

Abstract
Absent T lymphocytes were unexpectedly found in homozygotes of a transgenic mouse from an unrelated project. T cell development did not progress beyond double negative stage 1 thymocytes, resulting in a hypocellular, vestigial thymus. B cells were present, but NK cell number and B cell isotype switching were reduced. Transplantation of wild type hematopoietic cells corrected the defect, which was traced to a deletion involving 5 contiguous genes at the transgene insertion site on chromosome 12C3. Complementation using BAC transgenesis implicated zinc finger BTB-POZ domain protein 1 (Zbtb1) in the immunodeficiency, confirming its role in T cell development and suggesting involvement in B and NK cell differentiation. Targeted disruption of Zbtb1 recapitulated the T− B+ NK− severe combined immunodeficiency (SCID) phenotype of the original transgenic animal. Knockouts for Zbtb1 had expanded populations of bone marrow hematopoietic stem cells and also multipotent and early lymphoid lineages, suggesting a differentiation bottleneck for common lymphoid progenitors. Expression of mRNA encoding Zbtb1, a predicted transcription repressor, was greatest in hematopoietic stem cells, thymocytes and pre-B cells, highlighting its essential role in lymphoid development.
Introduction
Development of lymphocytes from hematopoietic stem cells (HSC) is incompletely understood. Current models suggest that T, B and natural killer (NK) cell precursors are predominantly derived from common lymphoid progenitors (CLPs) in bone marrow [1]. CLPs can differentiate into all-lymphoid progenitors (ALPs), which retain full lymphoid differentiation potential, forming both T cell progenitors that seed the thymus and B cell-biased lymphoid progenitors (BLPs) that become B cells [2]. Notch-1 signals are required to redirect common lymphoid progenitors from the default B lineage to the T lineage [3, 4]. After migration to the thymus, T lineage precursors proceed through distinct CD4− CD8− (double negative, DN) thymic stages, DN1 through DN4, after which they undergo T cell receptor (TCR) rearrangement and become CD4+ CD8+ double positive (DP) cells [5]. DP cells undergo selection, with surviving self-tolerant CD8+ or CD4+ single positive, naïve T cells emerging from the thymus to populate peripheral lymphoid organs. The zinc finger BTB-POZ (Zbtb) transcription factor 7 (Zbtb7/Th-POK/cKrox) is crucial for switching a fraction of DP cells from the default CD8+ pathway to form CD4+ cells [6, 7]. Identification of additional signaling factors at developmental branch points is required to understand lymphoid lineage commitment and differentiation in both mice and humans.
Human severe combined immunodeficiency (SCID) syndromes, defined by profound defects in both cellular and humoral immunity, comprise experiments of nature that have revealed many non-redundant, critical functions in human lymphoid developmental pathways. Human SCID is caused by defects in a wide variety of genes encoding proteins necessary and specific for lymphocyte differentiation, proliferation and activation [8–11]. About two thirds of SCID patients lack T cells but have B cells that fail to make specific antibodies (T− B+ phenotype); T− B+ SCID can be caused by defects in the X-linked common γ chain (γc) receptor for IL-2, -7, -9, -15 and -21; the IL-7 receptor α chain (IL-7Rα); intracellular cytokine signal transducers JAK3 or STAT5; protein tyrosine phosphatase receptor CD45; or components of the CD3 receptor. Patients with these forms of SCID are in contrast to those with T− B− SCID, whose defects may lie in mediators of T and B cell receptor gene rearrangement, including RAG1, RAG2, and Artemis; or in adenosine deaminase or purine nucleoside phosphorylase, which normally remove purine intermediates that are toxic to lymphocytes. Up to 10% of humans with SCID have other rare, or as yet unidentified, genetic defects.
In contrast to humans, the most studied mouse models for SCID have the T− B− phenotype, including the scid mouse deficient in the DNA dependent kinase DNA-PKc, and Rag1/Rag2 deficient mice, all of which fail to complete V(D)J rearrangement of antibody and T cell receptor genes during B and T cell development [12–17]. These and the athymic nude mouse lacking transcription factor Foxn1 [18, 19] have greatly facilitated basic studies of lymphoid cell maturation after the commitment to the T and B lineages has been made, but have not permitted dissection of pre-thymic lymphoid development. Moreover, lymphocyte phenotypes are discordant between mice and humans with disruption of most human SCID genes, limiting the utility of mouse knockouts for illuminating mechanisms for human T lineage commitment or to serve as faithful models for optimizing human SCID therapies. For example, while humans with γc and JAK3 defects have T− B+ NK− SCID, mice with targeted disruption of these genes have T cells, but lack B cells (T+ B− NK−) [20].
A T− B+ NK− SCID phenotype was unexpectedly observed in homozygotes of one of 5 mouse lines transgenic for a Fas cDNA bearing a dominant death domain mutation [21]. No offspring of other transgenic founders lacked T cells, suggesting a recessive defect related to the insertion site rather than to the expression of mutant Fas. This mouse presented a unique opportunity to develop a phenotypic model similar to human T− B+ NK− SCID, to dissect requirements for lineage differentiation of lymphoid progenitors, and to identify a critical step in the decision of CLPs and ALPs to become B vs. T vs. NK cells. Moreover, the underlying defect turned out to be in Zbtb1, a transcription repressor of the zinc finger BTB domain family very recently recognized to be involved in lymphoid cell development [22]. While the absence of peripheral T cells in our Zbtb1 knockout mouse confirms findings recently reported in the scanT mouse with a missense variant of Zbtb1 [22], our more detailed study of hematopoietic stem cells, lymphoid progenitors and lymphocyte subpopulations sheds light on Zbtb1 and its role in the development of T, B and NK cells.
Materials and Methods
Mice
Transgenic mouse line 26A with 6 tandem copies of mFas D231V has been described previously [21]. The SCID phenotype in homozygotes was achieved by heterozygous crosses. Wild type mouse strains in this study were from Jackson Lab (Bar Harbor, ME). Mice were fed autoclaved chow and antibiotics and were housed in sterile isolator cages, undergoing procedures according to approved protocols at the National Human Genome Institute at NIH, Bethesda, MD and UCSF, San Francisco, CA. Hpsa2 knockout mice were kindly provided by Dr. Mitchell Eddy, NIEHS, Research Triangle Park, NC.
Lymphocyte subset enumeration
Cell suspensions were prepared from blood, thymus, spleen and bone marrow and stained according to standard protocols with monoclonal antibodies (BD Biosciences, San Jose, CA; BioLegend and eBiosciences, San Diego, CA). Flow cytometry was performed on an LSRII FACS machine (BD Biosciences) and analyzed with FlowJo software (Tree Star, Inc, Ashland, OR). Complete and differential white blood counts were performed by the Clinical Pathology Department, NIH, Bethesda, MD.
Histopathology
Tissues collected in fixative according to standard protocols were sectioned and stained with hematoxylin and eosin at American Histolabs (Gaithersburg, MD) and analyzed with the kind assistance of Dr. Michael Eckhaus (Veterinary Resource Program, NIH). Testes from mice were fixed in Bouin’s solution. Peroxidase immunostaining with polyclonal rabbit anti-Hpsa2 antiserum 2A (kindly provided by Dr. Eddy, NIEHS) was performed by HistoServ Inc. (Germantown, MD).
T cell functional tests
Splenocytes were collected and cultured in Con A for 48 hours as described [21]. BrdU incorporation after 48 hours of incubation was analyzed using a BD Biosciences LSRII flow cytometer. Cytotoxicity was measured using the Promega Cytotox 96 kit (Promega Corp, Madison, WI) following the manufacturer’s instructions.
Bone Marrow Transplantation (BMT)
Donor bone marrow cell suspensions were harvested by flushing femurs and tibias with PBS and straining. HSC were enriched by depletion with biotinylated antibodies (BD Biosciences) to lineage markers CD3, CD4, CD8, B220, Gr-1 and Ter-119, followed by addition of streptavidin microbeads and purification using a MACS column (Miltenyi Biotec, Auburn, CA). Two million cells were injected by tail vein into 9 gray irradiated recipients. Lineage engraftment was determined in blood and tissues after 4 months. Experiments were performed in duplicate.
BAC isolation, cytogenetics and mapping
Metaphases from ConA stimulated splenocytes from heterozygous transgenic (tg/+) animals were prepared as described [23]. After fluorescence in situ hybridization (FISH) with the mFas plasmid labeled using cychrome (yellow) showed a signal on chromosome 12C-D, PCR-amplified segments from genes in this region were used to screen murine RPCI-23 BAC library filters [24]. DNA from positive BAC clones containing Sos2 (RP23-353H11), Esr2 (RP23-351E10), Hspa2 (RP23-901C4) and Psen1 (RP23-291K4) was prepared and labeled by standard methods. Cychrome labeled mFas DNA and BACs labeled with either rhodamine (red) or FITC (green) were used together as FISH probes. At least 20 cells with each combination were scored.
Additional mouse BAC clones containing Hpsa2 (RP23-191N23 and RP-90K4) were obtained and aligned by PCR content in wild type vs. homozygous transgenic (tg/tg) mice. Primer pairs from nearby flanking genes and intragenic segments were used to walk from centromeric and telomeric undisturbed regions towards the insertion/deletion locus. PCR was performed using RTG PCR beads (Amersham Pharmacia Biotech, Piscataway, NJ).
BAC transgenic mice for complementation
BAC DNA for transgenic injections was isolated by NucleoBond BAC Maxi Kit (Clontech, Mountain View, CA) and verified by pulse field gel electrophosesis after NotI digestion. BACs 90K4 and 191N23 were linearized with BsiWI and AscI, respectively. BAC DNA was injected into FVB/N fertilized eggs, and 3 resultant animals for each BAC with germline transmission were crossed to generate BAC-bearing tg/tg F2 mice for study. PCR with primers specific for genes located within the BACs confirmed BAC content. Animals were bred to determine fertility; immunophenotyping was carried out as above.
Retrovirus rescue of the SCID phenotype
A 2.3 kb EcoRI fragment containing Zbtb1 coding exon 2 was inserted into the 6.5 kb EcoRI-linearized MSCV-IRES-GFP vector (provided by Art Nienhuis, St. Jude Children’s Research Hospital, Memphis, TN). Full sequencing assured forward orientation and integrity. MSCV-Zbtb1-IRES-GFP was transfected into AmphoPack 293 cells (Clontech) using a CellPhect kit (GE Healthcare/Amersham Biosciences, Piscataway, NJ) and supernatant was used for transduction of GP+E-86 cells [25]. GP+E-86 clones expressing GFP were selected by FACS and expanded. Supernatants were titered on NIH 3T3 cells and analyzed by slot blot to determine the clone with the highest GFP expression.
Bone marrow was harvested from tg/tg mice and their wild type littermates 48 hours after injection with 5-fluorouracil (5-FU, Fluka-Sigma, St Louis, MO) [26]. Bone marrow cells were cultured and transduced with MSCV-Zbtb1-IRES-GFP as previously described [27]. Tg/tg mice irradiated with 5 gray received 2 × 106 cells in 200 µl by tail vein. Four months post-BMT, they were euthanized and immune phenotype was determined. DNA from ConA stimulated T cells and peripheral blood was extracted using a Puregene kit (Qiagen Inc, Valencia, CA). PCR was performed using vector primers MSCV-GFP 4128F TGCCCAGAAGGTACCCCATTGT and MSCV-IRES-4475R ACGCCGTAGGTCAGGATGGTCAC. Amplification conditions were 94°C 2m, (94°C 30s, 65°C 30s, 72°C 45s) × 35, 72°C 5m.
Production of Zbtb1 gene targeted mice
Zbtb1 coding exon 2 was replaced by eGFP using a pPNT-pgk-Neo vector. Homologous regions flanking Zbtb1 exon 2 were made from C57Bl/6 genomic DNA by PCR using Hi Fidelity Supermix (Invitrogen, Carlsbad, CA). The coding region of eGFP from pIRES2-eGFP (Clontech) was cloned into pBluescript SK+. For assembly, the 4.5 kb 5’ homologous segment released by XhoI and BspHI digestion was ligated into the pBluescript-eGFP digested with NcoI and XhoI. The 2.4 kb 3’ homologous segment was ligated into pPNT using KpnI and EcoRI sites. Finally, the 5’ arm and eGFP fragment were released from pBluescript with XhoI and SalI, gel purified, and ligated into the XhoI site of the pPNT construct, followed by electroporation into Genehog competent cells (Invitrogen). Sequence verified targeting construct DNA was linearized and injected at 1 µg/ µl into C57Bl/6 ES cells.
Injected ES clones under Neo selection were analyzed for homologous recombination by Southern blotting. Only 1 of 3 clones gave rise to germ line transmitting chimeras following injection of ES cells into C57Bl/6 blastocysts. Heterozygotes bred from this clone generated 7 wild type, 9 heterozygote and 8 homozygote offspring of both sexes, indicating that Zbtb1 is not necessary for embryonic development.
Real Time PCR for Zbtb1 expression
Expression levels of the 2 Zbtb1 splice variants were determined by quantitative real-time PCR. RNA was isolated from tissues and FACS sorted subpopulations of BM, thymus and spleen from 6 to 8 week old C57BL/6 mice. B cell subsets were sorted using markers described by Hardy and Hayakawa [28]. RNA was prepared using the Qiagen RNeasy kit (Qiagen Chatsworth, CA). All samples were treated with DNase I. RNA quality was assessed by A260/280 ratio (ND-100 spectrophotometer, NanoDrop Technologies, Wilmington, DE) and gel electrophoresis. A High Capacity RNA-to-cDNA kit (Applied Biosystems Inc, Foster City, CA) was used to prepare cDNA with 1ug of RNA from tissues or 200 ng from flow-sorted subpopulations. Absolute copy numbers of the two splice variant transcripts, Zbtb1713 and Zbtb1644, as well as those of housekeeping genes Pol2Ra (RNA polymerase II subunit A) and HPRT (Hypoxanthine-guanine phosphoribosyltransferase) were assessed for normalization (primer sequences available on request).
Amplified cDNA fragments were cloned into TOPO vector pCR2.1. Plasmids were prepared, sequence verified and serially diluted to generate standards to from 4 to 400 × 107 copies/well. Real time PCR was performed in TaqMan Universal PCR Master Mix using 100 nM of each primer and 200 nM of a FAM- and TAMRA-labeled oligonucleotide probe (primer and probe sequences available on request) in an ABI PRISM 7900HT system (Applied Biosystems Inc.). Cycle conditions were: 10 m 95°C, (95°C 30 s, 60°C 1 m) × 40. All samples were analyzed in duplicate. Copy numbers were determined by SDS 2.1 detection software (Applied Biosystems) and data was normalized to obtain test gene copies per 1,000 copies of HPRT and Polr2a.
B cell proliferation assays
Splenocytes were collected from Zbtb1 knockout, wild type and TCRα knockout mice and T cells were depleted using the Pan T cell isolation kit (Miltenyi Biotec). 1 × 105 B cells were added to each well of a 96-well round-bottomed plate and incubated with LPS (0.1 or 10 ng/µl), anti-IgM (0.1 or 10 ng/µl) or anti-CD40 (0.1 or 10 ng/µl) in RPMI with 10% FCS, Penicillin and Streptomycin, non-essential amino acids and 5 µM BrdU (Invitrogen). BrdU incorporation after 48 hours of incubation was analyzed using a BD Biosciences LSRII flow cytometer.
Serum immunoglobulin assays
Serum collected in BD Microtainer separator tubes (Becton, Dickenson, Franklin Lakes, NJ) had levels of IgM, IgG, and IgA assayed using an ELISA mouse Ig quantitation kit (Bethyl Laboratories, Montgomery, TX). Data were analyzed using an unpaired t test with Welch's correction (Prism 5, GraphPad Software, La Jolla, CA).
Western blotting and Immunoprecipitation
Whole cell, nuclear and cytoplasmic 293T cell extracts were prepared 24 and 48 hours following transfection of cloned human or murine cDNA encoding Zbtb1713 and Zbtb1644 isoforms with N- or C-terminal, FLAG or myc epitope tags. Aliquots were subjected to electrophoresis, transfer and immuno-blotting using reagents and methods from eBioscience (San Diego, CA). Anti-FLAG (Sigma) or anti-myc (Santa Cruz Biotechnology Inc, Sant Cruz, CA) mouse mAbs followed by HRP-anti-mouse IgG (Santa Cruz) were applied and detected by the SuperSignal Substrate Detection System (Pierce Protein Research Products, Invitrogen).
For immunoprecipitations, cell lysates from transfected 293T cells were mixed with primary antibody and rocked overnight with sepharose beads conjugated to Protein A. After washing the beads, protein was released by boiling in gel loading buffer. Electrophoresis and detection were carried out as above.
Results
Severe combined immunodeficiency (SCID) and infertility in a transgenic mouse
Of 5 lines of FVB/N mice transgenic for mutant Fas D231V [21], one line, 26A, exhibited male infertility and profound immunodeficiency when the transgene was bred to homozygosoity. Heterozygotes from all lines and homozygotes from lines other than 26A had mild hepatic infiltration with T cells, but did not have the phenotype of Fas deficient MRLlpr mice or humans with autoimmune lymphoproliferative syndrome- autoimmunity, excess CD4− CD8− DN T cells and defective apoptosis [29]. Instead, the transgenic animals were healthy and indistinguishable from wild type (+/+) mice. However, homozygotes of line 26A (tg/tg) had undetectable T and markedly reduced B and NK cells in peripheral blood, although B cells were present in spleens (Figure 1A, B). Peripheral blood myeloid cells were slightly decreased in tg/tg mice while erythrocytes and platelets were normal (Supplementary Figure 1A). Spleens in tg/tg mice were 50% of normal weight, with only 3% T cells but with an increased proportion of B cells to 60% (Figure 1B) and normal absolute B cell numbers. Tg/tg mice showed poor proliferation to ConA and absent T cell cytotoxicity (Figure 1C, D). They also had impaired isotype switching of the B cells, indicated by increased serum concentrations of IgM and decreased IgG and IgA (Figure 1E). The phenotype was invariant with age up to 8 months.
Figure 1. Characterization of homozygous transgenic mice.
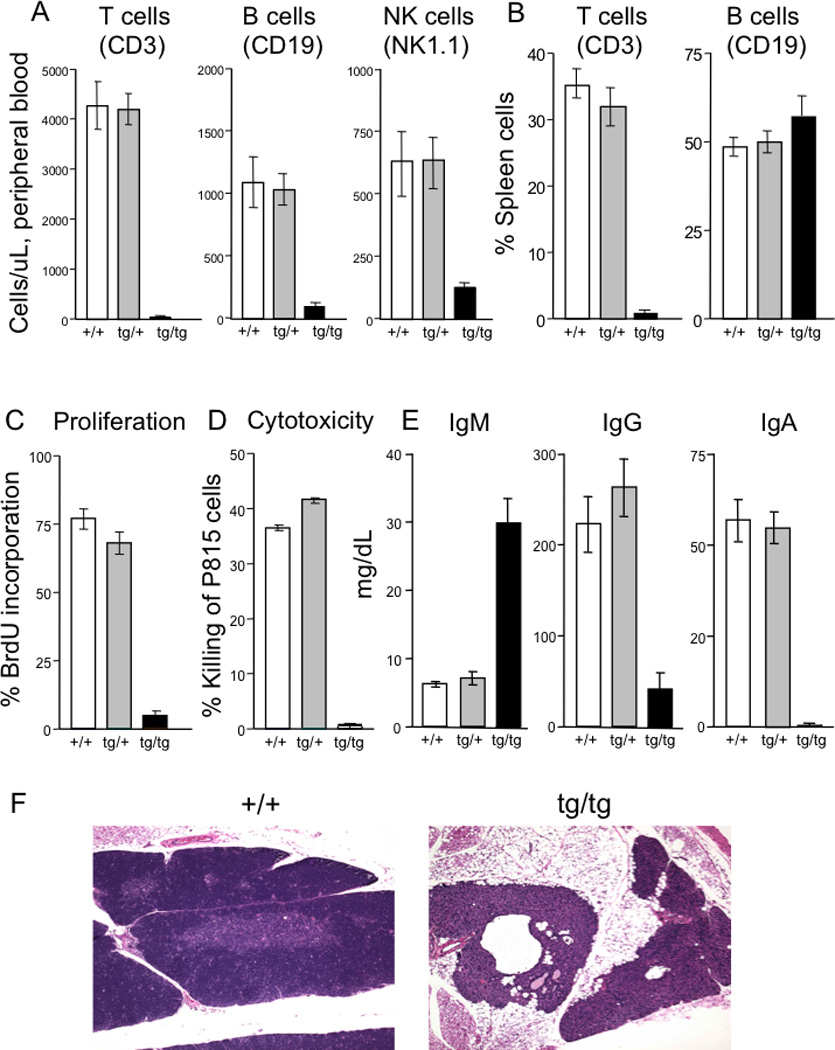
A, peripheral blood, and B, splenic, lymphocyte subsets from wild type (+/+), heterozygous (tg/+) and homozygous transgenic (tg/tg) mice.
C, proliferation of splenic T cells after ConA stimulation, measured by BrdU incorporation.
D, killing of P815 target cells.
E, serum immunoglobulin levels.
F, thymus histology of wild type (+/+) compared to homozygous transgenic (tg/tg) mice.
In contrast to their unaffected +/+ littermates, tg/tg mice had only a vestigial thymus (Figure 1F). Typical of most human SCID cases, thymic epithelial and stromal cells were present, but cortical areas were undeveloped and lymphoid cells were rare to absent. Mesenteric lymph nodes in tg/tg animals, when located, were small; axillary and inguinal lymph nodes were not found. The infertility of male tg/tg mice was due to arrested spermatocyte development (Supplementary Figure 1B).
Bone marrow transplantation to define the cellular lesion in tg/tg SCID mice
To determine whether the SCID phenotype of tg/tg mice was HSC autonomous or attributable to a dysfunctional thymic environment, lineage depleted bone marrow (BM) from +/+ mice was injected into irradiated tg/tg mice as well as into irradiated control heterozygous littermates. After 4 months, reconstitution in tg/tg mice was equivalent to that in controls (Supplementary Figure 1D, Supplementary Table 1A). Not only did CD3, CD4 and CD8 T cells appear in numbers equivalent to those of control mice, but also T cell proliferation to ConA and cytotoxicity against P815 target cells was normalized. In addition, serum IgM, IgG and IgA levels became normal, indicating recovery of isotype switching (data not shown).
When the reciprocal BMT was performed using lineage depleted FVB/N tg/+ or tg/tg SCID mouse cells to reconstitute irradiated C57Bl/6 X FVB/N F1 mice, the recipients developed fewer T and B cells than recipients of tg/+ heterozygous BMT. Even though hemoglobin analysis indicated 100% donor engraftment, lineage chimerism using PCR with primers specific for C57Bl/6 and FVB/N strains showed failure to engraft T and B cells (Supplementary Figure 1E, Supplementary Table 1B). Proliferation and cytotoxic function remained poor, and IgM levels rose even through IgA and IgG levels were normal, the latter attributed to radio-resistant recipient mouse plasma cells (data not shown). Thus the SCID defect was intrinsic to HSC, did not affect myeloid progenitors and was limited to lymphoid lineages, mainly affecting the generation of T and NK cells.
Mapping and identification of the genetic locus for immunodeficiency
Labeled plasmid containing the 6 kb mFas transgene was used as a fluorescence FISH probe to map the integration site on metaphase chromosome spreads from heterozygous tg/+ animals; a single hybridization signal was found on mouse chromosome 12qC–D. Bacterial artificial chromosome (BAC) clones containing known mouse genes were used as probes in 3-color hybridizations to narrow the region (Figure 2A). The transgene signal (yellow) was found between BACs containing Sos2 (green) and Psen1 (red) (Figure 2Ai). Additional BACs within this interval were further used to refine the transgene location. A BAC containing Esr2 (red) was centromeric to the transgene, while one with Hspa2 (green) was replaced by the plasmid signal (yellow) on one copy of chromosome 12 (Figure 2Aii), suggesting that partial or full deletion of Hspa2, perhaps with surrounding genes, had occurred when the transgene was integrated into mouse chromosome 12. We thus hypothesized that one or more genes disrupted by the transgene integration accounted for the SCID phenotype of tg/tg mice.
Figure 2. Mapping and identification of the genetic locus for immunodeficiency.
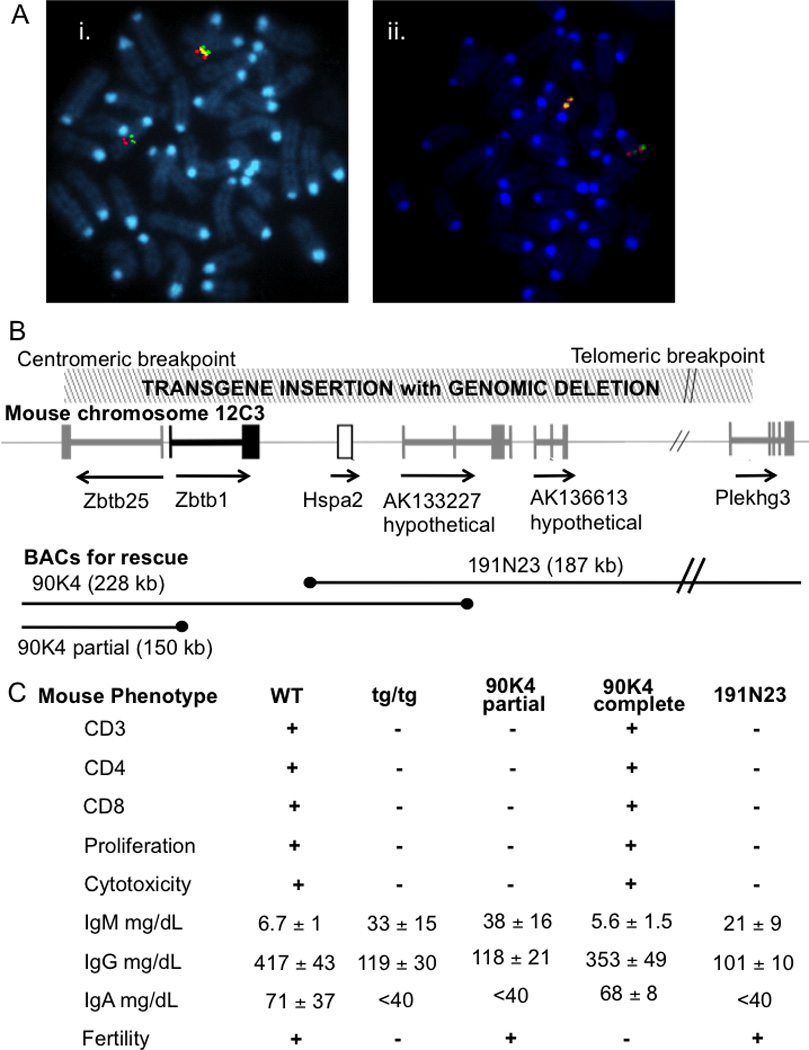
A, FISH localization of tg (yellow) insertion: i. telomeric to a BAC encoding Sos2 (green) and centromeric to Psen1 (red); and ii. telomeric to Esr2 (red) and co-localizing with and replacing Hspa2 (green).
B, map of mouse chromosome 12C3 showing order and direction of transcription of genes Hspa2 (open box) and Zbtb1 (black) and transcripts Zbtb25, AK133227, AK136613 and Plakhg3 (gray); hatched rectangle, above, transgene insertion/deletion region; black lines, below, BAC genomic clones (ends indicated by black dots) used for rescue transgenesis.
C, immune and fertility phenotypes of wild type, original mFas tg/tg and tg/tg BAC transgenic animals containing the indicated BAC sequences, 90K4 partial, 90K4 complete, and 191N23 (mean ± SD, 6 mice/group).
Hspa2, encoding a 70 kDa heat shock chaperone protein, had been knocked out in mice and found to be essential for male fertility [30, 31]. However, a role for Hpsa2 in the immune system had not been studied. Hspa2 knockouts, provided by M. Eddy, had normal T and NK cells and normal antibody levels (data not shown), ruling out Hpsa2 as a candidate gene for the tg/tg immunodeficiency. However, Hpsa2 protein was absent in tg/tg mice as expected, shown by lack of staining with polyclonal anti-Hpsa2 antibody, also provided by M. Eddy (Supplementary Figure 1C). Lack of Hspa2 thus explained the male infertility in tg/tg mice.
Boundaries of the tg insertion-deletion were established by PCR mapping with primer pairs selected from the mouse genome sequence (Figure 2B). Positive PCR amplification in +/+ and tg/+ mice with failure in tg/tg mice indicated deleted sequences (Figure 2B hatched area, top). The tg allele was deleted for hypothetical genes AK133227, AK136613 and a portion of Plekhg3 on the telomeric side of Hspa2 and two genes in the Zbtb family, Zbtb1 and Zbtb25, on the centromeric side.
Implication of Zbtb1 in immunodeficiency
With Hpsa2 ruled out as the immunodeficiency gene, we used a complementation approach with transgenesis of BAC clones 191N23 and 90K4, extending to the telomeric and centromeric sides of Hspa2, respectively (Figure 2B), to evaluate the ability of the candidate genes to rescue immunity and fertility. After injections into blastocysts from the original tg/tg mice, 3 lines of mice with each BAC were produced and crossed to mFas tg/+ mice to generate tg/tg BAC-bearing animals. Tg/tg mice carrying BAC clone 191N23 were fertile, as expected with restoration of Hpsa2, but retained the immunodeficient phenotype with absent T cells and T cell function, elevated IgM and low IgG and IgA (Figure 2C, right column). Only mice with the full-length 90K4 BAC were fertile with intact immunity, as shown in the “90K4 complete” column in Figure 2C. In one mouse line, spontaneous breakage of BAC 90K4 resulted in restoration of the coding portion of Zbtb25 (“90K4 partial,” Figure 2C). Crosses of the BAC transgenics with the mFas tg/tg mice revealed that triple transgenics homozygous for the original tg/tg insertion/deletion, and carrying the 191N23 BAC (restoring Hpsa2, AK133227, AK136613, and Plekhg3) plus the partial 90K4 BAC (possibly restoring Zbtb25, but not Zbtb1), were fertile, but were affected with SCID. These experiments suggested that loss of Zbtb1, or of both Zbtb1 and Zbtb25, were necessary and sufficient to produce T− B+ NK− SCID in the mouse.
To determine whether absent Zbtb1 alone was the cause of SCID, we used a retroviral vector containing the single large coding exon of Zbtb1 to transduce bone marrow stem cells from tg/tg or +/+ control donor mice and injected these transduced cells to rescue lethally irradiated tg/tg mice. At 4 months post-BMT, T cells were found in peripheral blood and spleen, although normal numbers were not completely restored (Supplementary Table 1C). However, serum immunoglobulin levels were comparable to wild type mice and T cell proliferation and cytotoxicity were normal (Supplementary Figure 1F), indicating that restoration of Zbtb1 restored immunity, curing the SCID phenotype.
Targeted disruption of Zbtb1
To prove that Zbtb1 was necessary and sufficient to cause the observed immunodeficiency of tg/tg mice, we replaced exon 2 of Zbtb1 with a GFP-pgk-Neo cassette to produce a gene-targeted mouse (Figure 3A). The first exon of the Zbtb1 gene is noncoding, with the ATG start of translation at nucleotide 19 in exon 2; therefore disruption of exon 2 would obliterate Zbtb1 gene expression. After transfection of C57BL6/J ES cells and injection into blastocysts, we obtained 3 founder mice, one of which passed the targeted disruption to offspring. Zbtb1 knockout F2 mice were generated and were fertile (but unfortunately failed to express GFP). While Zbtb1 heterozygous (+/−) and wild type (+/+) littermates had normal T, NK and B cell numbers in the periphery, Zbtb1 knockout (−/−) mice showed nearly absent T cells, reduced NK cells and normal B cells (Figure 3B). In addition, splenic T cells, which were very few in number, showed a complete lack of proliferative and cytotoxic function (Supplementary Figure 2A). The thymus and lymph nodes were vestigial in Zbtb1 knockout mice and the spleen was half the weight of spleens of littermate controls (data not shown). Non-lymphoid cells in the peripheral blood were unaffected (Supplementary Figure 2B). The phenotype of the Zbtb1 knockout mice therefore replicated that of the original transgenic mice, as well as the recently reported scanT mice with a missense mutation in Zbtb1 [22]. Detailed characterization of knockout mice enabled us to define the phenotype resulting from complete absence of the Zbtb1 protein vs. that resulting from published missense mutation C74R, therefore further delineating the role of Zbtb1 in hematopoietic stem cell differentiation and lymphoid development.
Figure 3. Targeted disruption of Zbtb1 and characterization of knockout mice.
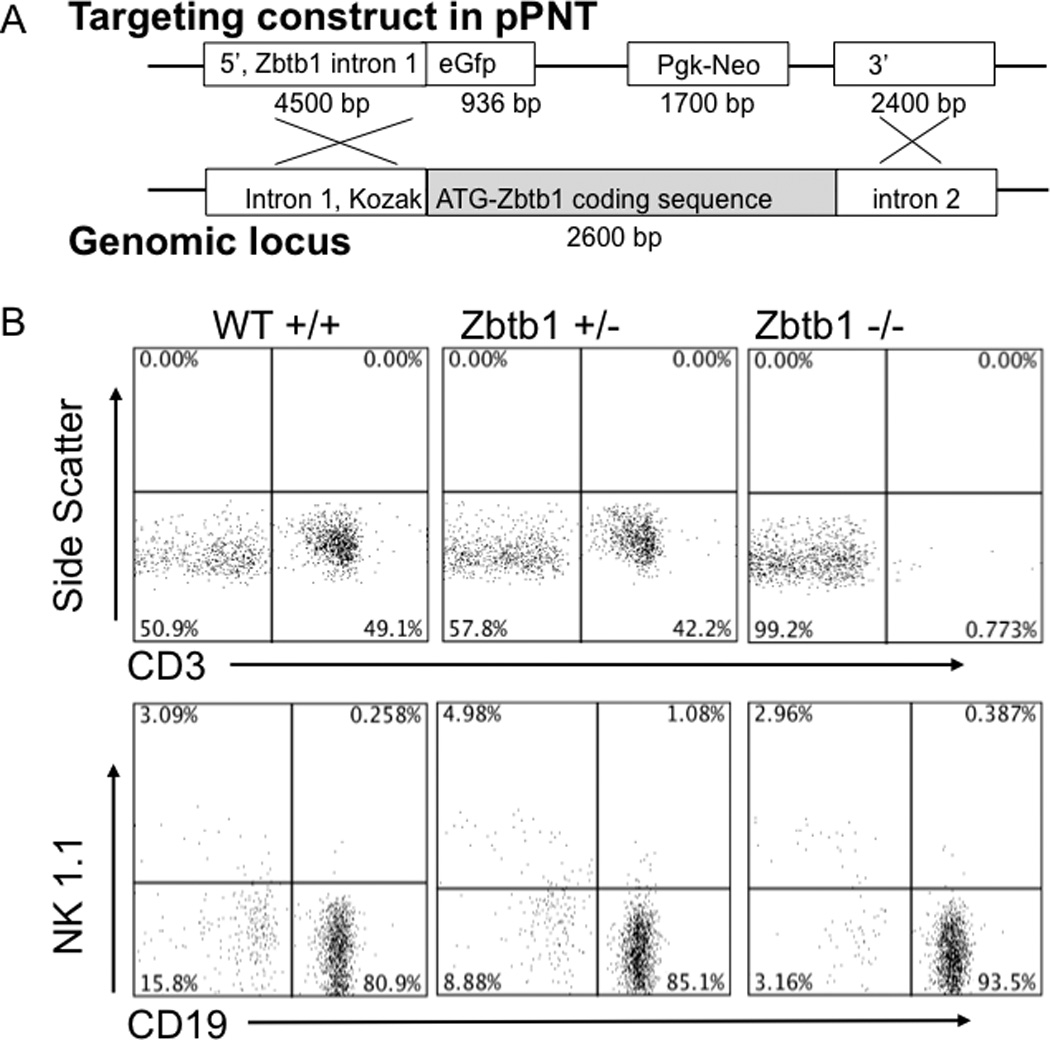
A, gene-targeting construct, pPNT-Zbtb1 5’-eGFP-Neo-Zbtb1 3’ (15.338 Kb, above) and homologous B6 genomic target locus (below).
B, peripheral blood lymphocyte phenotype of knockout transmitting founder offspring: wild type, Zbtb1 heterozygous (+/−) and Zbtb1 knockout (−/−) mice. T cells detected as CD3+; B cells, CD19+; NK cells, NK1.1+.
Investigation of embryonic thymi in Zbtb1 deficient mice
Due to the absence of mature T cells in the periphery and a hypocellular, vestigial thymus in adult knockout mice lacking Zbtb1, embryonic thymi were analyzed to determine at which stage T cell development was arrested. Thymocytes were identified according to their differentiation status, with the earliest arrivals from the BM being DN1 cells, CD4− CD8− TCRβ− CD25− CD44+; successive maturation to DN2 CD25+ CD44+, and then DN3 CD25+ CD44− cells was monitored by cell surface marker content [32], with progression beyond the DN3 stage indicated by rearrangement and expression of the TCRβ genes [33]. When day 15.5 and day 17.5 mouse embryos from the original tg insertion-deletion were genotyped by FISH and examined for thymocyte maturation, tg/tg mice had reduced but detectable DN1, but very few DN2 and DN3 cells (Figure 4A) and essentially no DN4, double positive or single positive T cells (data not shown). Further analysis of the DN1 population as described by Porritt et al [34] (Figure 4B, 4C) showed low, but detectable numbers of DN1a (ckit/CD117+, HSA/CD24−) and DN1b (ckit/CD117+, HSA/CD24lo) cells, thought to represent the major pathway for expansion and differentiation from bone marrow progenitors to DN2 thymocytes. DN1c cells (ckit/CD117+, HSA/CD24+), thought to retain B lineage developmental potential, were also detectable, but there were essentially no CD117− DN1d or DN1e cells, which have poor proliferative potential and do not mature into T cells. This pattern indicates that T cell progenitors lacking Zbtb1, while present in the thymus in small numbers, were unable to progress through thymic differentiation.
Figure 4. Analysis of thymocytes and bone marrow subsets in knockout mice.
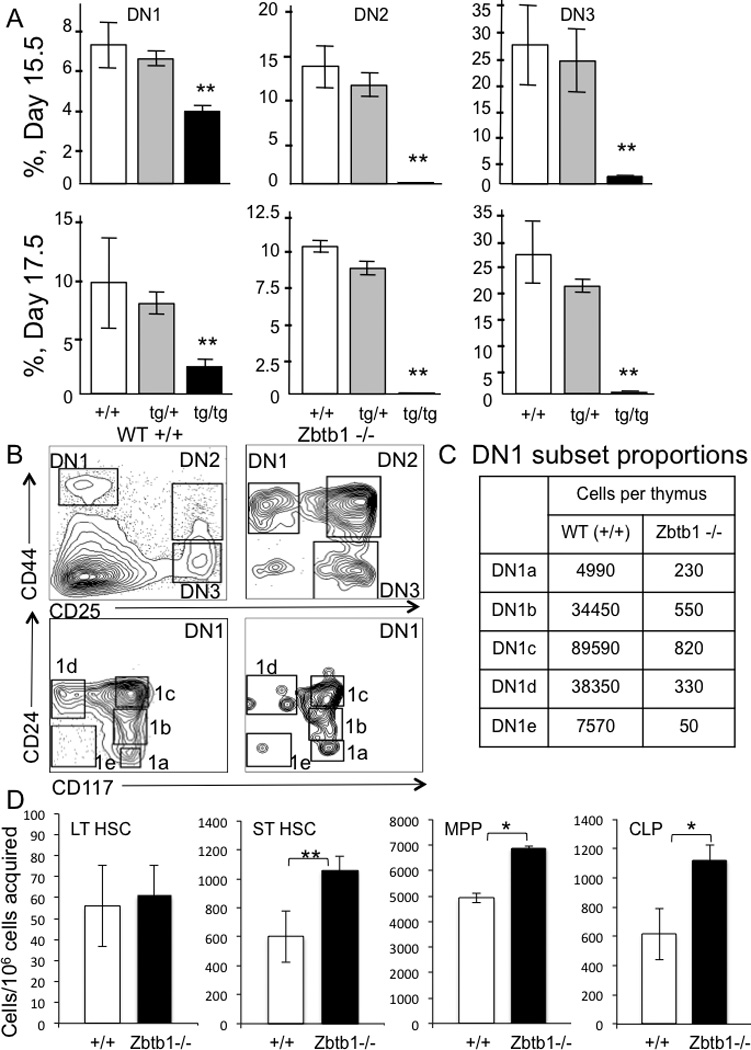
A, day 15.5 and 17.5 fetal TCRβ− CD4− CD8− double negative (DN) thymocytes in wild type +/+, heterozygous tg/+ and homozygous transgenic tg/tg mice. DN1, CD25− CD44+, DN2, CD25+ CD44+, DN3 CD25+ CD44−. Percentages are means ± SD of 10 fetuses/group.
B, DN subsets from adult wild type and Zbtb1 knockout mice; DN1-DN3, upper plots; subdivided DN1 (lower plots) with DN1a, CD117+ CD24−; 1b, CD117+ CD24lo; 1c, CD117+ CD24+; 1d, CD117− CD24+; and 1e, CD117− CD24− [34].
C, DN1 cell subsets per thymus (n=2 per genotype).
D, bone marrow cell subsets from wild type and Zbtb1 knockout mice; long term HSC (LT HSC, LSK CD34− CD48−), short term HSC (ST HSC, LSK CD34+ CD48−), multi potent progenitors (MPP, LSK CD34+ CD48+), common lymphoid precursors (CLP, LSK CD127+ CD135+). Data represent mean ± SD, 10 mice per group; * p < 0.05, ** p < 0.001.
Stem cell subpopulations in Zbtb1−/− mice
To determine pre-thymic effects of knocking out Zbtb1, absolute numbers of cells comprising the different BM stem cell subsets, long term HSC (LSK, CD34−, CD48−), short term HSC (LSK, CD34+, CD48−), multipotent progenitors (MPP) (LSK, CD34+, CD48+) and common lymphoid progenitors (CLP) (LSK, CD127+, CD135+) [35, 36], were determined from the Zbtb1 knockout vs. wild type mice. Zbtb1 knockout mice had equivalent numbers of long term HSC, but an increased number of cells of the other early subsets, short term HSC, MPP and CLP (Figure 4C). This could reflect feedback mechanisms in an attempt by the bone marrow to make up for the reduced number of mature T cells in the periphery. Although the bone marrow was able to produce lymphoid progenitors in the absence of Zbtb1, these progenitors did not appear in the thymus.
Our observations contrast with the report of scanT mice, in which numbers and percentages of all hematopoietic subsets were equal to those of wild type mice [22]. This could be because the Zbtb1 protein, although mutated in the scanT mice, could still retain residual function and influence HSC numbers and differentiation, while in our knockout mice, the complete absence of Zbtb1 protein affects the maintenance of stem cell numbers.
Bone marrow and spleen B cell subsets in Zbtb1 knockout mice
Since the absence of Zbtb1 resulted in lack of T cells in the periphery and spleen, but did not prevent B cell development, B cell subsets [28] in both BM and spleen were analyzed to determine if the absence of Zbtb1 affected their proportions. Wild type and TCRα knockout mice were used as controls to evaluate the absence of T cell help as a contributor to any alterations in B cell subsets. No differences in pro-B cell frequencies in the BM were observed across the three mouse phenotypes. However, Zbtb1 knockout mice had fewer pre-B cells compared to TCRα knockout and wild type mice (Figure 5A, Supplementary Figure 2Ci). When the B cell subsets in the spleens were analyzed, Zbtb1 knockout mice were found to exhibit higher proportions of marginal zone B cells and higher proportions of follicular B cells than either wild type or TCRα knockout mice (Figure 5B, Supplementary Figure 2Cii). These findings were consistent with the phenotype of the scanT mice [22]. Thus although B cell development appeared to be impaired at the pre-B cell stage in the absence of Zbtb1, suggesting that Zbtb1 is important during early B cell development, the number of mature B cells in the periphery was comparable to that in wild type controls. This could be due to redundancy of the function of Zbtb1 in peripheral B cells and/or compensation for its functions by other members of the same protein family or through different signaling pathways.
Figure 5. Analysis of B cells of knockout mice.
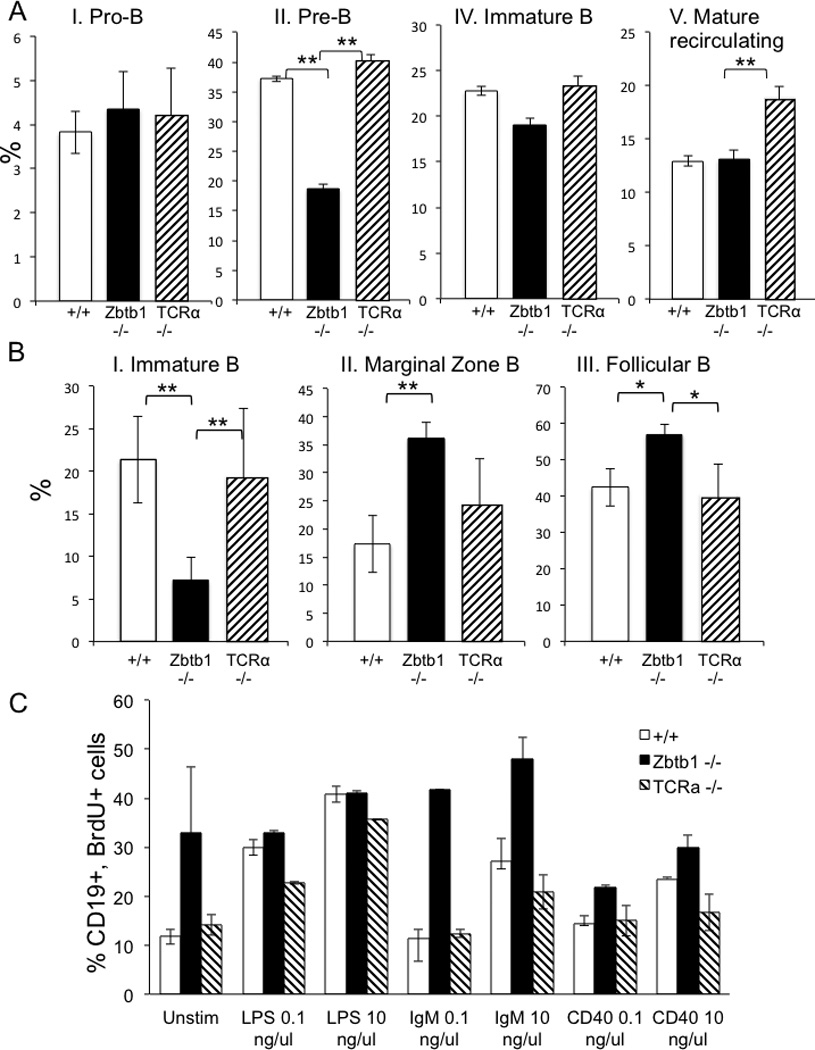
A, relative proportions of BM B cell subsets wild type, Zbtb1−/− and TCRα−/− mice, as defined by the Hardy and Hayakawa gating scheme: I, Pro-B (B220+ CD43+); II, Pre-B (B220+ CD43−); III, Pre-B Late (B220+ CD43− IgD− IgM−); IV, Immature B (B220+ CD43− IgD− IgM+) V, Mature recirculating B (B220+ CD43− IgD+ IgM+).
B, relative proportions of splenic B cell subsets in wild type, Zbtb1−/− and TCRα −/− mice: I, Immature B (B220+ IgDhi IgMlo CD23− CD21/35−); II, Marginal zone (B220+ IgDhi IgMlo CD23lo CD21/CD35hi); III, Follicular (B220+ IgDlo IgMhi CD43− CD5− CD23hi CD21/CD35mid).
Data represent mean ± SD, 4 mice per genotype; *, p< 0.1; **, p < 0.05.
C, wild type, Zbtb1−/− and TCRα −/− splenic B cell proliferation after stimulation with LPS (0.1 and 10 ng/µl), anti-IgM (0.1 and 10 ng/µl) or anti-CD40 (0.1 and 10 ng/µl), measured by BrdU incorporation at 48 h.
It is difficult to distinguish a B cell intrinsic defect from altered proportions of B cells at different stages of development. However, in vitro proliferation of B cells from Zbtb1 knockout mice, wild type littermates and TCRα knockout mice, in response to anti-IgM, LPS and anti-CD40 showed that B cells from the Zbtb1 knockout mice proliferated at least as much or more than those from wild type or TCRα knockout mice (Figure 5C). The brisk proliferation of B cells from the Zbtb1 knockout mice could be accounted for by the increased proportion of marginal zone cells in their spleens (Figure 5B), since marginal zone cells proliferate more than follicular B cells [37–39].
Tissue expression of Zbtb1
Copy number of Zbtb1 mRNA in lymphoid tissues of wild type mice was determined by quantitative real-time PCR and normalized to the housekeeping gene HPRT (Figure 6). Maximum tissue expression was found in thymus and spleen, followed by lymph nodes and PBMCs. Unfractionated and lineage-depleted BM had expression, but at lower levels (Figure 6A).
Figure 6. Zbtb1 mRNA expression measured by quantitative PCR in lymphoid populations in wild type mice.
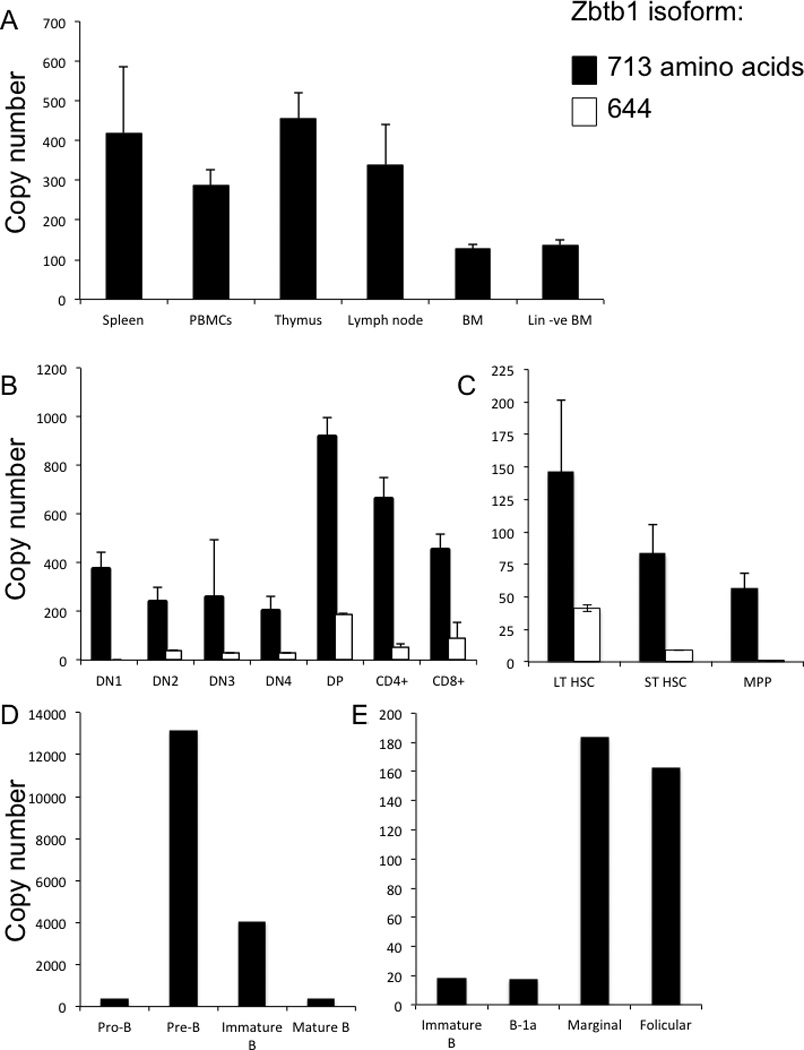
A, spleen, PBMC, thymus, lymph node, bone marrow (BM), and bone marrow depleted of mature lineages (Lin –ve BM);
B, thymocyte subsets, as described in Figure 4A;
C, bone marrow progenitor populations, as described in Figure 4B.
D and E, bone marrow and splenic B lineage subsets, as described in Figure 5;
Y-axis shows copy numbers of Zbtb1 per 1000 copies of housekeeping gene HPRT.
Data represent mean ± SD, 5 mice per group.
Two isoforms were found, Zbtb1713 and Zbtb1644, which share an initial noncoding exon 1 and the 5’ end of exon 2. Zbtb1713 is not further spliced and encodes a BTB/POZ domain followed by 8 zinc finger domains in a single open reading frame [40]. The shorter Zbtb1644, formed by a splice from within exon 2 to an alternative exon 3, lacks the last three C-terminal zinc finger domains found in Zbtb1713 and was less abundant in all cell types studied (Figure 6B, C). While DN1 through DN4 thymocytes expressed both Zbtb1 isoforms, maximum expression was observed in the CD4+ CD8+ DP population, in which Zbtb1644 was 20% as abundant as the Zbtb1713. CD4+ and CD8+ single positive cells had lower expression of both isoforms (Figure 6B).
Analysis of the expression of the two isoforms of Zbtb1 mRNA in bone marrow revealed maximum expression in long term HSCs (LSK, CD34−, CD48−), less in short term HSCs (LSK, CD34+, CD48−) and still less in multipotent progenitors (LSK, CD34+, CD48+) [35, 36] (Figure 6C).
B lymphoid subsets of the BM showed a striking increase in Zbtb1 expression from pro-B (B220+, CD43+) to pre-B (B220+, CD43−) cell stages, with levels then declining upon further differentiation into immature B cells (B220+, CD43−, IgD−, IgM+) and low levels in mature B cells (B220+, CD43−, IgD+, IgM+) (Figure 6D). The increased expression of Zbtb1 mRNA in pre-B cells suggests that Zbtb1 plays an important role at this stage of B cell development and could possibly explain the above-described decreased number of pre-B cells observed in BM of Zbtb1 deficient compared to wild type mice. In splenic B cells, overall Zbtb1 mRNA levels were 10-fold lower, with predominant expression in follicular (B220+, IgDlo, IgMhi, CD43−, CD5−, CD23hi, CD21/35med) and marginal zone (B220+, IgDhi, IgMlo CD23lo/−, CD21/35hi) fractions (Figure 6E).
Expression and nuclear localization of Zbtb1 protein
Expression vectors with human and mouse cDNAs encoding the 713 and 644 amino acid isoforms of Zbtb1 were constructed with epitope tags at either N or C terminal positions. Human Zbtb1713, with an N-terminal FLAG sequence following the initial methionine codon (N-FLAG-hZBTB1713) was expressed in 293T cells. To determine the cellular localization of the protein, Western blotting was carried out with the nuclear and cytoplasmic protein fractions and blots were probed with anti-FLAG antibody to detect ZBTB1, as well as anti-histone deacetylase-1 (HDAC1) and anti-α1a-tubulin antibodies to document purity of nuclear and cytoplasmic extracts, respectively (Figure 7A). The tagged protein was readily detectable in nuclear extracts (Figure 7Ai). Consistent with its structural similarity to transcriptional repressors and its previously described nuclear location [41], we found the ZBTB1 signal with the anti-FLAG antibody confined to the nuclear fraction, and not in the cytoplasmic fraction (Figure 7Aii).
Figure 7. Zbtb1 protein detection by Western blotting.
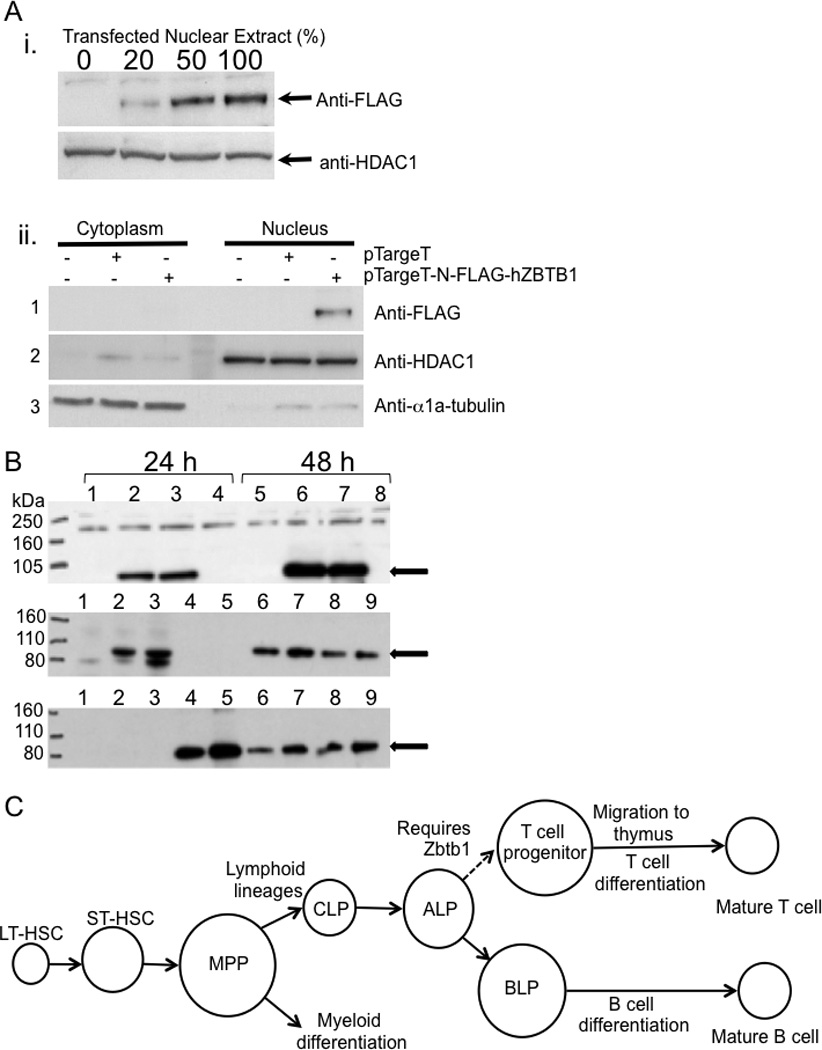
A, i. expression of tagged N-FLAG-hZBTB1 in nuclear extracts of transfected 293T cells. Upper panel: Anti-FLAG mAb (clone M2) detects the FLAG-tagged Zbtb1. Increasing proportions of nuclear extract from 293T cells transfected with the pTargeT-N-FLAG-hZBTB1 were mixed with extract from untreated cells, keeping the amount of protein loaded constant. The positive signal increased accordingly. Lower panel: anti-HDAC1 loading control. ii. nuclear localization of FLAG-tagged Zbtb1. Panel 1: Anti-FLAG in fractionated cytoplasmic vs. nuclear protein extracts of transfected 293T cells. Panels 2 and 3: HDAC1 and α1a-tubulin, nuclear and cytoplasmic controls, respectively.
B, Western blot of 293T cell lysates 24 h (left) and 48 h (right) after transfection. Upper panel, probed with anti-FLAG antibody. Lanes: 1&5, control insert pEF-BOS-EGFP (green fluorescence to ensure efficient transfection); 2&6, N-FLAG-mZBTB1713; 3&7, C-FLAG-mZBTB1713; 4&8, plasmid with no insert. Anti-FLAG signal is at the expected 83 kDa. Middle and lower panels, hetero-dimerization after co-transfection, demonstrated by immunoprecipitation (IP) and reciprocal detection of FLAG-Zbtb1713 and myc-Zbtb1644. Lanes: 1, pEF-BOS-EGFP; 2, N-FLAG-Zbtb1713; 3, C-FLAG- mZbtb1713; 4, N-myc- Zbtb1644; 5, C-myc-Zbtb1644; 6, N-FLAG-Zbtb1713 + N-myc-Zbtb1644; 7, N-FLAG- Zbtb1713 + C-myc-mZbtb1644; 8, C-FLAG-Zbtb1713 + N-myc-Zbtb1644; 9, C-FLAG-Zbtb1713 + C-myc-Zbtb1644.
C, schematic representation of an essential role of Zbtb1 during T lineage commitment.
Dimerization of Zbtb1 isoforms sharing identical BTB/POZ domains
We studied dimerization using Zbtb1713 tagged with FLAG and Zbtb1644 tagged with myc at either end to minimize the chance of steric hindrance [42]. After transfection into 293T cells, N- and C-FLAG-Zbtb1713 were detected at 83 kDa using mouse anti-FLAG and HRP-anti-mouse IgG (Figure 7B, upper panel); similarly, somewhat smaller N- and C-myc-Zbtb1644 were detected using mouse anti-myc (not shown). Upon reciprocal immunoprecipitation the N- and C-FLAG-Zbtb1713 proteins were co-precipitated with either the N- or C-myc-Zbtb1644, and vice-versa (Figure 7B, middle and lower panels).
Thus the epitope tags did not affect protein expression or interaction, and the short and long isoforms, with identical 5’ BTB dimerization domains were shown to undergo dimerization. Thus the short isoform, although lacking the three C-terminal zinc fingers and expressed at a considerably lower level than the predominant 713 aa isoform, may bind to the long form under physiologic conditions, potentially modifying the activity of Zbtb1 DNA-binding complexes.
ZBTB1 sequencing in human SCID patients
The phenotoype of mouse Zbtb1 deficiency suggested that deleterious mutations in human ZBTB1 could cause T− B+ immune deficiency in humans. We examined the sequence of ZBTB1 in 20 cases of human SCID with T− B+ phenotype, but without demonstrable mutations in previously reported SCID disease genes. However none were found to have variants in ZBTB1 that were predicted to be deleterious.
Discussion
Our results strengthen the evidence that Zbtb1 is essential for T cell development, as recently noted in scanT mice generated by chemical mutagenesis by Siggs et al [22]. The steps in T lineage differentiation from HSC are not fully understood, and identification of the role of Zbtb1 in pre-thymic lymphoid pathways and migration to the thymus will further elucidate this process. Analysis of fetal thymi of our mice lacking Zbtb1 showed decreased numbers of the earliest DN1 thymocytes and failure to progress beyond the DN1 stage. A lymphocyte-autonomous defect rather than defective thymic microenvironment was demonstrated by reciprocal transplants showing that knockout thymus supported generation of WT BM into T cells, while the converse transplant of knockout BM into WT mice did not. Zbtb1 mRNA was expressed in wild type thymocytes, increasing to a maximal level at the DP stage and then declining, and the total lack of maturing thymocytes in mice without Zbtb1 implies that the role of this factor in thymocyte differentiation is irreplaceable and cannot be compensated for by other members of the same protein family or by alternate signaling pathways. The much-reduced NK cell number in all mice lacking Zbtb1 indicates that it is also important for NK cell development or expansion.
The B cell phenotype of our knockout mice is similar to that of scanT mice [22]; neither the scanT C74R missense mutation nor the complete absence of Zbtb1 in our transgenic mouse affected the number of B cells. Mature B cells were found in the periphery of Zbtb1 deficient animals. Their in vitro proliferation in response to various stimuli was comparable to wild type B cells, arguing against a B cell-intrinsic defect and suggesting that the observed inability to class switch could be due to absence of T cell help.
There are 49 known mammalian Zinc finger BTB/POZ domain (Zbtb) gene family members [43]. BTB/POZ domains at the N termini of these proteins mediate either homo- or hetero-dimerization with related proteins [44]. All have multiple C2H2 zinc finger domains, which in canonical form have 2 Cys and 2 His residues in a C-2-C-12-H-3-H sequence. The 12 central residues, when stabilized by a Zn atom, form a projection that can interact with the major groove of a double-stranded DNA helix, recognizing around 5 specific base pairs of a G-rich sequence [45, 46]. Proteins belonging to the Zbtb family, including BCL6/ZNF51, Th-POK/ZFP67, PLZP, and PLZF/Zbtb16, to name a few, are transcription repressors implicated as switches at branch points between alternative developmental pathways. Their under-expression causes failure to branch out from a default pathway and alters homeostasis of hematopoietic cell compartments, while over-expression can also produce uncontrolled (often malignant) expansion of cells [6, 7, 44, 47, 48]. In addition, proteins with a similar structure have also been shown to mediate transcriptional repression and interact with components of histone deactylase co-repressor complexes [40, 48–51]. The Zbtb25 gene located immediately adjacent to Zbtb1 is also implicated in T cell development and negatively regulates nuclear factor of activated T cells, NF-AT [52].
Zbtb1 is a small gene now known to encode a transcriptional repressor that may act through control of chromatin remodeling, which in turn leads to changes in gene expression [40]. It is one of a few genes believed to be transcribed by spRNAP-IV, a single polypeptide nuclear polymerase expressed from an alternate transcript of the mitochondrial RNA polymerase gene POLRMT [53, 54]. We have shown that both predicted isoforms of the protein are expressed, a 713 amino acid protein encoded by an unspliced open reading frame in exon 2 and an alternate 644 amino acid isoform produced by a splice from within the distal coding region of exon 2 to exon 3. Both isoforms share the N-terminal BTB/POZ dimerization domain and proximal 5 zinc finger domains [44], but the last 3 zinc finger domains are truncated from the alternately spliced isoform. The longer Zbtb1713 is expressed at a higher level than Zbtb1644 in all tissues investigated. The two isoforms not only form homodimers, as expected for members of the Zbtb family [44], but also can dimerize with each other. Although the physiologic targets of Zbtb1 transcription repression are unknown, the reduced complement of zinc finger domains in the shorter isoform might confer reduced repressive activity upon a heterodimeric Zbtb1713-Zbtb1644 complex, potentially modulating or fine-tuning its effect. Further functional studies with the two isoforms of the protein may also shed light on the regulation of repressive activity of other proteins with similar structures.
Detailed analysis of our Zbtb1 knockout mice has revealed a contrast between the scanT mice and the Zbtb1 knockout mice in the numbers of the bone marrow progenitor subsets. While the numbers and percentages of bone marrow stem cell subsets in the scanT mice were reported to be comparable to wild type mice [22], our Zbtb1 knockout mice had significantly increased numbers of short term HSCs, MPPs and CLPs. This difference could be due to residual function of the protein bearing a C74R mutation in the BTB domain of Zbtb1 of scanT mice. Complete absence of Zbtb1 may result in a loss of repression of branching into differentiation pathways, causing an increase in numbers of the stem cell subsets in default pathways in our knockout mice. Analysis of the transcriptional activity of truncated Zbtb1 proteins by Liu et al. [40], each containing one of the 2 major functional domains (BTB and ZNF) revealed that the ZNF domain is responsible for its nuclear localization and exhibits the strongest repressive activity. This suggests that some residual activity may, therefore, still be present in the scanT mice due to their unaffected ZNF domains.
An explanation for the increased number of HSC in Zbtb1 knockouts could be that there is an attempt to compensate for increased apoptosis or otherwise decreased fitness that impairs lymphocyte development; our preliminary mixed chimerism studies have suggested the latter, as did similar experiments with scanT mice [22]. The increased number of HSC and progenitors in the bone marrow, but decreased thymocyte number in Zbtb1 deficient mice, could also reflect a defect in migration and homing to the thymus, a process facilitated by chemokine receptors CCR7 and CCR9 [55–57]. Indeed, our preliminary experiments (not shown) suggest a decrease in expression of CCR9 mRNA in Zbtb1 knockout HSC.
Activation of the cyclic AMP signaling pathway has been shown to modulate T cell development, and stimulation of cAMP dependent signaling in the thymus prevents differentiation of T cell precursors and results in increased apoptosis [58]. It has been reported that one function of Zbtb1 is a dose dependent repression of a cAMP response element (CRE), preventing it from binding to a CRE-binding protein (CREB) [40]. Moreover, a human multiple malformation syndrome with hypoplasia of the thymus was recently associated with CREB1 mutation [59]. Thus the absence of Zbtb1 mediated repression of c-AMP signaling pathways, could be one of the reasons why T cells are unable to develop in the thymus of both complete knockout and scanT mice.
The mechanism of action of Zbtb1 during early lymphoid determination could also be to repress the differentiation of B cell progenitors (BLP) from the all lymphocyte precursors (ALPs) in bone marrow [2], to favor the differentiation of ALPs into T cell precursors instead (Figure 7C). Further experiments, such as with RNA expression arrays and chromatin immunoprecipitation, will elucidate the molecular targets of Zbtb1.
A precise understanding of the development of lymphocytes from HSCs is critical for treating humans with immunodeficiencies, blood diseases and cancers by transplantation, gene therapy and discovery of new drugs. Zbtb1 is similar to transcription factors with proven roles in lymphocyte development and has now been shown repeatedly to be essential and specific for mouse thymocyte development [6, 7, 22]. Our ability to elucidate and manipulate Zbtb1 functions in mice and ultimately in humans will facilitate understanding and treating human immune disorders. SCID and related primary immunodeficiencies in humans are rare, but life threatening, and in many cases with genetic causes still unknown. Although to date we have not found mutations of human ZBTB1 in patients with typical T− B+ SCID phenotypes, characteristics of humans with ZBTB1 defects may be distinct, and examining a wider range of human phenotypes, including those with multiple malformations associated with thymic hypoplasia with deficient cortical T cells like the CREB1 defect reported by Kitazawa et al [59] may be fruitful. In any case, our new Zbtb1 deficient model will be useful to study the role of this transcription factor and its associations and targets in the development of T lymphocytes.
Supplementary Material
{kind=link}
Acknowledgements
Robert Nussbaum, Fabio Candotti, Mark Killian, Nigel Killeen and Pam Schwartzberg gave expert advice in mouse construction and flow cytometry; Art Nienhaus provided the MSCV-IRES-GFP vector; Mitchell Eddy provided Hspa2 mice and antibody; Michael Eckhaus helped with histology; Amy Chen, Beverly Hay and Marla Vacek provided skillful experimental assistance.
Footnotes
This research was supported by RO1 AI078248 to JMP and the Division of Intramural Research, National Human Genome Research Institute, NIH.
Authorship
DP, KS, YC, MN, CHS, JZ, DG, EP and AD designed and performed experiments, prepared figures and analyzed data; DB co-designed experiments and provided essential tools and guidance; JMP designed and supervised the research, performed experiments and with DP and KS wrote the paper.
The authors declare no competing financial interests.
References
- 1.Karsunky H, Inlay MA, Serwold T, Bhattacharya D, Weissman IL. Flk2+ common lymphoid progenitors possess equivalent differentiation potential for the B and T lineages. Blood. 2008;111(12):5562–5570. doi: 10.1182/blood-2007-11-126219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Inlay MA, Bhattacharya D, Sahoo D, Serwold T, Seita J, Karsunky H, Plevritis SK, Dill DL, Weissman IL. Ly6d marks the earliest stage of B-cell specification and identifies the branchpoint between B-cell and T-cell development. Genes & development. 2009;23(20):2376–2381. doi: 10.1101/gad.1836009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilson A, MacDonald HR, Radtke F. Notch 1-deficient common lymphoid precursors adopt a B cell fate in the thymus. The Journal of experimental medicine. 2001;194(7):1003–1012. doi: 10.1084/jem.194.7.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacDonald HR, Wilson A, Radtke F. Notch1 and T-cell development: insights from conditional knockout mice. Trends in immunology. 2001;22(3):155–160. doi: 10.1016/s1471-4906(00)01828-7. [DOI] [PubMed] [Google Scholar]
- 5.Shortman K, Wu L. Early T lymphocyte progenitors. Annual review of immunology. 1996;14:29–47. doi: 10.1146/annurev.immunol.14.1.29. [DOI] [PubMed] [Google Scholar]
- 6.He X, He X, Dave VP, Zhang Y, Hua X, Nicolas E, Xu W, Roe BA, Kappes DJ. The zinc finger transcription factor Th-POK regulates CD4 versus CD8 T-cell lineage commitment. Nature. 2005;433(7028):826–833. doi: 10.1038/nature03338. [DOI] [PubMed] [Google Scholar]
- 7.Kappes DJ, He X, He X. Role of the transcription factor Th-POK in CD4:CD8 lineage commitment. Immunological reviews. 2006;209:237–252. doi: 10.1111/j.0105-2896.2006.00344.x. [DOI] [PubMed] [Google Scholar]
- 8.Buckley RH. The multiple causes of human SCID. The Journal of clinical investigation. 2004;114(10):1409–1411. doi: 10.1172/JCI23571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ochs HSC, Puck J. Primary Immunodeficiency Diseases, A Molecular and Genetic Approach. :2006–2007. [Google Scholar]
- 10.Gaspar HB, Aiuti A, Porta F, Candotti F, Hershfield MS, Notarangelo LD. How I treat ADA deficiency. Blood. 2009;114(17):3524–3532. doi: 10.1182/blood-2009-06-189209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Al-Herz W, Bousfiha A, Casanova JL, Chapel H, Conley ME, Cunningham- Rundles C, Etzioni A, Fischer A, Franco JL, Geha RS, et al. Classification of Primary Immunodeficiency Diseases by the International Union of Immunological Societies (IUIS) Expert Committee on Primary Immunodeficiency 2011. Clinical & Experimental Immunology. 2012 doi: 10.1111/j.1365-2249.2012.04561.x. no-no. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Araki R, Fujimori A, Hamatani K, Mita K, Saito T, Mori M, Fukumura R, Morimyo M, Muto M, Itoh M, et al. Nonsense mutation at Tyr-4046 in the DNA-dependent protein kinase catalytic subunit of severe combined immune deficiency mice. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(6):2438–2443. doi: 10.1073/pnas.94.6.2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blunt T, Gell D, Fox M, Taccioli GE, Lehmann AR, Jackson SP, Jeggo PA. Identification of a nonsense mutation in the carboxyl-terminal region of DNA-dependent protein kinase catalytic subunit in the scid mouse. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(19):10285–10290. doi: 10.1073/pnas.93.19.10285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bosma GC, Davisson MT, Ruetsch NR, Sweet HO, Shultz LD, Bosma MJ. The mouse mutation severe combined immune deficiency (scid) is on chromosome 16. Immunogenetics. 1989;29(1):54–57. doi: 10.1007/BF02341614. [DOI] [PubMed] [Google Scholar]
- 15.Miller RD, Hogg J, Ozaki JH, Gell D, Jackson SP, Riblet R. Gene for the catalytic subunit of mouse DNA-dependent protein kinase maps to the scid locus. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(23):10792–10795. doi: 10.1073/pnas.92.23.10792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huppi K, Siwarski D, Shaughnessy J, Jr, Klemsz MJ, Shirakata M, Maki R, Sakano H. Genes associated with immunoglobulin V(D)J recombination are linked on mouse chromosome 2 and human chromosome 11. Immunogenetics. 1993;37(4):288–291. doi: 10.1007/BF00187456. [DOI] [PubMed] [Google Scholar]
- 17.Schlake T, Schorpp M, Nehls M, Boehm T. The nude gene encodes a sequence-specific DNA binding protein with homologs in organisms that lack an anticipatory immune system. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(8):3842–3847. doi: 10.1073/pnas.94.8.3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nehls M, Pfeifer D, Schorpp M, Hedrich H, Boehm T. New member of the winged-helix protein family disrupted in mouse and rat nude mutations. Nature. 1994;372(6501):103–107. doi: 10.1038/372103a0. [DOI] [PubMed] [Google Scholar]
- 19.Segre JA, Nemhauser JL, Taylor BA, Nadeau JH, Lander ES. Positional cloning of the nude locus: genetic, physical, and transcription maps of the region and mutations in the mouse and rat. Genomics. 1995;28(3):549–559. doi: 10.1006/geno.1995.1187. [DOI] [PubMed] [Google Scholar]
- 20.Thomis DC, Lee W, Berg LJ. T cells from Jak3-deficient mice have intact TCR signaling, but increased apoptosis. J Immunol. 1997;159(10):4708–4719. [PubMed] [Google Scholar]
- 21.Choi Y, Ramnath VR, Eaton AS, Chen A, Simon-Stoos KL, Kleiner DE, Erikson J, Puck JM. Expression in transgenic mice of dominant interfering Fas mutations: a model for human autoimmune lymphoproliferative syndrome. Clin Immunol. 1999;93(1):34–45. doi: 10.1006/clim.1999.4767. [DOI] [PubMed] [Google Scholar]
- 22.Siggs OM, Li X, Xia Y, Beutler B. ZBTB1 is a determinant of lymphoid development. J Exp Med. 2011;209(1):19–27. doi: 10.1084/jem.20112084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karkera JD, Izraeli S, Roessler E, Dutra A, Kirsch I, Muenke M. The genomic structure, chromosomal localization, and analysis of SIL as a candidate gene for holoprosencephaly. Cytogenetic and genome research. 2002;97(1–2):62–67. doi: 10.1159/000064057. [DOI] [PubMed] [Google Scholar]
- 24.Osoegawa K, Tateno M, Woon PY, Frengen E, Mammoser AG, Catanese JJ, Hayashizaki Y, de Jong PJ. Bacterial artificial chromosome libraries for mouse sequencing and functional analysis. Genome research. 2000;10(1):116–128. [PMC free article] [PubMed] [Google Scholar]
- 25.Onorato IM, Markowitz LE, Oxtoby MJ. Childhood immunization, vaccine-preventable diseases and infection with human immunodeficiency virus. The Pediatric infectious disease journal. 1988;7(8):588–595. [PubMed] [Google Scholar]
- 26.Otsu M, Anderson SM, Bodine DM, Puck JM, O'Shea JJ, Candotti F. Lymphoid development and function in X-linked severe combined immunodeficiency mice after stem cell gene therapy. Molecular therapy : the journal of the American Society of Gene Therapy. 2000;1(2):145–153. doi: 10.1006/mthe.1999.0020. [DOI] [PubMed] [Google Scholar]
- 27.Aviles Mendoza GJ, Seidel NE, Otsu M, Anderson SM, Simon-Stoos K, Herrera A, Hoogstraten-Miller S, Malech HL, Candotti F, Puck JM, et al. Comparison of five retrovirus vectors containing the human IL-2 receptor gamma chain gene for their ability to restore T and B lymphocytes in the X-linked severe combined immunodeficiency mouse model. Molecular therapy : the journal of the American Society of Gene Therapy. 2001;3(4):565–573. doi: 10.1006/mthe.2001.0292. [DOI] [PubMed] [Google Scholar]
- 28.Hardy RR, Hayakawa K. B cell development pathways. Annual review of immunology. 2001;19:595–621. doi: 10.1146/annurev.immunol.19.1.595. [DOI] [PubMed] [Google Scholar]
- 29.Sneller MC, Wang J, Dale JK, Strober W, Middelton LA, Choi Y, Fleisher TA, Lim MS, Jaffe ES, Puck JM, et al. Clincal, immunologic, and genetic features of an autoimmune lymphoproliferative syndrome associated with abnormal lymphocyte apoptosis. Blood. 1997;89(4):1341–1348. [PubMed] [Google Scholar]
- 30.Dix DJ, Allen JW, Collins BW, Mori C, Nakamura N, Poorman-Allen P, Goulding EH, Eddy EM. Targeted gene disruption of Hsp70-2 results in failed meiosis, germ cell apoptosis, and male infertility. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(8):3264–3268. doi: 10.1073/pnas.93.8.3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dix DJ, Allen JW, Collins BW, Poorman-Allen P, Mori C, Blizard DR, Brown PR, Goulding EH, Strong BD, Eddy EM. HSP70-2 is required for desynapsis of synaptonemal complexes during meiotic prophase in juvenile and adult mouse spermatocytes. Development. 1997;124(22):4595–4603. doi: 10.1242/dev.124.22.4595. [DOI] [PubMed] [Google Scholar]
- 32.Godfrey DI, Kennedy J, Suda T, Zlotnik A. A developmental pathway involving four phenotypically and functionally distinct subsets of CD3-CD4-CD8− triple-negative adult mouse thymocytes defined by CD44 and CD25 expression. J Immunol. 1993;150(10):4244–4252. [PubMed] [Google Scholar]
- 33.Balciunaite G, Ceredig R, Fehling HJ, Zuniga-Pflucker JC, Rolink AG. The role of Notch and IL-7 signaling in early thymocyte proliferation and differentiation. European journal of immunology. 2005;35(4):1292–1300. doi: 10.1002/eji.200425822. [DOI] [PubMed] [Google Scholar]
- 34.Porritt HE, Rumfelt LL, Tabrizifard S, Schmitt TM, Zuniga-Pflucker JC, Petrie HT. Heterogeneity among DN1 prothymocytes reveals multiple progenitors with different capacities to generate T cell and non-T cell lineages. Immunity. 2004;20(6):735–745. doi: 10.1016/j.immuni.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 35.Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121(7):1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 36.Osawa M, Hanada K, Hamada H, Nakauchi H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science. 1996;273(5272):242–245. doi: 10.1126/science.273.5272.242. [DOI] [PubMed] [Google Scholar]
- 37.Oliver AM, Martin F, Gartland GL, Carter RH, Kearney JF. Marginal zone B cells exhibit unique activation, proliferative and immunoglobulin secretory responses. European journal of immunology. 1997;27(9):2366–2374. doi: 10.1002/eji.1830270935. [DOI] [PubMed] [Google Scholar]
- 38.Srivastava B, Quinn WJ, 3rd, Hazard K, Erikson J, Allman D. Characterization of marginal zone B cell precursors. The Journal of experimental medicine. 2005;202(9):1225–1234. doi: 10.1084/jem.20051038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meyer-Bahlburg A, Bandaranayake AD, Andrews SF, Rawlings DJ. Reduced c-myc expression levels limit follicular mature B cell cycling in response to TLR signals. J Immunol. 2009;182(7):4065–4075. doi: 10.4049/jimmunol.0802961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Q, Yao F, Wang M, Zhou B, Cheng H, Wang W, Jin L, Lin Q, Wang JC. Novel human BTB/POZ domain-containing zinc finger protein ZBTB1 inhibits transcriptional activities of CRE. Molecular and cellular biochemistry. 2011;357(1–2):405–414. doi: 10.1007/s11010-011-0911-5. [DOI] [PubMed] [Google Scholar]
- 41.Matic I, Schimmel J, Hendriks IA, van Santen MA, van de Rijke F, van Dam H, Gnad F, Mann M, Vertegaal AC. Site-specific identification of SUMO-2 targets in cells reveals an inverted SUMOylation motif and a hydrophobic cluster SUMOylation motif. Molecular cell. 2010;39(4):641–652. doi: 10.1016/j.molcel.2010.07.026. [DOI] [PubMed] [Google Scholar]
- 42.Rowan AJ, Bodmer WF. Introduction of a myc reporter tag to improve the quality of mutation detection using the protein truncation test. Human mutation. 1997;9(2):172–176. doi: 10.1002/(SICI)1098-1004(1997)9:2<172::AID-HUMU10>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 43.Seal RLGS, Lush MJ, Wright MW, Bruford EA. genenames.org: HGNC Database, HUGO Gene Nomenclature Committee (HGNC) Nucleic Acids Res. 2011;39:D519–D519. doi: 10.1093/nar/gkq892. (Database) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Collins T, Stone JR, Williams AJ. All in the family: the BTB/POZ, KRAB, and SCAN domains. Molecular and cellular biology. 2001;21(11):3609–3615. doi: 10.1128/MCB.21.11.3609-3615.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Evans RM, Hollenberg SM. Zinc fingers: gilt by association. Cell. 1988;52(1):1–3. doi: 10.1016/0092-8674(88)90522-3. [DOI] [PubMed] [Google Scholar]
- 46.Murre C. Helix-loop-helix proteins and lymphocyte development. Nature immunology. 2005;6(11):1079–1086. doi: 10.1038/ni1260. [DOI] [PubMed] [Google Scholar]
- 47.Piazza F, Costoya JA, Merghoub T, Hobbs RM, Pandolfi PP. Disruption of PLZP in mice leads to increased T-lymphocyte proliferation, cytokine production, and altered hematopoietic stem cell homeostasis. Molecular and cellular biology. 2004;24(23):10456–10469. doi: 10.1128/MCB.24.23.10456-10469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li W, Wang F, Menut L, Gao FB. BTB/POZ-zinc finger protein abrupt suppresses dendritic branching in a neuronal subtype-specific and dosage-dependent manner. Neuron. 2004;43(6):823–834. doi: 10.1016/j.neuron.2004.08.040. [DOI] [PubMed] [Google Scholar]
- 49.Melnick A, Carlile G, Ahmad KF, Kiang CL, Corcoran C, Bardwell V, Prive GG, Licht JD. Critical residues within the BTB domain of PLZF and Bcl-6 modulate interaction with corepressors. Molecular and cellular biology. 2002;22(6):1804–1818. doi: 10.1128/MCB.22.6.1804-1818.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li X, Peng H, Schultz DC, Lopez-Guisa JM, Rauscher FJ, 3rd, Marmorstein R. Structure-function studies of the BTB/POZ transcriptional repression domain from the promyelocytic leukemia zinc finger oncoprotein. Cancer research. 1999;59(20):5275–5282. [PubMed] [Google Scholar]
- 51.Huynh KD, Bardwell VJ. The BCL-6 POZ domain and other POZ domains interact with the co-repressors N-CoR and SMRT. Oncogene. 1998;17(19):2473–2484. doi: 10.1038/sj.onc.1202197. [DOI] [PubMed] [Google Scholar]
- 52.Benita Y, Cao Z, Giallourakis C, Li C, Gardet A, Xavier RJ. Gene enrichment profiles reveal T-cell development, differentiation, and lineage-specific transcription factors including ZBTB25 as a novel NF-AT repressor. Blood. 2010;115(26):5376–5384. doi: 10.1182/blood-2010-01-263855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kravchenko Iu E, Chumakov PM. Alternative transcripts from POLRMT responsible for synthesis of nuclear RNA polymerase IV. Mol Biol (Mosk) 2005;39(1):67–71. [PMC free article] [PubMed] [Google Scholar]
- 54.Kravchenko JE, Rogozin IB, Koonin EV, Chumakov PM. Transcription of mammalian messenger RNAs by a nuclear RNA polymerase of mitochondrial origin. Nature. 2005;436(7051):735–739. doi: 10.1038/nature03848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Uehara S, Grinberg A, Farber JM, Love PE. A role for CCR9 in T lymphocyte development and migration. J Immunol. 2002;168(6):2811–2819. doi: 10.4049/jimmunol.168.6.2811. [DOI] [PubMed] [Google Scholar]
- 56.Zlotoff DA, Sambandam A, Logan TD, Bell JJ, Schwarz BA, Bhandoola A. CCR7 and CCR9 together recruit hematopoietic progenitors to the adult thymus. Blood. 115(10):1897–1905. doi: 10.1182/blood-2009-08-237784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Svensson M, Marsal J, Uronen-Hansson H, Cheng M, Jenkinson W, Cilio C, Jacobsen SE, Sitnicka E, Anderson G, Agace WW. Involvement of CCR9 at multiple stages of adult T lymphopoiesis. Journal of leukocyte biology. 2008;83(1):156–164. doi: 10.1189/jlb.0607423. [DOI] [PubMed] [Google Scholar]
- 58.Lalli E, Sassone-Corsi P, Ceredig R. Block of T lymphocyte differentiation by activation of the cAMP-dependent signal transduction pathway. The EMBO journal. 1996;15(3):528–537. [PMC free article] [PubMed] [Google Scholar]
- 59.Kitazawa S, Kondo T, Mori K, Yokoyama N, Matsuo M, Kitazawa R. A p.D116G mutation in CREB1 leads to novel multiple malformation syndrome resembling CrebA knockout mouse. Human mutation. 2012 doi: 10.1002/humu.22027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.