

Abstract
Purpose
Duodenal atresia in humans has been hypothesized to arise from a failure of the duodenal lumen to recanalize after formation of an endodermal plug. Recently, mutations in the Fibroblast Growth Factor Receptor 2 gene have been shown to cause atretic defects of the duodenum in mice (Fgfr2IIIb). However, work in rats suggests that murine species do not form an endodermal plug during normal duodenal development. These lines of data led us to hypothesize that mice are able to form a duodenal atresia in the absence of an endodermal plug. To test this hypothesis we examined duodenal development in wild-type and Fgfr2IIIb-/- embryos.
Methods
Paraffin sections were generated for either hematoxylin and eosin, E-cadherin or terminal deoxynucleotidyl transferase mediated X-dUTP nick end labeling (TUNEL) staining from Fgfr2IIIb-/- and wild-type embryos between Embryonic Days (E) 10.5 and E14.5. Sections were photographed and reconstructed into 3-dimensional display using Adobe Photoshop and Amira Visage software.
Results
Normal mouse duodenum does not form an endodermal plug, although a plug does form in the pyloric region of the stomach at E14.5. Fgfr2IIIb-/- embryos experience significant apoptosis in the duodenal region at E10.5, followed by the disappearance of the endoderm in the atretic precursor by E11.5. Thereafter, the mesoderm of the atretic precursor involutes over the next 2 days in the absence of further apoptosis. Interestingly, an endodermal plug was not observed at any point during the formation of a duodenal atresia.
Conclusions
These results suggest that duodenal atresia in the Fgfr2IIIb-/- model does not arise from persistence of an epithelial plug. Rather it appears to result from the loss of the endoderm due to apoptosis very early in development.
Keywords: Duodenal atresia, Fgfr2IIIb, aberrant morphogenesis, mouse, apoptosis
Duodenal atresia is hypothesized to result from the persistence of a “solid core” of tissue arising from a failure of the proximal duodenum to recanalize its lumen after a period of endodermal hyperplasia [1]. This hypothesis arose from the observations of Julius Tandler, a Viennese anatomist, who performed a detailed histological analysis of normal human duodenal development. He noted that the endoderm of the proximal duodenum undergoes, “A remarkable thickening of the epithelium… until finally the duodenum becomes a solid string, in which no lumen at all is to be found.” This occurs at day 42 in development or Carnegie Stage 17. He then went on to state: “If one keeps in mind the fact that on one hand the epithelial occlusion of the duodenum represents a normal event, but on the other hand that it is exactly in this place that most pathologic occlusions of the intestine occur, the question does not appear unjustified to ask whether these processes relate to each other, that is, whether they are causally related. It would not be impossible that in rare cases the physiologic atresia remains and develops into a congenital atresia.” Since that time, there has been little experimental evidence to support his hypothesis and it remains unclear whether the developmental events described by Tandler are of etiologic significance in duodenal atresia.
The histological analysis of human duodenal atresia may never be described. First, the defect is rare, occurring in approximately 1/5,000 births. Second, the number of human embryos available for study is small with the largest collection (approximately 30,000) housed at Kyoto University in Kyoto, Japan. Even in this sizable collection there are possibly only 4 to 6 specimens with duodenal atresia leaving very few specimens for analysis. And the likelihood of expanding the number of embryos for study is remote in the current medical-legal environment. Third, in spite of Tandler's work, the timing of the formation of this defect remains unknown. Thus, because the events prior to the involution of the atretic segment have not been described, there is no way of identifying specimens that will form an atresia in their pre-atretic phase. Given all these factors, the prospect of being able to assemble a series of specimens to sequentially detail the aberrant morphogenesis of duodenal atresia in humans appears infinitesimally low. Therefore, we set out to thoroughly examine the aberrant morphogenesis of this defect in an established genetic animal model of duodenal atresia.
Mutations within the Fgfr2 gene leading to the loss of Fgfr2IIIb function in mice result in duodenal atresia [2]. Fgfr2IIIb is a membrane bound tyrosine kinase receptor, and one of two known receptor isoforms generated from the Fgfr2 gene [3]. Mutant embryos are generated by deleting the exon encoding the extracellular Fgf ligand-binding domain of the receptor. These embryos survive to term but are non-viable at birth due to the absence of lungs [4]. Forty-two percent of the embryos develop a duodenal atresia [5] with over 95% being a type III atresia [2]. Mutant embryos have additional defects including the heart, lungs, pituitary, skin, eye lids and limbs [4].
We set out to determine whether the normal mouse duodenum undergoes a solid core stage with formation of an endodermal plug. A step that is critical in defining the mechanism and characterizing the aberrant morphogenesis of duodenal atresia formation in Fgfr2IIIb-/- embryos. We performed a comparative analysis of normal mouse and human duodenum at matching Carnegies Stages (CS14-20) [6] with complete 3-dimensional reconstruction of normal mouse histological sections at all stages. A similar analysis of duodenal atresia formation was performed in Fgfr2IIIb-/- embryos. We observed that unlike human duodenal development, the mouse duodenum does not form a solid core in the second or third portions during normal development. Furthermore, the formation of duodenal atresia is associated with a complete loss of endoderm by E11.5, or CS 16 in the anatomic region where the atresia will form: the atretic precursor. In contrast, the formation of an endodermal plug in humans occurs at CS17.
1. Methods
1.1 Analysis of human specimens
IRB approval was obtained (P.F.N. protocol # m2010-1077). The human embryos examined in this study were from the Kyoto Collection of Human Embryos held in the Congenital Anomaly Research Center of Kyoto University [7, 8]. A series of preserved human embryos were selected for Carnegie Stages (CS) 13, 14, 15, 16, 17, 18, and 20 based on availability of transverse histological sections. Prior to our study, all embryos had been serially sectioned at 10 μm thickness and stained with Hematoxylin and Eosin, Periodic Acid Schiff, and Trichrome stains on alternating sections. Photomicrographs of existing slides were taken at 100× using an Olympus® BX41 microscope and an Olympus® DP20 camera (Tokyo, Japan).
1.2 Animals
Approval from the Institutional Animal Care and Use Committee (IACUC) was obtained from the University of Wisconsin School of Medicine and Public health for these studies (P.F.N. protocol # M02258). All animals were maintained in a clean facility with access to fresh food and water, and kept on a 12 hour alternating light/dark cycle. Fgfr2IIIb-/-- embryos were generated by breeding Fgfr2IIIb-/+; HprtCre/+ to Fgfr2IIIbflox/flox males [5, 9]. Genotyping of embryos was performed as described previously [4, 9].
1.3 Histology
Wild-type embryos were harvested at E10.5, E11.0 E11.5, E12.0, E12.5, E13.0, E13.5, and E14.5 (corresponding to Carnegie Stages 14, 15, 16, 17, 18, 19, 20, and 23 respectively). Fgfr2IIIb-/- embryos were harvested at E10.5, E 11.5, E12.5 and E13.5. Embryos were fixed in Bouin's fixative overnight at 4°C and dehydrated through a series of escalating Phosphate Buffered Saline (PBS)/Ethanol steps, then embedded into Paraffin. Sections were taken at 7μm thickness and dried overnight at 37 °C. Sections were de-waxed, stained with Hematoxylin and Eosin and cover slipped. Photomicrographs were taken at 100× and 200× using an Olympus® BX41 light microscope and an Olympus® DP20 camera (Tokyo, Japan).
1.4 3-dimensional reconstructions
Photomicrographs of transverse sections of the wild-type and Fgfr2IIIb-/- duodenal region were uploaded sequentially into Amira™ (Berlin, Germany), aligned with the auto align tool, and labeled using the segmentation editor. Following the markup of each image, a 3-dimensional structure of the duodenal region was generated for each embryonic stage using the surface generator tool. A final JPEG image file was produced at 600 pixels per square inch and imported into Adobe Photoshop® for labeling.
1. 5 Immunohistochemistry
Embryos were harvested at E12.0 (CS17) and fixed in 4% Paraformaldehyde overnight at 4°C. Embryonic tissues were dehydrated through a series of escalating PBS-Tween (PBST)/Methanol steps, and embedded into Paraffin. Sections were generated at 7 μm and dried overnight at 37 °C. Sections were then deparaffinized and rehydrated. Antigen retrieval was performed with HIER-citrate at a pH of 6.0 using a decloaking chamber at 100°C for 20 minutes. Following this, they were blocked with 10% Bovine serum albumin for 60 minutes. Sections were then incubated with a rabbit monoclonal anti-E-cadherin antibody (Cell Signaling Technology cat# 3195, Beverly, MA) at 1:50 dilution overnight at 4°C. Specimens were then washed and endogenous peroxidase was quenched with 1.0% hydrogen peroxide in methanol for 10 minutes. The specimens were incubated with goat anti-rabbit HRP polymer (Vector Laboratories, Burlingame, CA) for 30 minutes at room temperature. Specimens were washed with 3,3′-diaminobenzidine (DAB) (Vector Laboratories, Burlingame, CA) for 2 minutes and counterstained with hematoxylin.
1.6 TUNEL staining
Fgfr2IIIb-/-- and Fgfr2IIIb+/+ embryos were harvested at E10.5 and 11.5, fixed in 4% paraformaldehyde overnight at 4°C, then dehydrated and embedded in paraffin as described above. Transverse sections were generated at 5 μm and processed for detection of apoptosis. The DeadEnd Fluorometric TUNEL System (Promega, Madison, WI) was used per supplied instructions with appropriate positive and negative controls. Slides were cover slipped and processed sections were then photographed with a fluorescent microscope.
2. Results
2.1 Analysis of human duodenum CS13-20
We examined human duodenal histology to demonstrate the morphogenesis of the endodermal plug prior to undertaking similar studies in the mouse. As early as CS 13, the endodermal lining of the human duodenum appears to be stratified pseudo-columnar (Figure 1A). At this stage, the lumen is circular when transversely sectioned, and becomes elliptical by CS14 (Figure 1B). By CS 16 the lumen narrows dramatically and the surrounding mesoderm appears to be organizing into layers (Figure 1D). By CS 17 the lumen is obliterated by a solid core of endodermal tissue, as previously described by Tandler (Figure 1E). The boundary between the endoderm and the mesoderm is easily distinguished at this point. At CS 18 channels can be seen forming at multiple points in the lumen (Figure 1F). By CS 20 these channels coalesce into a single lumen and the surrounding mesoderm is organized into concentric layers of tissue (Figure 1G).
Figure 1. Histology of endodermal plug formation and recanalization during normal human duodenal development.
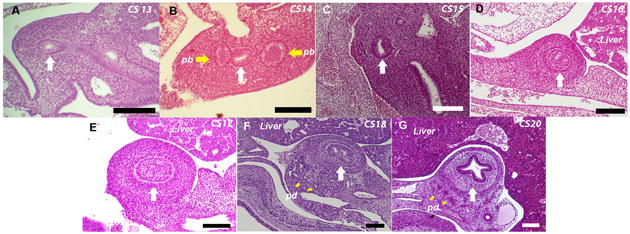
Transverse 10um sections of human duodenum. A. Carnegie Stage (CS) 13. B. CS 14. C. CS 15. D. CS 16. E. CS17 an occlusive plug has formed. F. CS 18 recanalization of the duodenal lumen begins G. CS 20. White arrow indicates the duodenum. Yellow arrows indicate Pb-Pancreatic buds and Pd-pancreatic ducts (Bars indicate 100μM).
2.2 Histology of the second portion of the duodenum in wild-type mice
Formation of duodenal atresia in Fgfr2IIIb-/- mice occurs slightly distal to the region where the pancreatic and hepatic ducts join the duodenum [2]. We examined the histology of wild-type mouse embryos in this region to determine if an endodermal plug forms during normal duodenal development (Figure 2). Similar to the human, endodermal cells of the mouse duodenum becomes stratified pseudo-columnar by E11.0. Thereafter, the lumen becomes elliptical (E11.5), and even crescent shaped at E12.5. By E13.0 the lumen starts to widen and the mesoderm is seen organizing into concentric rings. This continues through the final time point examined: E14.5. At no point in the development of the second portion of the wild-type mouse duodenum is an endodermal plug seen.
Figure 2. Histology of wild-type mouse duodenal development.
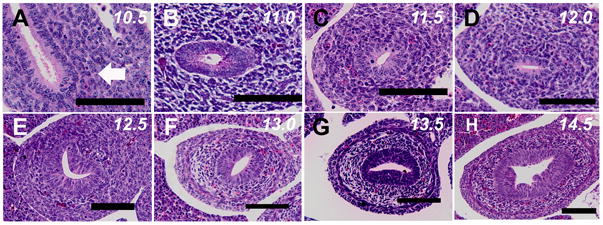
Transverse 7um sections of mouse duodenum taken through the duodenum distal to the pancreatic and hepatic buds. A. Embryonic Day (E) 10.5 (CS14). B. E11.0 (CS 15). C. E11.5 (CS 16). D. E12.0 (CS 17). E. E12.5 (CS18). F. E13.0 (CS 19). G. E13.5 (CS20). H. E14.5 (CS 23). At no point does an occlusive endodermal plug form in this portion of the duodenum (Bars indicate 100μM).
2.3 Three dimensional reconstruction of the mouse Duodenum between E10.5 and E14.5 (CS 14 to 20)
To confirm our histological observations that mice do not form a solid core of endodermal tissue during normal duodenal development, 7μm histological sections were taken from the pylorus though the C-loop of the duodenum and reconstructed in 3-dimensions. In these views, the pancreatic (Yellow) and hepatic ducts (Green) are easily seen. At E10.5, or the equivalent of CS 14, the lumen of the stomach widens in the ventral-dorsal dimension and there is no bend in the duodenum (Figure 3A). At E11.0, or CS 15, the duodenum initiates dorsal bending and the lumen remains continuous (Figure 3B). By E11.5, or CS16, a distinct duodenal C-Loop begins to appear (Figure 3C). C-Loop formation continues into E12.0 (CS17) (Figure 3D). From E12.5 to E13.5 (CS18 to 20), the duodenum begins to elongate between the pylorus and C-loop (Figure 3E-G). Interestingly, vacuoles or lacunae can be seen forming in the endodermal layers around the pylorus at E13.5 (Figure 3G, White area adjacent to lumen). By E14.5, the diameters of the duodenal endodermal tube and lumen increase considerably relative to the mesoderm. At this same time point, the diameter of the pyloric endoderm is reduced (Figure 3H). Closer inspection of the pyloric endoderm reveals a very narrow lumen with multiple channels (Figure 3I). Histological examination of pyloric endoderm demonstrates a nearly occlusive plug (Figure 3J). However, at no point during duodenal development is there a discontinuity of the pyloric lumen and the lumen in the region beyond the pancreatic and hepatic ducts remains widely patent.
Figure 3. Three dimensional reconstructions of histology sections of wild-type mouse duodenum.
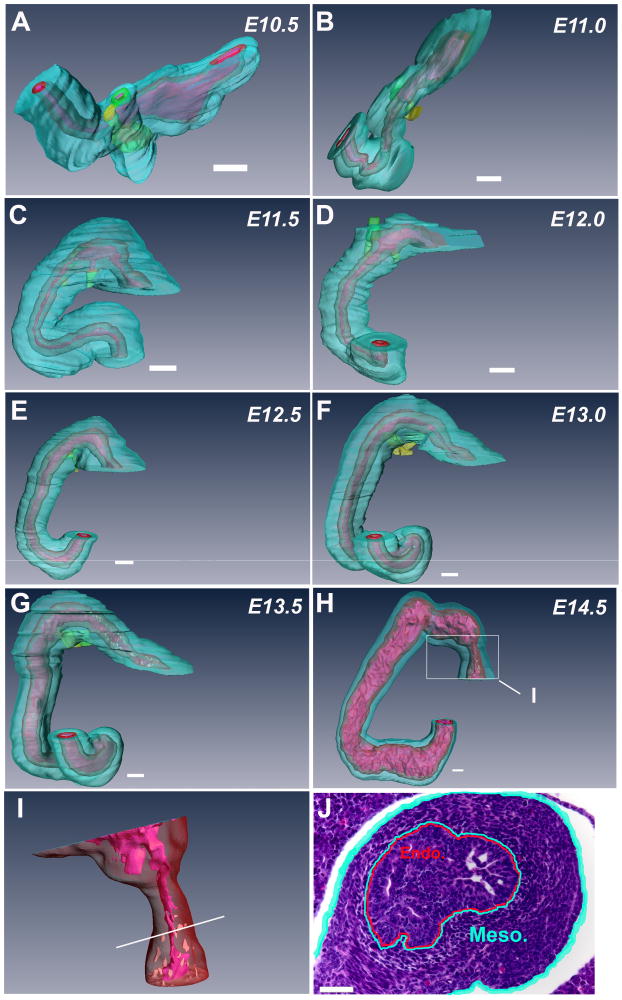
A. Embryonic Day (E) 10.5 (CS14). B. E11.0 (CS 15). C. E11.5 (CS 16). D. E12.0 (CS 17). E. E12.5 (CS18). F. E13.0 (CS 19). G. E13.5 (CS20). H. E14.5 (CS 23). I. Magnification of endoderm in the pyloric region (boxed area from Figure H). J. Section through pylorus at E14.5 (plane of section demonstrated by white line in Figure I). Mesoderm is aqua blue, endoderm is red and the lumen is pink. Hepatic buds/ducts are green and pancreatic buds/ducts are yellow. Reconstructions illustrate that normal duodenal development proceeds without the formation of an occlusive endodermal plug at all stages examined. In contrast, a nearly occlusive plug forms in the pyloric region (Figure H-White box, Figure I and J) indicating the mouse endoderm in this region retains the capacity to form such a structure. (Bars indicate 100μM).
2.4 Histology of duodenal atresia formation in Fgfr2IIIb-/- embryos (E10.5 to E13.5)
Having observed that mice do not form an endodermal plug in the duodenum during normal development, we next characterized the histology and aberrant morphogenesis of duodenal atresia formation in Fgfr2IIIb-/- embryos. At E10.5, the endoderm of the proximal duodenum is present and the lumen is patent (Figure 4A) and 3-dimensional reconstruction verified the continuity of the lumen beyond the hepatic and pancreatic ducts (Figure 4B). Notably, the mutant embryo's gastric region is much smaller in the ventral-dorsal and rostral-caudal dimensions than that of the normal embryo (Compare Figure 3A to Figure 4B). By E11.5, the human equivalent of CS 16, the duodenal endoderm is completely absent just beyond the hepatic and pancreatic ducts: the region termed the atretic precursor. This is demonstrated both histologically (Figure 4C) and on 3-dimensional reconstruction. (Figure 4D). The endoderm is visualized in the distal duodenum, just before the C-loop. The diameter of the mesoderm of the duodenal atretic precursor is modestly narrowed. Twenty-four hours later (E12.5), the atretic precursor region has narrowed considerably (Figure 4E and F), and this region is absent by E13.5 (Figure 4G and H). Formation of duodenal atresia has gone to completion a full 12 hours before the normal mouse duodenum will form an endodermal plug in the pyloric region.
Figure 4. Histology and aberrant morphogenesis of atretic duodenal development in Fgfr2IIIb-/- mouse embryos E10.5 to E13.5.
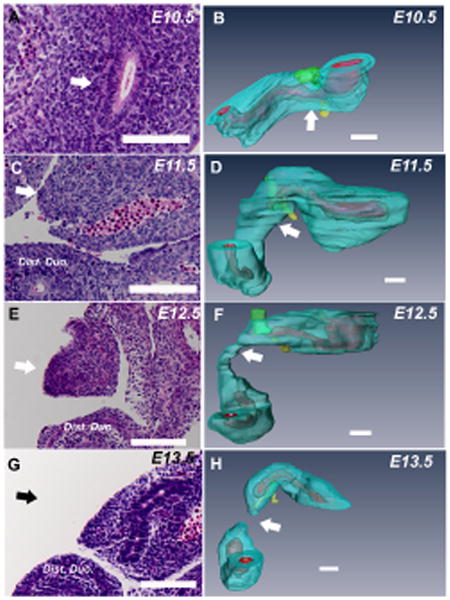
Transverse 7um sections of mouse duodenum taken through the atretic precursor (A, C, E and G) and corresponding three dimensional reconstructions (B, D, F and H). White arrows indicate the atretic precursor (AP) region in histological sections in A, C and E and the black arrow in G indicates the approximate area of where the AP had been at the previous time point. White arrows in B, D, F and H indicate location of the corresponding section in from A, C, E and G. Mesoderm is aqua blue, endoderm is red and the lumen is pink. Hepatic ducts are green and pancreatic ducts are yellow. Endoderm is absent in the duodenal atretic precursor region distal to the pancreatic buds by E11.5 (C and D). Involution of the mesoderm is evident at E12.5 (E and F) in this region and the defect goes to completion by E13.5 (G and H). (Bars indicate 100μM).
2.5 The atretic region of the duodenum is devoid of E-cadherin positive cells at E12.0 (CS17)
To confirm our observation that endodermal cells are absent in the atretic precursor after E11.5 and rule out the possibility that we had lost the ability to distinguished endodermal cells from mesodermal cells, we performed immunohistochemical staining of the duodenum for E-cadherin. E-cadherin is an epithelial specific marker. Within the intestine E-cadherin is only expressed by endodermal cells. No detectable E-cadherin staining was observed in the atretic precursor at E12.0 (Figure 5A, Blue arrow), whereas endodermal staining was preserved distal to this anatomic region (Figure 5B-D). This indicates an absence of endodermal cells in the duodenal atretic precursor of Fgfr2IIIb-/- mouse embryos at a stage when the human duodenum would have formed a solid core of endodermal tissue. In contrast E-cadherin positive cells were seen lining a continuous luminal structure throughout the duodenum proximal and distal to the C-loop in the wild-type control (5E-H).
Figure 5. E-cadherin positive cells are not detected within the duodenal atretic precursor at E12.0 (CS17).
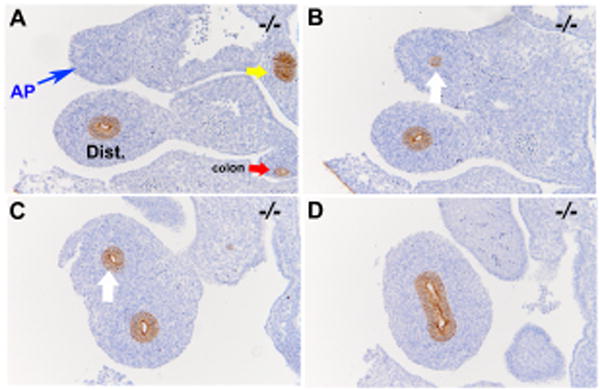
E-cadherin staining of the duodenal atretic precursor of an Fgfr2IIIb-/- embryo at E12.0 (A-D) and in a stage matched wild-type embryo (E-H). Images are organized in a rostral to caudal fashion. A. E-cadherin positive cells are absent in the atretic precursor (AP) region (blue arrow) and present in the region distal to this (B and C, white arrow) and in the C- loop of the duodenum (D). In contrast E-cadherin positive cells are seen lining a continuous luminal structure throughout the duodenum proximal and distal to the C-loop in the wild-type control (E-H). (Bars indicate 100μM). Yellow arrows indicate pancreatic duct tissue. Red arrow indicates colon. Pink arrow indicates pylorus. Dist. Indicates distal duodenum beyond the C-loop.
2.6 Fgfr2IIIb-/- embryos exhibit high levels of apoptosis in the proximal duodenal region at E10.5
It has previously been reported that formation of colonic atresia in Fgfr2IIIb-/- embryos is preceded by massive apoptosis in the endoderm at E10.5 [10, 11]. These results implicate endodermal apoptosis as a central event in atresia formation. To assess whether cell death occurs in the duodenal atretic precursor at E10.5 in Fgfr2IIIb-/- embryos, we performed TUNEL staining prior to the disappearance of the endodermal tube (Figure 6A). We observed robust (greater than 50%) TUNEL staining in the proximal duodenal endoderm of Fgfr2IIIb-/- embryos (Table 1). In contrast, there is almost no TUNEL staining (<2%) in wild-type embryos at this time point (Figure 6B) (Table 1). These results suggest that endodermal cell loss during duodenal atresia formation is due to apoptosis. We then examined Fgfr2IIIb-/- embryos at E11.5: the time-point of endodermal tube involution during atresia formation (Figure 6C). At E11.5, no TUNEL staining was observed. The transient nature of TUNEL staining suggests that subsequent mesodermal involution occurs via a mechanism independent of mesodermal apoptosis.
Figure 6. Loss of the endoderm in the atretic precursor of Fgfr2IIIb-/- embryos is preceded by robust endodermal apoptosis at E10.5 (CS 14). Subsequent mesodermal involution occurs in the absence of further apoptosis.
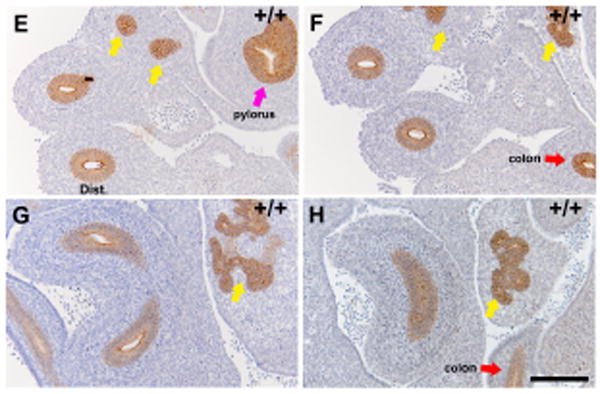
TUNEL staining of an A. Fgfr2IIIb-/- and B. Wild-type duodenum in the atretic precursor region at E10.5. TUNEL staining of the C. Fgfr2IIIb-/- duodenum at E11.5 in which the endodermal lumen of the atretic precursor is absent and D. Wild-type duodenum both at E11.5. White arrow indicates the atretic precursor region in mutants (A and C) and the corresponding region in wild-type embryos (B and D) (Bars indicate 100μM).
Table 1. Duodenal endodermal apoptosis at E10.5.
| Sample | Wild-type | Fgfrtlllb-/- |
|---|---|---|
| 1 | 1.62% | 64.96% |
| 2 | 1.52% | 53.31% | |
| 3 | 1.68% | 64.47% |
| Average | 1.61% | 60.98% |
| STD | 0.08% | 6.47% |
| p= | 0.0000459 |
Numbers indicate percentage of TUNEL staining cells/section (10 sections per sample)
3. Discussion
Our results indicate that during normal development, the murine duodenum does not undergo a period of luminal occlusion with an endodermal plug. Furthermore, our analysis demonstrates that Fgfr2IIIb-/- embryos experience a period of robust endodermal apoptosis early in development that precedes the disappearance of the endoderm in the atretic precursor at E11.5. Thereafter, involution of the mesodermal component of the atretic precursor appears to proceed in the absence of further apoptosis. The disparities in both timing and formation of atresia formation in this mouse model compared to normal human duodenal formation are illustrated as a developmental clock (Figure 7). These results do not support the original hypothesis of Julius Tandler: that atresias form as a result of a failure of the endoderm to recanalize its lumen after a period of vigorous proliferation.
Figure 7. Time line of normal and atretic duodenal development in Fgfr2IIIb-/- mice.
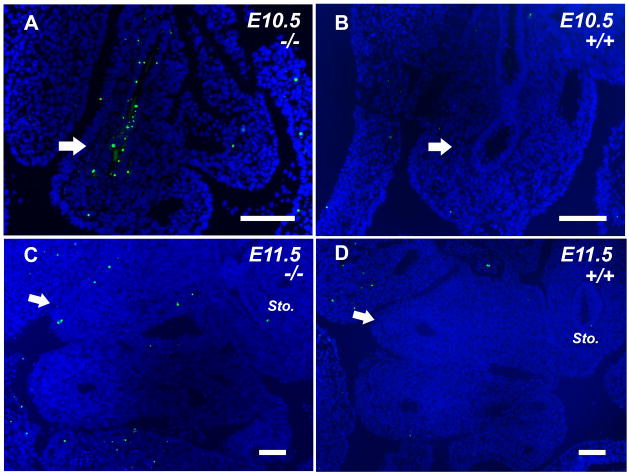
Based on our data we assembled a developmental clock for normal duodenal development in humans and mice and atretic duodenal development in mice. In humans, excessive proliferation of the endoderm becomes evident at CS 16. By CS17 the lumen is completely occluded and by CS 18 canals have formed within the endodermal plug. In the mouse, formation of a duodenal plug does not occur. By E14.5, which is equivalent of CS 23 in a human, a nearly occlusive plug has formed in the pyloric region. In contrast, during duodenal atresia formation, endodermal apoptosis is seen at E10.5 or the equivalent of CS14 in the human. By E11.5 or CS16, the endoderm in the atretic precursor is absent and involution of the mesoderm has begun. The involution of the mesoderm of the atretic precursor is complete by E13.5.
Extrapolating from our results it would appear that duodenal atresia in Fgfr2IIIb-/- embryos arise from loss of the endoderm during a critical phase of duodenal development. In the absence of viable endoderm, the atretic precursor mesoderm involutes by a mechanism other than cell death. Recent work by Nyeng et al. indicates that the loss of Fgf10 expression results in the premature differentiation of the progenitor niche in this region whereas over expression result in the expansion of this niche [12]. Together, these data suggest a more complex mechanism of duodenal atresia formation whereby a combination of endodermal cell death and terminal differentiation of progenitor cells results in formation of this defect. We have subsequently attempted to identify the endodermal lineages during the formation of duodenal atresia with a Pdx1-Cre [13] and a Pdx-1 specific antibody but have been unsuccessful in distinguishing pancreatic from duodenal endoderm during this temporal window.
It is unclear why loss of Fgfr2IIIb function results in such a discrete defect of the distal foregut as it is thought to be expressed throughout the endoderm of the entire intestine [14]. Fgf10, on the other hand, has a very restricted pattern of expression that is limited to the stomach, duodenum, cecum and proximal colon [14] at E10.5. Thus it is easy to understand why loss of Fgf10 expression can result in duodenal atresias. We are currently undertaking a temporal analysis of Fgfr2IIIb expression in an attempt to better understand why the loss of this receptor results in such focal defects.
Penetrance of duodenal atresia in Fgfr2IIIb-/- mouse embryo model is only 38 [2] to 42% [5]. This suggests that 58% of the time development proceeds normally. Our TUNEL studies demonstrate that apoptosis is occurring in the atretic precursor at E10.5. Previous studies of colonic atresia in the same model suggest that apoptosis is a mechanistically critical, early event in atresia formation [11]. It is unclear whether apoptosis occurs with equal intensity and anatomical distribution in the duodenums of all Fgfr2IIIb-/- embryos or if it is restricted to those embryos that will form and atresia, thus accounting for the partial penetrance of this defect. We are currently conducting studies to map apoptosis in the atretic precursor of Fgfr2IIIb-/- embryos using three-dimensional imagining software in an attempt to answer this question.
In summary, our results suggest an alternative mechanism of duodenal atresia formation in Fgfr2IIIb-/- embryos than previously proposed by Tandler [1]. In this model, duodenal atresia appears to result from a loss of endoderm, as opposed to the persistence of an endodermal plug. This represents a significant break from the previous hypothesis on the formation of this defect. Further work will be required to determine the signaling pathways involved in the formation of duodenal atresia in this animal model.
Acknowledgments
We would like to thank Drew Roenenberg for his assistance with preparing the E-cadherin stained slides and Dr. Silke Neiderhaus for her translation of Tandler's original publication, and Ms. Eden Tefera for her assistance with photomicrographs of histological sections. We also thank Dr. Richard Grose (Queen Mary University London, England) for kindly providing the Fgfr2IIIb c/c mice.
Funding PFN: Society for Surgery of the Alimentary Tract Career Development Award 2010-2012; American Pediatric Surgical Association Foundation Scholarship 2011-2012, NIH 2K08DK087854
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Robert Botham, Section of Pediatric Surgery, Department of Surgery, University of Wisconsin SMPH Madison, WI.
Marta Franco, Section of Pediatric Surgery, Department of Surgery, University of Wisconsin SMPH Madison, WI.
Amy Reeder, Section of Pediatric Surgery, Department of Surgery, University of Wisconsin SMPH Madison, WI.
Anastasia Lopukhin, Section of Pediatric Surgery, Department of Surgery, University of Wisconsin SMPH Madison, WI.
Kohei Shiota, Formerly: Director of the Congenital Anomaly Research Center and Professor and Chairman of the Department of Anatomy and Developmental Biology, Kyoto University Graduate School of Medicine, Kyoto, Japan. Currently: Executive Vice President of Kyoto University, Kyoto, Japan.
Shigehito Yamada, Director of the Congenital Anomaly Research Center and Professor and Chairman of the Department of Anatomy and Developmental Biology, Kyoto University Graduate School of Medicine, Kyoto, Japan.
Peter F. Nichol, Assistant Professor, Section of Pediatric Surgery, Department of Surgery, University of Wisconsin SMPH Madison, WI.
References
- 1.Tandler J. Zur Entwicklungsgeschichte des menschlichen Duodenum in fruhen Embryonalstadien. Morphol Jahrb. 1900;29:187. [Google Scholar]
- 2.Fairbanks TJ, Kanard R, Del Moral PM, et al. Fibroblast growth factor receptor 2 IIIb invalidation--a potential cause of familial duodenal atresia. J Pediatr Surg. 2004;39:872–874. doi: 10.1016/j.jpedsurg.2004.02.026. [DOI] [PubMed] [Google Scholar]
- 3.Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-3-reviews3005. REVIEWS3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Moerlooze L, Spencer-Dene B, Revest JM, et al. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development. 2000;127:483–492. doi: 10.1242/dev.127.3.483. [DOI] [PubMed] [Google Scholar]
- 5.Nichol PF, Botham RA, Saijoh Y, et al. A more efficient method to generate null mutants using Hprt-Cre with floxed alleles. J Pediatr Surg. 2011 doi: 10.1016/j.jpedsurg.2011.01.023. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O'Rahilly R. MF: Developmental stages in human embryos: including a revision of Streeter's “Horizons” and a survery of the Carnegie collection. Washington, D.C.: Carnegie Institution of Washington publication; 1987. [Google Scholar]
- 7.Nishimura H, Takano K, Tanimura T, et al. Normal and abnormal development of human embryos: first report of the analysis of 1,213 intact embryozs. Teratology. 1968;1:281–290. doi: 10.1002/tera.1420010306. [DOI] [PubMed] [Google Scholar]
- 8.Yamada S, Uwabe C, Nakatsu-Komatsu T, et al. Graphic and movie illustrations of human prenatal development and their application to embryologic education based on the human embryos specimens in the Kyoto Collection. Dev Dyn. 2006;235:468–477. doi: 10.1002/dvdy.20647. [DOI] [PubMed] [Google Scholar]
- 9.Grose R, Fantl V, Werner S, et al. The role of fibroblast growth factor receptor 2b in skin homeostasis and cancer development. EMBO J. 2007;26:1268–1278. doi: 10.1038/sj.emboj.7601583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fairbanks TJ, Kanard RC, Del Moral PM, et al. Colonic atresia without mesenteric vascular occlusion. The role of the fibroblast growth factor 10 signaling pathway. J Pediatr Surg. 2005;40:390–396. doi: 10.1016/j.jpedsurg.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 11.Fairbanks TJ, Sala FG, Kanard R, et al. The fibroblast growth factor pathway serves a regulatory role in proliferation and apoptosis in the pathogenesis of intestinal atresia. J Pediatr Surg. 2006;41:132–136. doi: 10.1016/j.jpedsurg.2005.10.054. discussion 132-136. [DOI] [PubMed] [Google Scholar]
- 12.Nyeng P, Bjerke MA, Norgaard GA, et al. Fibroblast growth factor 10 represses premature cell differentiation during establishment of the intestinal progenitor niche. Dev Biol. 2011;349:20–34. doi: 10.1016/j.ydbio.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gannon M, Herrera PL, Wright CV. Mosaic Cre-mediated recombination in pancreas using the pdx-1 enhancer/promoter. Genesis. 2000;26:143–144. doi: 10.1002/(sici)1526-968x(200002)26:2<143::aid-gene13>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 14.Burns RC, Fairbanks TJ, Sala F, et al. Requirement for fibroblast growth factor 10 or fibroblast growth factor receptor 2-IIIb signaling for cecal development in mouse. Dev Biol. 2004;265:61–74. doi: 10.1016/j.ydbio.2003.09.021. [DOI] [PubMed] [Google Scholar]