

Abstract
Inflammation in the eye is tightly regulated by multiple mechanisms that together contribute to ocular immune privilege. Many studies have shown that it is very difficult to abrogate the immune privileged mechanism called anterior chamber associated immune deviation (ACAID). Previously, we showed that retinal laser burn (RLB) to one eye abrogated immune privilege (ACAID) bilaterally for an extended period of time. In an effort to explain the inflammation in the non-burned eye, we postulated that neuronal signals initiated inflammation in the contralateral eye. Here, we test the role of substance P, a neuroinflamatory peptide, in RLB-induced loss of ACAID. Histological examination of the retina with and without RLB revealed an increase of the substance P-inducible neurokinin 1 receptor (NK1-R) in the retina of first, the burned eye, and then the contralateral eye. Specific antagonists for NK1-R, given locally with antigen within 24h, but not 3,5, or 7 days post RLB treatment, prevented the bilateral loss of ACAID. Substance P Knockout (KO) mice retained their ability to develop ACAID post RLB. These data support the postulate that substance P transmits early inflammatory signals from the RLB eye to the contralateral eye to induce changes to ocular immune privilege and has a central role in the bilateral loss of ACAID. The possibility is raised that blocking of the substance P pathway with NK1-R antagonists post ocular trauma may prevent unwanted and perhaps extended consequences of trauma-induced inflammation in the eye.
Keywords: Ocular Immune Privilege, Retinal Laser Burn, substance P
Introduction
The eye has a unique environment and contains both neuronal and non-neuronal tissues. Inflammation in the eye is heavily regulated by a wide variety of immunosuppressive factors, cytokines and neuropeptides. Previously, we reported that retinal laser burn (RLB) altered the immune homeostasis/regulation of both the treated and non-treated eye because antigens injected into the anterior chamber (a.c.) of either eye were unable to induce tolerance (1). Further testing showed that after RLB treatment, the aqueous humor from either eye was unable to induce tolerogenic antigen presenting cells in vitro, suggesting that the immunosuppressive environment was altered. The burned eye had evidence of injury but the non-burned eye was not physically compromised, retained its blood ocular barrier and lacked inflammatory cell infiltration (1). Since there are no known physical connections between the eyes, we posed that neuropeptides might relay signals from the RLB-treated eye to the non-treated eye to upset immune regulation. Such signals could either turn off the existing tolerogenic immune regulation or turn on a strong immune inflammatory response that disturbs immune homeostasis in the eye.
In the eye, family members of the vasoactive intestinal polypeptide (VIP) and Calcitonin gene-related peptide (CGRP) receptor family are immunosuppressive (2). Neuronal signaling throughout the body is carried out by a variety of neuropeptide families including tachykinins. Recently, the tachykinin family member, substance P, was reported to function outside the central nervous system as a proinflammatory molecule in the immune response (3, 4). Moreover, substance P and its receptor, Neurokinin 1 receptor (NK1-R), are expressed by eosinophils, lymphocytes, dendritic cells, macrophages and lymphocytes (4, 5). Binding of substance P to NK1-R on immune cells causes increased production of proinflammatory molecules such as MIP-1β, IL-6, and TNF-α and exacerbates the inflammatory response (6–8). The ability of substance P to exacerbate inflammation as well as the fact that it is expressed in cells of the amacrine layer of the retina, the cornea, and nerve fibers ending in the ciliary body, iris, and cornea epithelial (9) suggested to us that it may be involved in the loss of immune privilege after RLB.
Immune privileged sites were originally defined as areas of the body in which foreign tissue grafts survive for extended or indefinite periods of time (10). Since the first report of immune privileged sites by Sir Peter Medawar (11), many reports have explored the mechanisms of immune privilege (12–17). A popular experimental model for the study of immune privilege is called Anterior Chamber Associated Immune Deviation (ACAID). In the ACAID model, antigen that is introduced in the anterior chamber of the eye is taken up by indigenous F4/80+APC antigen presenting cells (F4/80+APC) processed, and transported to the secondary lymphoid organ (in this case, the spleen) (18). Subsequently, antigen specific T regulatory (Treg) cells are generated. F4/80+ cells that leave the eye recruit more F4/80+ cells to the spleen (18). Mice that receive antigen in the anterior chamber show a suppressed response to the subcutaneously inoculated antigen by exhibiting little or no swelling post antigenic challenge in the ear (18).
Immune privilege homeostasis in the eye is due in part, to regulatory mechanisms of the resident stromal cells(11, 19–22), indigenous antigen presenting cells (23), the dominance of soluble immunosuppressive molecules in the fluids (24) and the structural barriers of the tissues and outer borders of the eye (25). These multiple overlapping mechanisms contribute to immune homeostasis and control inflammation in the eye and define ocular immune privilege as we know it (26, 27). Immune privilege is difficult to disrupt even after the direct injection of LPS (28) or the induction of experimental autoimmune uveitis (EAU) (29). However, we found that multiple aspects of immune privilege were abrogated in both eyes of mice that receive unilateral RLB treatment (1). The goal of this study was to test the hypothesis that the release of substance P after RLB contributes to the loss of immune regulation in both the burned and non-burned eye.
Materials and Methods
Animals
Female C57BL/6J (B6) mice and B6.Cg-Tac1tm1Bbm/J female mice (substance P Knockout, B6 background), and B6.129S2-IL6tm1Kopf/J female mice (IL-6 Knockout, B6 background) were purchased from Jackson Laboratory (Bar Harbor, ME). Substance P Knockout mice lack the tac-1 gene and thus lack substance P and neurokinin A. Mice used in all experiments were 8–12-wk-old, unless otherwise stated. All animals were treated humanely and in accordance with the Schepens Eye Research Institute Animal Care and Use Committee and National Institutes of Health guidelines
Reagents and antibodies
Substance P peptide, Spantide I and Spantide II were purchased from Bachem, (Torrance, CA). Ovalbumin (OVA) and Complete Fruend’s Adjuvant were purchased from Sigma Aldrich (St. Louis, MO). Neurokinin 1 receptor antibody was purchased from Millipore (Billerica, MA). Alexa Fluor 488 conjugated goat anti rabbit IgG assays was purchased from Invitrogen (Carlsbad, CA).
Retnal laser burn (RLB)
Mice were anesthetized with ketamine (120 mg/kg) and xylazine (12 mg/kg), and pupils were dilated with 1% tropicamide. A hand-held cover slip was used as a contact lens. A slit lamp was used to deliver a laser beam (wave length 810nm, diameter 200nm, power 50mW, and duration time 50ms) from a diode laser (IRIDEX, Mountain View, CA) to the retina. Four spots (200 nm diameter) were burned on the retina in the right eye of B6 mice.
ACAID Assay
ACAID was induced in mice by inoculating OVA (50 μg/2 μl in HBSS) into the anterior chamber (a.c.) seven days before sensitizing, subcutaneously, with OVA (100 μg/ml in HBSS, 50 μl) emulsified in CFA (50 μl). One week after sensitization, mice were challenged in the right ear pinnae of the with OVA-pulsed peritoneal exudate cells (PEC: 2 × 105 in10μl). The thickness of the ear was measured 24 h later with an engineer’s micrometer (Mitutoyo, Paramus, NJ), as increased ear thickness was an indication of delayed hypersensitivity (DH).
Histological analyses
Eyes were enucleated from euthanized mice at various times post RLB treatment. Eyes were placed into Tissue-Tek Optimal Cutting Temperature Compound (Sakura Finetek, Torrance, CA), frozen on dry ice before storing at −85 °C. Horizontal cryosections (10μm) were prepared by Schepens Eye Institute’s Morphology Core.
Frozen cryosections were thawed and rehydrated in PBS for 20 minutes at 4 °C. Slides were than incubated in blocking solution (1% goat serum + .1% Saponin in PBS) for 1 hour at 4 °C. Primary staining antibody was diluted in blocking solution (1:100) was added and slides were incubated overnight at 4 °C in a humidification chamber. On day 2, slides were washed in PBS and incubated for 1h at 4 °C in Alexa Fluor 488 conjugated goat anti rabbit Ig in blocking solution. After washing, cover slips were mounted and sealed. Photomicrographs were taken using a Leica TCS-SP5 confocal microscope (Leica Microsystems, Bannockburn, IL), and images shown are 400× magnification.
Data analysis and statistics
Three dimensional (3D) images, 3D cartoons, analysis and quantification of confocal data as well as calculation of antibody binding intensity were made using Image Pro Plus v.7 (Media Cybernetics; Bethesda, MD). Integrated Optical Density (IOD) was determined by calculating the binding intensity of NK1-R antibody binding in the slices (Image Pro Plus version 6.3, Media Cybernetics Inc. Bethesda, MD). At least three confocal z-series were used at each time point and their average was plotted and analyzed. Statistical data were analyzed by analysis of variance (ANOVA) and Scheffe's test. (The data are presented as mean ± SEM). A value of P≤0.05 was considered significant.
Results
Retinal laser burn (RLB) induced extended loss in the contralateral eye
Previously, we reported that the eye that received RLB lost immune privilege for as long as 56 days post trauma (1), but the time course in the contralateral eye was not studied. Here, we extended the time course to 77 days in the burned (ipsilateral) eye and tested how long ACAID abrogation lasted in the non-burned (contralateral) eye. ACAID was initiated in non-treated or RLB treated mice 1, 7, 14, 21, 68, or 77 days post RLB by injecting antigen into the anterior chamber (a.c.). As expected, immune privilege was present in non-treated mice (Figure 1). Mice that received RLB treatment 1 day through 77 days prior to antigen injection into the burned eye had no significant difference in ear swelling compared to immunized mice and therefore were unable to develop ACAID (Figure 1). Mice that received RLB treatment followed by a.c. injection of antigen into the non-burned eye at all time points through 68 days post RLB also, did not develop ACAID. These data show that the loss of immune privilege after RLB treatment is long lasting in both eyes.
Figure 1. DH response after ACAID induction in RLB treated mice.
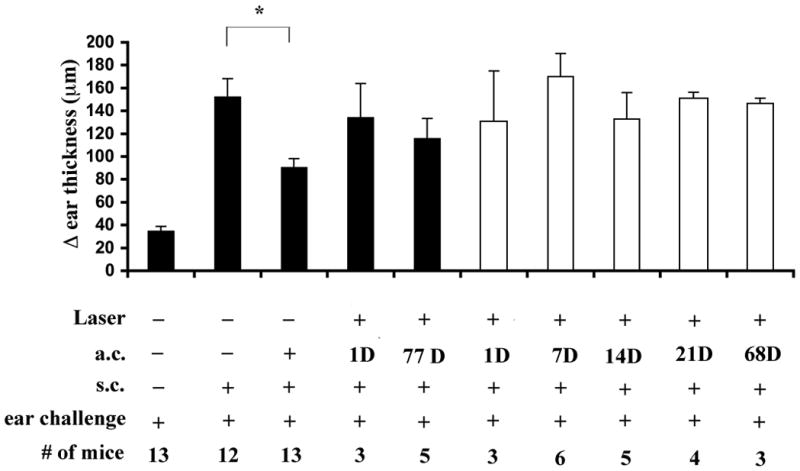
Change (Δ) in ear thickness is indicated on the ordinate. Experimental treatment of the mice in each group is indicated below each bar, on the abscissa. Mice received RLB treatment or not at the time indicated prior to ACAID induction, one week later, mice were immunized with OVA and the following week, the ear pinnae was challenged with OVA. Ipsilateral = black bars; and Contralateral = white bars. An asterisk, *, indicates a significant difference P≤0.05 between the groups. The time course to 21 days was preformed three times. Experiment at 68 days and 77 days was performed once.
Inflammatory changes to the ocular environment
A logical explanation for the inability of the eyes of RLB treated mice to develop ACAID is that the local immunosuppressive environment was altered toward inflammation by the trauma. To test if inflammatory cytokines were increased, eyes were enucleated 1, 3, and 7 days post RLB and after dissecting away the lens, the tissue RNA was extracted by standard measures. We observed increases in IL1 β, IL-6 and TNF α in both the burned and non-burned eye but there was no consistent pattern to suggest that any of the obvious inflammatory cytokines caused the loss of ACAID (data not shown). Since IL-6 was associated with loss of ACAID in experimental Autoimmune Uveitis, (29) and LPS induced inflammation in the eye (28), we tested if IL-6 were involved in RLB induced loss of ACAID (28, 33). However, IL-6 KO mice who received RLB like the IL-6 sufficient B6 mice, were unable to develop ACAID post RLB (Figure 2). Thus, IL-6 was not the cause of the RLB induced loss of immune privilege. We wondered if the RLB induced inflammatory molecule might be a neuropeptide.
Figure 2. Immune privilege in C57BL/6 or IL-6KO mice.

Change (Δ) in ear thickness is indicated on the ordinate. Experimental treatment of the mice in each group is indicated under the abscissa (anterior chamber inoculation = a.c.; subcutaneous inoculation = s.c.; ear challenge = e.c.). Panel A. B6 mice. Panel B. IL-6KO mice. Briefly mice were treated with OVA 1 day after RLB treatment. One week later, mice were immunized with antigen and mice were challenged in the ear pinnae with OVA the following week. This experiment was performed twice with similar results.
Immunohistochemistry analysis of Neurokinin 1 receptor (NK1-R) expression after RLB treatment
In the quiescent retina, 20–30% of the amacrine cells express NK1-R (30) and about one third of NK1-R positive cells express substance P (31). However, after RLB trauma the relative expression of substance P or NK1-R is unknown. Substance P is a neuropeptide of the tachykinin family that exerts biologic activities in the central nervous system and in peripheral organs and is known to modulate parameters of the immune response(32). Because substance P has a short half-life (33), substance P protein is difficult to measure. Therefore, monitoring its receptor, NK1-R, is an effective measurement for the presence of substance P. In brief, eyes were enucleated from mice at 6h, 72h, or 7 days post RLB. Eyes from untreated mice were used for controls. Cyrosections were stained with anti-NK1-R, Alexa Fluor 488 conjugated with goat anti rabbit Ig and the nuclear stain, TO-PRO 3, before analyzing by confocal microscopy. Analysis of retinal sections showed that by 72h, the overall expression of NK1-R in the ipsilateral eye increased in RLB treated mice compared to untreated mice (Figure 3). Seven days post RLB treatment, the ipsilateral eye showed an eight-fold increase of NK1-R and the contralateral eye had a ten-fold increase over the NK1-R expression in the retina of naive mice (Figure 3). The amount of NK1-R staining in the retina of both eyes at 72h and 7 days is statistically significant P≤0.05 compared to the amount of staining seen in the naïve retina. NK1-R staining of the cornea from RLB mice remained similar to the corneas in naïve mice (Figure 3). These data suggest that the substance P inflammatory pathway is activated in both eyes after RLB to one eye. NK1-R expression in the RLB treated eye preceded the increase of expression in the contralateral eye, suggesting that the substance P originates in the burned eye, and is the messenger for the loss of immune privilege in the contralateral eye and has a critical role in interfering with immune homeostasis in both eyes.
Figure 3. Photomicrograph of retina sections stained with Neurokinin 1 receptor of mice that received RLB treatment or not.

A. 3D Photomicrograph B. 3D cartoon representation. For figure 3A and B, green indicates NK1-R staining, and red indicates TOPRO 3 as a nuclear stain. Sections are from the eyes of either non-treated mice or mice that received RLB 6h, 72h, or 7 days prior to enucleation. All images are 400× magnification. The layers of the eye are indicated to the left of the photomicrograph. C. Green indicates NK1-R staining, and red indicates DAPI 3 as a nuclear stain. Cornea of naive or RLB treated mice 6h post-RLB. D. Quantification of NKR-1 expression in retina of RLB treated mice. Integrated Optical Density (IOD) is shown in fold increase for each group. All groups were repeated at least three times and where normalized to naïve (control) mice. Each IOD value from the individual groups was normalized to each of the controls and the fold increase calculated and plotted. Ipsilateral = black bars; Contralateral = white bars. The confocal analysis was performed three times.
Effect of blocking substance P receptor of RLB induced abrogation of ACAID
To test the idea that substance P signaling induced the loss of ACAID, we used commercially available substance P receptor antagonists to block binding signaling by substance P. Spantide I is a full-length modified amino acid sequence of substance P and a selective stereotactic inhibitor for NK1-R(34). In this experiment, ACAID was initiated by injecting (a.c.) OVA with or without Spantide I (10−4M), 1, 3, 5, or 7 days post-RLB treatment (Figure 4). As before, we were unable to induce ACAID by injection (a.c.) of OVA into either eye of mice that received a unilateral RLB. RLB mice that received Spantide I and OVA (a.c) in either the ipsilateral, or the contralateral eye, 1 day after RLB developed ACAID. Interestingly, ACAID was not restored in mice that received Spantide I with OVA in either the ipsilateral or contralateral eye if given after 24h after RLB treatment. After becoming aware that the NK1-R was increased by a log 7 days post RLB, we did a dose response curve for Spantide I treatment at 7 days. Even at a 3-log increase in the dose, the antagonist was unable to interfere with the loss of ACAID a week after RLB (Figure 4). Since early (1 day), but not delayed treatment with Spantide I blocked the abrogation of ACAID in both eyes, the possibility arises that substance P is critical for initiating, or exaggerating the inflammatory response that mitigates the immune privilege response.
Figure 4. DH response after ACAID induction with or without substance P antagonist in RLB treated mice.
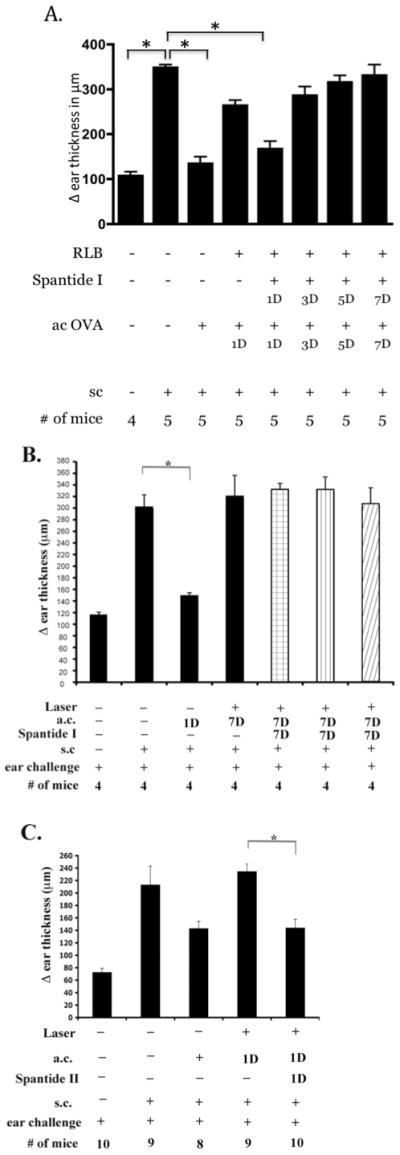
Change (Δ) in ear thickness is indicated on the ordinate (anterior chamber inoculation = a.c.; subcutaneous inoculation = s.c.). Experimental treatment of the mice in each group is indicated under the abscissa. A) Spantide I treatment over time. Mice were treated with OVA and substance P antagonist 1, 3, 5, or 7 days after RLB treatment. One week later, mice were immunized with antigen and mice were challenged in the ear pinnae with OVA the following week. Experiments on 1 and 7 days post RLB were performed a minimum of 3 times; induction of ACAID with Spantide I on 3 and 5 days post RLB was performed once. B. Spantide I treatment at different concentrations. Ipsilateral = black bars; Contralateral = white bars. Spantide I treatment 10−4 M= bar with check pattern, the dose curve was performed once. Spantide I treatment 10−3 M= bar with vertical line pattern, Spantide I treatment 10−2 M= bar with diagonal line pattern. C) Spantide II treatment. Spantide II given with OVA a.c. 1 day post RLB. Figure shows combined results of two experiments. An asterisk, *, indicates a significant difference P≤0.05 between the groups.
Since Spantide I treatment antagonizes the substance P pathway but also induces the release of histamine from mast cells (34), the possibility was raised that histamine, rather than the substance P pathway may be protective for immune privilege. To test the possibility that histamine contributed to the protection of immune privilege, NK1-R antagonist, Spantide II was tested. Spantide II is also a full-length stereotactic inhibitor of NK1-R, but did not cause the release of histamine. Thus, if Spantide II did not prevent the loss of ACAID, histamine would be involved. We found that the a.c. inoculation of OVA with Spantide II (10−4M) also mitigated the RLB induced loss of ACAID (Figure 4). Thus, the blockade of signaling via the NK1-R, and not histamine release, is responsible for the restoration of ACAID by Spantide post RLB. These observations support the postulate that substance P signaling through NK1-R abrogates immune privilege in the eye post RLB.
Effect of RLB on ACAID in substance P knockout vs. wild-type mice
Another approach to evaluating substance P’s role in the loss of ACAID was to use genetically modified mice or mice that are deficient in substance P. In brief, antigen was inoculated (a.c.) into either eye of B6 or substance P KO mice 1 day after the right eye received RLB. As before, B6 mice that received RLB prior to antigen, (a.c.) did not develop ACAID, but substance P KO mice that received RLB developed ACAID post (a.c.) inoculation of antigen (Figure 5). These data further support the idea that substance P is the neuronal message that disrupts the ability to induce ACAID post RLB in both eyes.
Figure 5. DH response after ACAID induction in RLB treated B6 or substance P KO mice.

Panel A. B6 mice. Panel B. Substance P KO mice. Change (Δ) in ear thickness is indicated on the ordinate. Experimental treatment of the mice in each group is indicated under the abscissa. Briefly, B6 or substance P KO mice were inoculated with OVA 1 day after RLB treatment. One week later, mice were immunized with antigen and mice were challenged in the ear pinnae with OVA the following week. Ipsilateral = black bars; Contralateral = white bars. An asterisk, *, indicates a significant difference P≤0.05 between the bars. The figure represents the combined results of two experiments.
Effect of substance P treatment in the abrogation immune privilege
Finally, we reasoned that if RLB induced substance P, and it, in turn, induced the inflammatory signals that interfered with ACAID induction, direct a.c. injection of substance P would mitigate ACAID induction in non-burned mice. In brief, antigen was injected (a.c.) with or without the full-length substance P peptide (10−12M). We observed that mice that received OVA into the anterior chamber developed ACAID, but mice that received OVA with the substance P peptide were unable to develop ACAID (Figure 6). These data show that substance P has the capacity to abrogate ACAID.
Figure 6. DH response after ACAID induction in substance P treated mice.
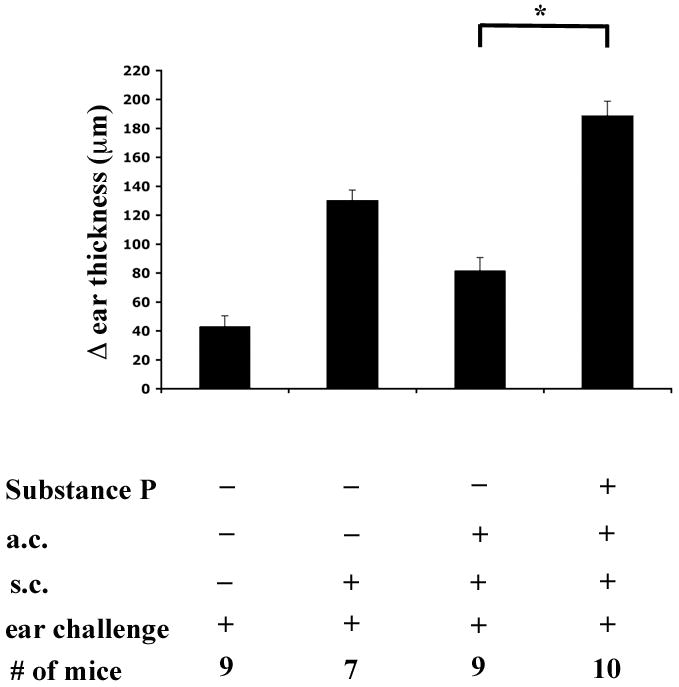
Change (Δ) in ear thickness is indicated on the ordinate. Experimental treatment of the mice in each group is indicated under the abscissa. Briefly, mice were treated with OVA and substance P peptide one week prior to being immunized with antigen. One week later mice were challenged in the ear pinnae with OVA. An asterisk, *, indicates a significant difference of P≤0.05 between the bars. The figure represents the combined results of two experiments.
Discussion
In this study, we show that RLB compromised immune privilege (ACAID) bilaterally for an extended period of time. The extended loss of immune privilege is remarkable and has not been shown before. Other studies attempting to abrogate ACAID show only short-term losses (28, 29, 35). One possibility to explain the bilateral loss of immune privilege after RLB is that cells from the periphery induce inflammatory changes in the ocular environment of both eyes. However, if the loss of immune privilege were due to a systemic signal, one would expect to observe cells infiltrating the contralateral eye, but infiltrating cells were only observed in the ipsilateral eye of mice post RLB (1). Thus, it is improbable that inflammatory cells recruited from the periphery were responsible for the loss of ACAID in the contralateral eye post RLB. Furthermore, we did not observe consistent increases in inflammatory cytokines and IL-6 KO mice still lost ACAID post RLB.
Here, we focused on the notion that a neuroinflamatory signal disrupted immune privilege and the ocular environment in both eyes post RLB. Neuropeptides are polypeptide molecules that act as neurotransmitters to carry information between neuronal tissues. The fluids in the eye contain a variety of factors, including immunosuppressive neuropeptides that contribute to immune suppression (36, 37).
Substance P is an inflammatory neuropeptide that is found in the naïve quiescent eye and was shown to contribute to the loss of ACAID in dark-adapted mice (38). Substance P binds exclusively to NK1-R. Furthermore, the binding of substance P to NK1-R induces the upregulation of the NK1-R receptor, thus an increase is a strong indication for the presence of substance P. Immunohistochemistry analysis showed a bilateral increase of NK1-R in both the ipsilateral and contralateral eye after RLB, suggesting a role for substance P in the neurological communication between the two eyes. The fact that the increase in NK1-R in the contralateral eye followed the appearance of NK1-R in the burned eye suggests that the burn initiated the neuronal signal. This observation is consistent with other reports that show a unilateral injury to the CNS leads to a bilateral response (39, 40).
This is the first report to show that neuropeptides are involved in the loss of immune regulation post ocular trauma. It is known that in other parts of the body, substance P facilitates an array of morphological and functional deficits following injury. Substance P increases vasodilatation, increases permeability of blood-brain barriers, and increases the activity of other cytotoxic neurons, post non-ocular trauma (41, 42). Substance P could be released from amacrine cells to change the immunosuppressive ocular environment to support inflammation bilaterally. Ligation of substance P to NK1-R initiates production of inflammatory cytokines from resident macrophages and microglia and recruits immune cells, including eosinophils, lymphocytes, dendritic cells, macrophages and lymphocytes from the periphery (4, 5). Substance P ligation to NK1-R has been shown to initiate downstream cascade, resulting in increased production of proinflammatory molecules such as MIP-1β, IL-6, and TNF-α (6–8). Thus, substance P has the potential to promote an inflammatory environment that in turn activates resident macrophages and microglia. A change in the suppressive microenvironment of the eye could interfere with the immune homeostasis and ocular immune privilege.
The possibility arises that targeting substance P pathway with NK1-R antagonists may be therapeutically advantageous for treatment of patients post ocular trauma to prevent or curb the subsequent inflammation. NK1-R antagonists have the potential to prevent the ability of substance P to amplify itself in an autocrine and paracrine fashion, interfere with substance P initiated recruitment of peripheral inflammatory cells, and prevent the subsequent production of many inflammatory cytokines.
Acknowledgments
We thank Mr. Thomas Flynn for preparation and submission of this manuscript. We are also appreciative of the technical training or staff received from Mr. Don Pottle on the Confocal Microscope. We thank Mr. Peter Mallen for his expert help with figures and graphs in this manuscript.
Support: This work was supported in part by grants to JSS: NIH EY11983, EY016476, NIH EY020614, and DOD W81XWH; KL: NIH F32 EY018983.
Abbreviations
- ACAID
anterior chamber associated immune deviation
- RLB
retinal laser burn
- NK1-R
neurokinin 1 receptor
- KO
Knockout
- a.c
anterior chamber
- DH
delayed hypersensitivity
References
- 1.Qiao H, Lucas K, Stein-Streilein J. Retinal laser burn disrupts immune privilege in the eye. Am J Pathol. 2009;174:414–422. doi: 10.2353/ajpath.2009.080766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taylor AW. Neuroimmunomodulation and immune privilege: the role of neuropeptides in ocular immunosuppression. Neuroimmunomodulation. 2002;10:189–198. doi: 10.1159/000068325. [DOI] [PubMed] [Google Scholar]
- 3.Maggi CA. The effects of tachykinins on inflammatory and immune cells. Regul Pept. 1997;70:75–90. doi: 10.1016/s0167-0115(97)00029-3. [DOI] [PubMed] [Google Scholar]
- 4.O'Connor TM, O'Connell J, O'Brien DI, Goode T, Bredin CP, Shanahan F. The role of substance P in inflammatory disease. J Cell Physiol. 2004;201:167–180. doi: 10.1002/jcp.20061. [DOI] [PubMed] [Google Scholar]
- 5.Douglas SD, Lai JP, Tuluc F, Schwartz L, Kilpatrick LE. Neurokinin-1 receptor expression and function in human macrophages and brain: perspective on the role in HIV neuropathogenesis. Ann N Y Acad Sci. 2008;1144:90–96. doi: 10.1196/annals.1418.007. [DOI] [PubMed] [Google Scholar]
- 6.Sio SW, Puthia MK, Lu J, Moochhala S, Bhatia M. The neuropeptide substance P is a critical mediator of burn-induced acute lung injury. J Immunol. 2008;180:8333–8341. doi: 10.4049/jimmunol.180.12.8333. [DOI] [PubMed] [Google Scholar]
- 7.Arsenescu R, Blum AM, Metwali A, Elliott DE, Weinstock JV. IL-12 induction of mRNA encoding substance P in murine macrophages from the spleen and sites of inflammation. J Immunol. 2005;174:3906–3911. doi: 10.4049/jimmunol.174.7.3906. [DOI] [PubMed] [Google Scholar]
- 8.Lotz M, Vaughan JH, Carson DA. Effect of neuropeptides on production of inflammatory cytokines by human monocytes. Science. 1988;241:1218–1221. doi: 10.1126/science.2457950. [DOI] [PubMed] [Google Scholar]
- 9.Tervo T, Tervo K, Eranko L. Ocular neuropeptides. Med Biol. 1982;60:53–60. [PubMed] [Google Scholar]
- 10.Streilein JW. Ocular immune privilege: therapeutic opportunities from an experiment of nature. Nat Rev Immunol. 2003;3:878–889. doi: 10.1038/nri1224. [DOI] [PubMed] [Google Scholar]
- 11.Medawar PB. Immunity to homologous grafted skin. III. The fate of skin homografts transplanted to the brain, to subcutaneous tissue and to the anterior chamber of the eye. Br J Exp Pathol. 1948;29:58–69. [PMC free article] [PubMed] [Google Scholar]
- 12.Li J-H, Rosen D, Sondel P, Berke G. Immune privilege and FasL: two ways to inactivate effector cytotoxic T lymphocytes by FasL-expressing cells. Immunology. 2002;105:267–277. doi: 10.1046/j.1365-2567.2002.01380.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Streilein JW, Ohta K, Mo JS, Taylor AW. Ocular immune privilege and the impact of intraocular inflammation. DNA Cell Biol. 2002;21:453–459. doi: 10.1089/10445490260099746. [DOI] [PubMed] [Google Scholar]
- 14.Suter T, Biolaz G, Gatto D, Bernasconi L, Herren T, Reith W, Fontana A. The brain as an immune privileged site: dendritic cells of the central nervous system inhibit T cell activation. European Journal of Immunol. 2003;33:2998–3006. doi: 10.1002/eji.200323611. [DOI] [PubMed] [Google Scholar]
- 15.Li X, Taylor S, Zegarelli B, Shen S, O'Rourke J, Cone RE. The induction of splenic suppressor T cells through an immune-privileged site requires an intact sympathetic nervous system. J Neuroimmunol. 2004;153:40–49. doi: 10.1016/j.jneuroim.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 16.Dai Z, I, Nasr W, Reel M, Deng S, Diggs L, Larsen CP, Rothstein DM, Lakkis FG. Impaired recall of CD8 memory T cells in immunologically privileged tissue. J Immunol. 2005;174:1165–1170. doi: 10.4049/jimmunol.174.3.1165. [DOI] [PubMed] [Google Scholar]
- 17.Sonoda KH, Sakamoto T, Qiao H, Hisatomi T, Oshima T, Tsutsumi-Miyahara C, Exley M, Balk SP, Taniguchi M, Ishibashi T. The analysis of systemic tolerance elicited by antigen inoculation into the vitreous cavity: vitreous cavity-associated immune deviation. Immunology. 2005;116:390–399. doi: 10.1111/j.1365-2567.2005.02239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Faunce DE, Sonoda KH, Stein-Streilein J. MIP-2 recruits NKT cells to the spleen during tolerance induction. J Immunol. 2001;168:313–321. doi: 10.4049/jimmunol.166.1.313. [DOI] [PubMed] [Google Scholar]
- 19.Sugita S, Usui Y, Horie S, Futagami Y, Aburatani H, Okazaki T, Honjo T, Takeuchi M, Mochizuki M. T-cell suppression by programmed cell death 1 ligand 1 on retinal pigment epithelium during inflammatory conditions. Invest Ophthalmol Vis Sci. 2009;50:2862–2870. doi: 10.1167/iovs.08-2846. [DOI] [PubMed] [Google Scholar]
- 20.Sugita S, Horie S, Nakamura O, Maruyama K, Takase H, Usui Y, Takeuchi M, Ishidoh K, Koike M, Uchiyama Y, Peters C, Yamamoto Y, Mochizuki M. Acquisition of T regulatory function in cathepsin L-inhibited T cells by eye-derived CTLA-2alpha during inflammatory conditions. J Immunol. 2009;183:5013–5022. doi: 10.4049/jimmunol.0901623. [DOI] [PubMed] [Google Scholar]
- 21.Zamiri P, Sugita S, Streilein JW. Immunosuppressive properties of the pigmented epithelial cells and the subretinal space. Chem Immunol Allergy. 2007;92:86–93. doi: 10.1159/000099259. [DOI] [PubMed] [Google Scholar]
- 22.Ishida K, Panjwani N, Cao Z, Streilein JW. Participation of pigment epithelium in ocular immune privilege. 3. Epithelia cultured from iris, ciliary body, and retina suppress T-cell activation by partially non-overlapping mechanisms. Ocul Immunol Inflamm. 2003;11:91–105. doi: 10.1076/ocii.11.2.91.15914. [DOI] [PubMed] [Google Scholar]
- 23.Wilbanks GA, Streilein JW. Macrophages capable of inducing anterior chamber associated immune deviation demonstrate spleen-seeking migratory properties. Regional Immunology. 1992;4:130–137. [PubMed] [Google Scholar]
- 24.Taylor AW. Neuropeptides, aqueous humor, and ocular immune privilege. In: Troger J, Kieslbach G, Bechrakis N, editors. Neuropeptides in the Eye. Kerala, India: 2009. pp. 79–91. [Google Scholar]
- 25.Crane IJ, Liversidge J. Mechanisms of leukocyte migration across the blood-retina barrier. Semin Immunopathol. 2008;30:165–177. doi: 10.1007/s00281-008-0106-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stein-Streilein J, Streilein JW. Anterior chamber associated immune deviation (ACAID); regulation, biological relevance, and implications for therapy. Int Rev Immunol. 2002;21:123–152. doi: 10.1080/08830180212066. [DOI] [PubMed] [Google Scholar]
- 27.Stein-Streilein J. Immune regulation and the eye. Trends Immunol. 2008;29:503–586. doi: 10.1016/j.it.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 28.Mo JS, Streilein JW. Immune privilege persists in eyes with extreme inflammation induced by intravitreal LPS. Eur J Immunol. 2001;31:3806–3815. doi: 10.1002/1521-4141(200112)31:12<3806::aid-immu3806>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 29.Ohta K, Wiggert B, Taylor AW, Streilein JW. Effects of experimental ocular inflammation on ocular immune privilege. Invest Ophthalmol Vis Sci. 1999;40:2010–2018. [PubMed] [Google Scholar]
- 30.Casini G, Rickman DW, Sternini C, Brecha NC. Neurokinin 1 receptor expression in the rat retina. J Comp Neurol. 1997;389:496–507. [PMC free article] [PubMed] [Google Scholar]
- 31.Catalani E, Gangitano C, Bosco L, Casini G. Expression of the neurokinin 1 receptor in the mouse retina. Neuroscience. 2004;128:519–530. doi: 10.1016/j.neuroscience.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 32.Ansel JC, Brown JR, Payan DG, Brown MA. Substance P selectively activates TNF-alpha gene expression in murine mast cells. J Immunol. 1993;150:4478–4485. [PubMed] [Google Scholar]
- 33.Matsas R, Kenny AJ, Turner AJ. The metabolism of neuropeptides. The hydrolysis of peptides, including enkephalins, tachykinins and their analogues, by endopeptidase-24.11. Biochem J. 1984;223:433–440. doi: 10.1042/bj2230433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Folkers K, Hakanson R, Horig J, Xu JC, Leander S. Biological evaluation of substance P antagonists. Br J Pharmacol. 1984;83:449–456. doi: 10.1111/j.1476-5381.1984.tb16506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ohta K, Yamagami S, Taylor AW, Streilein JW. IL-6 antagonizes TGF-beta and abolishes immune privilege in eyes with endotoxin-induced uveitis. Invest Ophthalmol Vis Sci. 2000;41:2591–2599. [PubMed] [Google Scholar]
- 36.Streilein JW, Cousins SW. Aqueous humor factors and their effect on the immune response in the anterior chamber. Current Eye Research. 1990;9:175–182. doi: 10.3109/02713689008999439. [DOI] [PubMed] [Google Scholar]
- 37.Taylor AW, Streilein JW, Cousins SW. Immunoreactive vasoactive intestinal peptide contributes to the immunosuppressive activity of normal aqueous humor. J Immunol. 1994;153:1080–1086. [PubMed] [Google Scholar]
- 38.Ferguson TA, Fletcher S, Herndon J, Griffith TS. Neuropeptides modulate immune deviation induced via the anterior chamber of the eye. Journal of Immunology. 1995;155:1746–1756. [PubMed] [Google Scholar]
- 39.Jeglinski W, Koczyk D, Zaremba M, Oderfeld-Nowak B. Bilateral gliosis in unilaterally lesioned septohippocampal system: changes in GFAP immunoreactivity and content. J Neurosci Res. 1995;41:394–402. doi: 10.1002/jnr.490410312. [DOI] [PubMed] [Google Scholar]
- 40.Decaris E, Guingamp C, Chat M, Philippe L, Grillasca JP, Abid A, Minn A, Gillet P, Netter P, Terlain B. Evidence for neurogenic transmission inducing degenerative cartilage damage distant from local inflammation. Arthritis Rheum. 1999;42:1951–1960. doi: 10.1002/1529-0131(199909)42:9<1951::AID-ANR22>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 41.Donkin JJ, Turner RJ, Hassan I, Vink R. Substance P in traumatic brain injury. Prog Brain Res. 2007;161:97–109. doi: 10.1016/S0079-6123(06)61007-8. [DOI] [PubMed] [Google Scholar]
- 42.Stjernschantz J, Sears M, Mishima H. Role of substance P in the antidromic vasodilation, neurogenic plasma extravasation and disruption of the blood-aqueous barrier in the rabbit eye. Naunyn Schmiedebergs Arch Pharmacol. 1982;321:329–335. doi: 10.1007/BF00498522. [DOI] [PubMed] [Google Scholar]