio L, Martin-de-saavedra MD, Egea J, Leon R, Villarroya M, Lopez MG, Garcia AG, Conde S, Rodriguez-Franco MI. Eur J Med Chem. 2011;46:2224. doi: 10.1016/j.ejmech.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 30.Uversky VN, Li J, Fink AL. J Biol Chem. 2001;276:10737. doi: 10.1074/jbc.M010907200. [DOI] [PubMed] [Google Scholar]
- 31.Celej MS, Jares-Erijman EA, Jovin TM. Biophy J. 2008;94:4867. doi: 10.1529/biophysj.107.125211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ye L, Velasco A, Fraser G, Beach TG, Sue L, Osredkar T, Libri V, Spillantini MG, Goedert M, Lockhart AJ. Neurochem. 2008;105:1428. doi: 10.1111/j.1471-4159.2008.05245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Honson NS, Johnson RL, Huang W, Inglese J, Austin CP, Kuret J. Neurobiol Dis. 2007;28:251. doi: 10.1016/j.nbd.2007.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levine H., III Protein Sci. 1993;2:404. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lockhart A, Ye L, Judd DB, Merritt AT, Lowe PN, Morgenstern JL, Hong G, Gee AD, Brown J. J Biol Chem. 2005;280:7677. doi: 10.1074/jbc.M412056200. [DOI] [PubMed] [Google Scholar]
- 36.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Ann Neurol. 2004;55:306. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 37.Mathis CA, Wang Y, Holt DP, Huang GF, Debnath ML, Klunk WEJ. Med Chem. 2003;46:2740. doi: 10.1021/jm030026b. [DOI] [PubMed] [Google Scholar]
- 38.Giasson BI, Uryu K, Trojanowski JQ, Lee VM. J Biol Chem. 1999;274:7619. doi: 10.1074/jbc.274.12.7619. [DOI] [PubMed] [Google Scholar]
- 39.Huang C, Ren G, Zhou H, Wang CC. Protein Expr Purif. 2005;42:173. doi: 10.1016/j.pep.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 40.Cheng Y, Prusoff WH. Biochem Pharmacol. 1973;22:3099. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.