

Abstract
Very-Long-Chain Acyl-CoA Dehydrogenase deficiency (VLCADD) is an autosomal recessive disorder considered as one of the more common β-oxidation defects, possibly associated with neonatal cardiomyopathy, infantile hepatic coma, or adult-onset myopathy. Numerous gene missense mutations have been described in these VLCADD phenotypes, but only few of them have been structurally and functionally analyzed, and the molecular basis of disease variability is still poorly understood. To address this question, we first analyzed fourteen disease-causing amino acid changes using the recently described crystal structure of VLCAD. The predicted effects varied from the replacement of amino acid residues lining the substrate binding cavity, involved in holoenzyme-FAD interactions or in enzyme dimerisation, predicted to have severe functional consequences, up to amino acid substitutions outside key enzyme domains or lying on near enzyme surface, with predicted milder consequences. These data were combined with functional analysis of residual fatty acid oxidation (FAO) and VLCAD protein levels in patient cells harboring these mutations, before and after pharmacological stimulation by bezafibrate. Mutations identified as detrimental to the protein structure in the 3-D model were generally associated to profound FAO and VLCAD protein deficiencies in the patient cells, however, some mutations affecting FAD binding or monomer-monomer interactions allowed a partial response to bezafibrate. On the other hand, bezafibrate restored near-normal FAO rates in some mutations predicted to have milder consequences on enzyme structure. Overall, combination of structural, biochemical, and pharmacological analysis allowed to assess the relative severity of individual mutations, with possible applications for disease management and therapeutic approach.
Keywords: fatty acid β-oxidation, Very-Long-Chain-AcylCoA Dehydrogenase, 3-Dmodeling, structure-function analysis, VLCAD deficiency, pharmacological therapy
INTRODUCTION
The mitochondrial fatty acid β-oxidation represents a major source of energy in a number of tissues including liver, heart, and skeletal muscle, and, accordingly, genetic defects in this metabolic pathway are often associated with complex and severe clinical manifestations. Very-Long-Chain-AcylCoA Dehydrogenase (VLCAD), one of the five acyl-CoA dehydrogenases (ACAD) that catalyze the first step of mitochondrial fatty acid β-oxidation (FAO), plays a prominent role in the use of long-chain fatty acids as energy substrates in human [1]. VLCAD deficiency is an autosomal recessive disorder considered as one of the more common FAO defect, with an estimated incidence of about 1/31500 births [2]. It is characterized by three major phenotypes: a generally fatal form with cardiomyopathy and hepatic failure in the neonate, an intermediate infantile presentation with liver insufficiency, and a mild adolescent onset myopathic form [3]. In addition to clinically ill patients, asymptomatic VLCAD-deficient newborns have now been characterized, as a result of newborn screening programs for this disorder [4].
The molecular basis of VLCAD deficiency is complex, with about one hundred mutations described [5]. Mutations of null type, which result in complete loss of enzymatic function, are always associated to severe disease presentation [3, 6]. However, the majority of diagnosed patients, and all asymptomatic newborns so far, harbor missense mutations in the VLCAD gene. Despite marked progress in characterization of this disease, VLCAD deficiency is still considered as a relatively insidious disease because of its variable phenotype, and because of the lack of clear phenotype-genotype correlations for missense mutations [2]. Functional analysis in various expression systems can provide valuable data on the cellular effects and severity of individual missense mutations. However, this approach is too complex to be applied to the whole panel of missense mutations presently known in this disease. In line with this, there is a direct clinical need to define approaches for analysis of individual VLCAD missense mutations, in order to improve management of diagnosed patients and to aid the prognosis and treatment of VLCAD-deficient asymptomatic newborns [2].
To address this question, we evaluated an approach based on combining predictive analysis of mutation effects in a 3-D VLCAD model, and biochemical analysis of patient cells using relatively widespread methods. The crystal structure of VLCAD has recently been elucidated, and the 3-dimentional model shows that the enzyme forms a homodimer associated with the inner mitochondrial membrane, in contrast with the majority of ACADs, which are soluble homotetrameric proteins [7]. The N-terminal domain of VLCAD (~400 amino acid residues) presents a high sequence identity and homology with MCAD, and has the same polypeptide fold as the soluble ACADs. On the other hand, the C-terminal domain composed of about 180 residues is unique to VLCAD, and forms a helical bundle that interacts with the N-terminal domain in a similar manner as the dimer-dimer interaction in the tetrameric ACADs. In the C-terminal domain, it is proposed that the fragment including residues 485 to 519, which is disordered in the crystal structure, forms an amphipathic helix with six positively charged residues, responsible for anchoring of VLCAD by interacting with negatively charged lipid head groups in the inner mitochondrial membrane [7]. Glu462 is the catalytic residue in VLCAD [8] and is conserved in several members of ACAD family [9].
In our study, fourteen disease-causing amino acid substitutions were reported in the 3-D VLCAD model, with the aim to predict their possible deleterious effects on structure and function of the enzyme. In parallel, biochemical studies of VLCAD protein levels and β-oxidation flux were performed in patient cells harboring these mutations, before and after treatment by bezafibrate. Indeed, we recently showed that treatment by bezafibrate efficiently reveals maximal metabolic capacities in VLCAD-deficient patient cells, in contrast to basal measurements performed in the absence of pharmacological stimulation [10]. We thus analyzed the correlations between predictions of mutation severity from the 3-D model, and residual FAO capacities and protein levels in patient cells, allowing a comparative analysis ofindividual mutation effects in our panel of missense mutations.
MATERIALS and METHODS
Patient fibroblasts
VLCAD-deficient human skin fibroblasts used for this study have been described previously [10]. Mutations and genotypes harbored in these cell lines are mentioned in Table 1. Patients were either homozygous for a missense mutation (N122D, V174M, G222R, R453Q), or carried a missense mutation (G185S, T260M, R286G, R366H, K382Q, A416T, G441D) associated to a null mutation on the other allele, or were compound heterozygous for two missense mutations (A304T/G439D, D405H/R450H). Fibroblasts were grown in standard Ham-F10 medium (Invitrogen, Cergy-Pontoise, France) with glutamine, 12% fetal bovine serum and 100 U/ml penicillin and 0.1 mg/ml streptomycin under standard conditions, as previously described [11]. For treatment, cells were incubated in fresh media containing 400 μM bezafibrate (Sigma) or the equivalent amount of DMSO (vehicle, 0.05%) for 48h.
Table 1.
VLCAD patient genotypes and corresponding FAO flux.
| Groups | Amino acid change | Nucleotide change | FAO flux | Fold-increase | |||
|---|---|---|---|---|---|---|---|
| Allele 1 | Allele 2 | Non treated | Treated | FAO-flux | Protein level | ||
| Control | 4.8 ± 1.0 | 7.6 ± 0.35 | 1.6 | 2 | |||
| A | G185S/N252_H293del42 | c.553G>A | IVS8-2A>C | 0.24 ± 0.02 | 0.41 ± 0.05 | 1.7 | 7 |
| A | G222R/G222R | c.664G>A | c.664G>A | 0.46 ± 0.02 | 0.81 ± 0.09 | 1.8 | 5.6 |
| A | R366H/R453X | c.1097G>A | c.1358G>T | 0.71 ± 0.1 | 1.85 ± 0.3 | 2.6 | 10 |
| A | K382Q/E130fsX216 | c.1144A>C | c.388_391del4 | 0.60 ± 0.17 | 2.27 ± 0.33 | 3.8 | 9 |
| A | G441D/N252_H293del42 | c.1322G>A | IVS8-2A>C | 0.26 ± 0.15 | 0.50 ± 0.37 | 1.9 | 7 |
| A | R453Q/R453Q | c.1358G>A | c.1358G>A | 0.24 ± 0.01 | 0.23 ± 0.01 | 1 | NA |
| B | N122D/N122D | c.364A>G | c.364A>G | 0.32 ± 0.04 | 0.21 ± 0.3 | 1 | 1 |
| B | T260M/A640fsX679 | c.779C>T | c.1918_1921del4 | 0.32 ± 0.15 | 0.54 ± 0.37 | 1.7 | 1 |
| B | R286G/Q13X | c.856A>G | c.37C>T | 0.7 ± 0.01 | 1.28 ± 0.13 | 1.8 | 1 |
| C | V174M/V174M | c.520G>A | c.520G>A | 1.29 ± 0.07 | 4.14 ± 0.28 | 3.2 | 2.9 |
| C | A416T/K600fsX679 | c.1246G>A | c.1798delA | 1.49 ± 0.54 | 2.7 ± 0.75 | 1.8 | 2.3 |
| C/A | A304T/G439D | c.910G>A | c.1316G>A | 1.43 ± 0.18 | 3.30 ± 0.1 | 2.3 | 3 |
| C/C | D405H/R450H | c.1213G>C | c.1349G>A | 1.90 ± 0.7 | 3.35 ± 1 | 1.8 | 2.4 |
A to C corresponds to the predicted effects of disease-causing missense mutations from in silico analysis. A: missense mutations affecting substrate- or FAD-binding, or monomer-monomer interactions, B: missense mutations inducing major tertiary structure re-arrangements outside catalytic or dimerization domains, C: missense mutations with minor structural consequences.
The numbering of 1/nucleotides starts at the first adenine of the translation initiation codon and 2/amino acids starts at the first methionine encoded by the translation initiation codon. FAO (Fatty Acid Oxidation) flux was measured in nmol 3H FA / h / mg protein from deficient patient fibroblasts, before and after bezafibrate treatment (400 μM, 48h).
NA: cells not available
Fatty acid oxidation (FAO)
FAO flux was determined by quantifying the production of 3H2O from (9,10-3H) palmitate(Perkin Elmer), as previously described [12]. Palmitate bound to fatty acid-free albumin wasused at the final concentration of 125 μM (60 Ci/mmol). For each cell line, FAOmeasurements were performed in triplicate, and repeated in two to three independent experiments. The oxidation rates were expressed as nmol of [3H] fatty acid oxidized per hour per mg of cell protein (nmol [3H] FA h−1. mg−1 protein).
Western blot analysis
Control and VLCAD fibroblasts were harvested in RIPA buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1%NP40, 0.25% sodium deoxycholate, 0.1% SDS, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 1 mM phenylmethylsulfonyl fluoride) to prepare protein extracts. 20 μg of total protein per lane were resolved by 10% SDS-PAGE and transferred to Hybond-P PVDF membrane (Amersham Biosciences, Freiburg, Germany). The following antibodies were used: rabbit polyclonal anti-VLCAD (kindly provided by Dr S. Yamagushi, Japan), and mouse monoclonal anti-actin (Chemicon International, Temecula, USA). The primary antibodies were used at a dilution of 1/2000 and 1/15000 respectively, and detected using horseradish peroxidase (HRP) conjugated anti-rabbit or anti-mouse IgG, and the chemiluminescent reagent, ECL (Amersham Biosciences, Freiburg, Germany). Immunoreactive bands corresponding to the two proteins were scanned by densitometry with a computerized video densitometer.
In Silico Mutational Analysis
Selected mutations were analyzed based on the recently determined structure of VLCAD [7]. Specific mutations were introduced to the wild-type structure (PDB ID, 3B96) in silico using the model-building program COOT [13]. When applicable, the most appropriate rotamer was chosen to fit the space and surrounding for the mutation site, maximizing hydrogen bonding, ionic, and/or hydrophobic interactions. The resulting structure was compared with the wild-type for assessment of the protein integrity, including cofactor binding, active site configuration, and protein stability. All figures were generated using the graphic program PyMOL [14]. The location of analyzed mutations in the VLCAD 3-D structure is depicted in Figure 1.
Figure 1. Ribbon diagram of the human VLCAD dimer (PDB 3B96) with various mutation sites.

Each subunit is represented in gray or blue. Stick representations of the FAD cofactors have CPK coloring with yellow carbons. Mutation sites are shown in only one subunit.
RESULTS
The 3-D analysis revealed that six of the mutations considered in our study, namely G185S, G222R, R366H, K382Q, G441D, and R453Q could induce major structural alterations in substrate-binding, FAD- binding, or in enzyme monomer-monomer interactions (figure 1). The G185S mutation is predicted to affect the conformation of substrate binding cavity. Indeed, the conserved Gly185 residue lies in a very hydrophobic pocket surrounded by Ile176, Leu178, and Ile184, all of which are lining the acyl-CoA binding cavity. Furthermore, homolog of Ile184 in the MCAD (Leu103) or SCAD structure (Leu98) plays a role in the fatty acyl moiety binding [15, 16]. Accordingly, introduction of a polar serine residue in position 185 (G185S) could directly affect the substrate binding. The R453Q substitution is another example of mutation that might affect substrate binding and catalysis, by inducing changes in the positioning of catalytic glutamate (Glu462, see figure 2A). Interestingly, mutation of the equivalent residue in the SCAD enzyme (R359C) was also shown to be disease-causing in human, and cDNA expression studies of this SCAD mutation suggested major protein folding alterations leading to aggregation of the mutated protein [17]. Gly222 is the first residue of a conserved Gly-Ser-Asp (GSD) segment located at the loop between strands 1 and 2 (see figure 3A), which was shown to be directly involved in the binding of FAD in both MCAD and SCAD [15, 16, 18]. Detailed analysis of G222R mutation effects is provided in figure 3A. Gly441 is also part of a highly conserved motif (Gly-Gly-x-Gly) close to the FAD binding pocket [19]. Substitution of Gly441 for aspartate could affect both FAD binding and monomer-monomer interaction (see figure 3B). The conserved residue Arg366 participates to monomer-monomer interactions through making salt bridges with the FAD pyrophosphate group of the neighboring subunit (see figure 3C). When mutated to histidine (R366H), these salt bridges would be replaced by hydrogen bonds, weakening FAD binding and the association between enzyme monomers (see figure 3C). K382Q was also predicted to affect FAD binding. Indeed, Lys382 located on helix H makes a salt bridge with Glu432 on helix I (figure 3D). The K to Q substitution would break this salt bridge and affect the position of helix I, which could negatively affect FAD binding (figure 3D).
Figure 2. Comparison of mild and severe VLCAD human mutation sites.

Wild-type amino acid residues (magenta) are overlayed with the corresponding residues in the clinical variant (cyan). Potential steric hindrances are depicted with red dashes. The cartoon carbon backbone representation is colored either grey or blue depending on the location of the mutation site in Figure 1. A) The severe mutation R453Q would most certainly directly affect substrate binding and catalysis. The arginine to glutamine mutation would break a salt bridge between residue Arg453 on helix J and Asp123 on helix A. The positioning of the catalytic glutamate (Glu462, not shown in the Figure), which is on a loop between helices J and K, would likely be affected by the loss of this salt bridge. B) The severe clinical variant R286G is also not near FAD or substrate binding. However, the mutation would lead to the breaking of hydrogen bonds with the main chain carbonyl oxygens of Glu253 and Ala256. This would lead to major tertiary structural rearrangement and protein instability. C) V174M (mild) mutation site. No hydrogen bonds would be broken by this mutation. However, potential steric hindrances with Thr132 and Leu136 would lead to some structural instability. D) The A304T (mild) mutation site also results potentially in only minor steric hindrance. The distal location of this mutation from FAD and substrate binding combined with only slight structural perturbation result in a mild clinical phenotype.
Figure 3. Three dimensional dimeric structure of the VLCAD: ribbon diagram (PDB 3B96) with mutation sites.

The two monomers are colored gray and blue. Helices and beta-strands are labeled as described [7] and those in the blue monomer are labeled with asterisks Stick representations of the FAD cofactors have CPK coloring with yellow carbons. The boxes indicate mutation sites associated with FAD binding, whose vicinities are shown in greater detail in panels A–D. In each panel, amino acid residues that are mutated in patients are shown with CPK coloring with magenta carbons and are labeled in magenta. Other neighboring residues depicted with CPK coloring grey carbons and labeled in black. Hydrogen bonds and salt bridges are marked with dotted lines. A) G222R mutation site. Gly222 is located on the loop between β-strands 1 and 2 close to the FAD phosphate and thesubstitution for a bulky charged arginine may change the loop conformation, and disrupt binding of the cofactor, including a hydrogen bond between the FAD pyrophosphate and the neighboring residue Ser223. B) G439D and G441D mutation sites are part of a conserved Gly-Gly-x-Gly motif on the loop between helices I and J of neighboring monomer in the VLCAD dimeric molecule. Again, substitution of either Gly to an acidic residue would be detrimental to the interaction with the acidic pyrophosphate moiety of FAD in the neighboring monomer, resulting in a weakening of the dimer interaction. C) R366H mutation site. Arg366 forms salt bridges with the FAD pyrophosphate of the other monomer. Substitution of positively charged arginine 366 for a neutral histidine could lose this ionic interaction, instead forming a hydrogen bond between histidine 366 and the pyrophosphate, weakening FAD binding and the association between enzyme monomers. D) K382Q mutation site. Lys382 on helix H forms a salt bridge with Glu432 on helix I, and another salt bridge with Asp466 on helix K of the other monomer, which hydrogen bonds with the adenosine 2′-OH of the FAD. Since glutamine is almost isosteric as lysine, Gln382 can make hydrogen bonds with both Glu432 and Asp466, which are much weaker interactions compared to the salt bridges. The VLCAD catalytic glutamate (Glu462) is proximal to the site of this salt bridge, and thereforethe K382Q mutation may also affect its positioning
Fibroblasts harboring one of the aforementioned mutations (genotypes G185S/N252_H293del42, G222R/G222R, R366H/R453X, K382Q/E130fsX216 and G441D/N252_H293del42, R453Q/R453Q) exhibited a marked FAO deficiency under basal conditions, with residual palmitate oxidation rates ranging from 0.24 to 0.71 nmol 3H. FA h−1. mg−1 protein, versus 4.8±1.0 nmol 3H. FA h−1. mg−1 protein in control cells (i.e. 85 to 95% FAO deficiency in the patient cells) (Table 1). After treatment by bezafibrate, cells harboring G185S, G222R, or G441D exhibited quite limited changes in FAO capacities, and therefore remained extremely FAO-deficient (0.41 to 0.81 nmol 3H. FA h−1. mg−1 protein) (Table 1). In contrast, bezafibrate induced a significant FAO increase in cells harboring R366H (from 0.71 to 1.85 nmol 3H. FA h−1. mg−1 protein) or K382Q (from 0.6 to 2.27 nmol 3H. FA h−1. mg−1 protein) (Table 1).
Western-blot analysis revealed barely detectable VLCAD protein levels in untreated cells harboring G185S, G222R, R366H, K382Q, or G441D (figure 4). Exposure to bezafibrate resulted in an induction of VLCAD protein for G185S (7-fold), G222R (5.6-fold), and G441D (7-fold), however the absolute levels remained considerably lower than those found in control cells. On the other hand, residual VLCAD protein was induced ten to nine-fold in patient cells harboring R366H or K382Q in response to bezafibrate, resulting in relatively high protein levels compared to other missense mutations in this group.
Figure 4. VLCAD protein levels in untreated (DMSO) or bezafibrate-treated (BZ) patient fibroblasts.
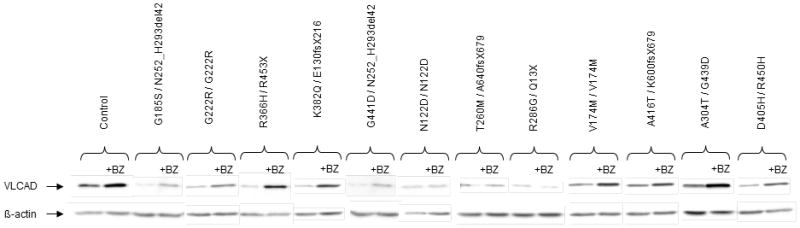
Western-blot of protein extracts from patient or control cells were incubated with anti-VLCAD antibody, and re-probed with anti-β-actin antibody.
3-D analysis also pointed to amino acid substitutions like T260M, N122D, or R286G that do not directly affect catalytic enzyme domains, but were predicted to disrupt local hydrogen bond networks and to generate strong repulsive forces, leading to major tertiary structure rearrangement and protein instability (figure 1). Thus, substitution of conserved Thr260 for a bulky hydrophobic methionine (T260M) induces steric hindrance and disruption of the hydrogen bond normally formed between Thr260 and the main chain amide nitrogen of Gly185. Similarly, replacement of Asn122 by a negatively charged aspartate (N122D) could disrupt hydrogen bonds formed between Asn122 and three other residues, namely Asp171, Asn117, and Glu129, and also cause strong charge repulsion among the three carboxylate groups of these residues, predicted to induce severe protein instability. Finally, R286G is another example of a severe variant targeting a residue outside catalytic domains, leading to disrupt local hydrogen bonds (see figure 2B).
In keeping with structural analysis, FAO studies in cells carrying N122D (N122D homozygous), or T260M (T260M/null), revealed very low levels under basal conditions (0.32 nmol [3H]FA h−1. mg−1 protein, i.e. FAO deficiency >93% compared to control cells), which did not vary in response to bezafibrate (Table 1). Fibroblasts harboring R286G/Q13X showed slightly higher FAO rates, but remained markedly deficient when treated by bezafibrate. In parallel, western-blot studies revealed very low protein levels in patient cells, which remained unchanged after treatment by bezafibrate (Figure 4).
Two missense mutations, V174M and A416T, were predicted to have relatively minor consequences. Indeed, V174M appears as a mild mutation since introduction of a methionine would not break existing hydrogen bonds (see figure 2C). Patient fibroblasts homozygous for V174M exhibited FAO capacities equivalent to 27% of normal level (1.29 nmol 3H. FA h−1. mg−1 protein) under basal conditions, which markedly increased after bezafibrate treatment (4.14 nmol [3H]FA h−1. mg−1 protein, i.e. 86% of normal level) (Table 1). A416T also appears as a mild mutation since Ala416 is directly across from a phenylalanine on helix K, which forms a loop around the catalytic Glu462, however, alanine substitution for a threonine, with a slightly larger polar side chain, is predicted to induce minor changes in the folding and positioning of helix K. Patient cells harboring A416T exhibited palmitate oxidation rate reaching 31% of control values under basal conditions (1.49 nmol [3H]FA h−1. mg−1 protein), and 56% (2.7 nmol [3H]FA h−1. mg−1 protein) after bezafibrate treatment (Table 1). Western-blot studies showed that cells carrying the V174M or A416T mutation already exhibited significant VLCAD protein levels under basal conditions. These were clearly up-regulated (2.9- or 2.3-fold, respectively) in bezafibrate-treated cells, resulting in VLCAD protein levels close to those observed in control cells (figure 4).
We included in this study compound heterozygous genotypes carrying two missense mutations, which could not be individually analyzed on the basis of functional data. Thus, the genotype A304T/G439D, which exhibited residual FAO close to 30% of control values under basal conditions, raising to 65% after bezafibrate treatment (Table 1), might represent the association of two mild mutations or, alternatively, of a mild and of a severe one. 3-D analysis showed that the highly conserved Gly439 is involved in the same Gly-Gly-x-Gly motif previously discussed in the case of the severe G441D mutation (see figure 3B). In line with this, G439D substitution, which also led to the substitution of a small apolar glycine for a bulky charged aspartate, could alter both FAD binding and dimer formation. Accordingly, the associated A304T mutation is likely responsible for the expression of significant residual FAO flux. In fact, in the 3-D model, residue Ala304 is located in a hydrophobic region in the loop between strands 6 and 7 (see figure 2D), and introduction of a hydrophilic threonine at this position would decrease protein stability without major changes in the enzyme structure. Western-blot analysis revealed relatively high VLCAD protein levels in untreated A304T/G439D cells, increasing (3-fold) after bezafibrate treatment. In contrast, the genotype D405H/R450H most likely associates two mild mutations. Indeed, previous studies using transient expression in SV-40 fibroblasts demonstrated that R450H is a mild temperature-sensitive mutation [20]. Furthermore, modeling of D405H showed that the conserved Asp405 is located on the surface of the molecule and that substitution to a hydrophilic His would probably have mild consequences on protein structure and stability. In this genotype, FAO levels were significantly increased after bezafibrate treatment. VLCAD protein abundance was clearly lower than normal in the D405H/R450H genotype under basal conditions, but protein levels increased 2.4 fold in response to bezafibrate.
DISCUSSION
Progress in the diagnosis of VLCAD deficiency led to the identification of a large number of disease-causing missense mutations, and raised the question of elucidation of the molecular and cellular effects of these various mutations. In our study, we used the recently published crystal structure of VLCAD to perform 3-D mapping of fourteen missense mutations, in order to analyze their potential deleterious effects on architecture of the active site, enzyme-cofactor interaction, enzyme dimerisation, or folding of polypeptide chain. One of our objectives was to evaluate the level of correlation between information gathered from 3-D analysis and biochemical data obtained from patient cells harboring these mutations. Six of the mutations studied (G185S, G222R, R366H, K382Q, G441D, and R453Q) were predicted to induce major conformational changes in enzyme domains structurally essential for catalysis. Fibroblasts harboring one of the three glycine substitutions (G185S, G222R, G441D) all exhibited barely detectable FAO levels and were unresponsive to bezafibrate, in good correlation with the 3-D mapping conclusions. Patient cells with the K382Q or R366H mutations exhibited detectable FAO flux after bezafibrate treatment, though much lower than normal values. Interestingly, structural analysis predicted that K382Q or R366H mutations led to substitute salt bridge interactions by hydrogen bonds, consistent with lesser consequences on enzyme structure and stability. In contrast, substitutions of glycine residues lead to disrupt existing bonds with the FAD group, and to create strong repulsive forces, and, accordingly, are predicted to induce loss-of-function and protein misfolding, especially if the amino acid is buried deep inside the normal structure. Misfolded proteins due to severe missense mutations are often degraded by the cellular protein quality system [5], resulting in virtually no immunodetectable enzyme in the patient cells. Consistent with this, missense mutations predicted to induce major structural rearrangements (groups A and B) were all associated to drastically reduced VLCAD protein levels under standard conditions. Interestingly, mutant protein levels were found up-regulated by bezafibrate for some of these severe mutations, particularly in the case of R366H and K382Q, and this was associated with relatively high FAO levels in drug-treated cells, compared to other severe mutations.
The other eight mutations considered in our study did not affect residues lying in enzyme domains essential for catalysis or monomer-monomer association. More detailed analysis in the 3-D model predicted that three of them could alter local bonding networks by abolishing one (T260M), two (R286G), or three (N122D) hydrogen bonds with nearby amino acids, in addition to causing steric hindrance and strong charge repulsion. Altogether, 3-D analysis clearly supports the hypothesis that these mutations could induce major protein instability. Consistent with this, cells harboring N122D, T260M, or R286 G showed extremely low residual VLCAD protein levels and were found severely FAO-deficient, whether or not treated by bezafibrate. Analysis of four other missense mutations (V174M, A304T, D405H, A416T) pointed to residues that were not engaged in charge-charge interactions or hydrogen bonding networks with other amino acids in the 3-D structure of the enzyme. This is illustrated by V174M or A416T, involving residues that take part to hydrophobic interactions outside key domains, or near the surface, of the protein. Accordingly, these mutations can be predicted to have relatively milder structural consequences on protein folding. In line with this, fibroblasts carrying V174M or A416T exhibited significant amounts of protein and FAO levels at the basal state, which were markedly increased in response to bezafibrate. In the case of V174M, the high residual protein and FAO capacities might also be due to the fact that this patient cell line carries two V174M alleles, whereas most other cell lines harbor one missense mutation associated to a null allele.
Altogether, this study shows that analysis of individual mutations by 3-D mapping in the enzyme crystal structure can provide initial indications on mutation effects that appear consistent with functional data obtained from patient cells. Although structural analysis can obviously not predict the importance of all interactions involved in the mature protein, our data suggest that 3-D mapping can help to distinguish between mutations that are likely to severely affect key structural elements of the enzyme, thereby inducing complete loss-of-function, and other types of mutations targeting non-critical domains or non-critical interaction networks, with milder functional impact. On the other hand, the fatty acid oxidation assay, which can easily be applied to serial studies of patient cells, provides a valuable tool for analysis of biochemical phenotype when predictions of mutation severity could not be firmly established by in silico analysis. In addition, this study further illustrates the potential of FAO measurements to evaluate the response to bezafibrate in various genotypes, and the correlation between mutation severity and drug-induced changes in FAO in the patient cells [10].
Our results are in good agreement with the literature data on analysis of T260M, K382Q, and A416T, which are the only missense mutations previously analyzed in functional studies, among the panel included in our study. In particular, these published data showed that expression of cDNA encoding K382Q or A416T resulted in significant residual enzyme activity [21, 22], whereas very low levels were observed in the case of T260M [23]. It is worth mentioning that combination of structural, biochemical and pharmacological analysis, as performed in the present study allowed not only to analyze the relative severity of individual missense mutations, but also to assess the capacity of patient cells to up-regulate their FAO capacity under bezafibrate stimulation. This approach could therefore bring relevant information to aid prognosis and to consider therapeutic options in clinically affected or asymptomatic individuals.
Finally, analysis of structural consequences of missense mutations could also be helpful to provide rationale for the research of new treatments. For example, it can be proposed that riboflavin supplementation might further improve VLCAD residual enzyme activity in the case of mutations affecting the FAD binding, but which do not fully abolish residual FAO, such as R366H or K382Q. In line with this, it has been recently reported that pharmacological doses of riboflavin could improve patient condition in the mitochondrial electron transfer flavoprotein-ubiquinone oxidoreductase (ETF-QO) deficiency, suggesting that increased FAD supply to the mitochondria might reduce the impact of mutations affecting its binding [24, 25]. Additionally, therapeutic approaches based on chaperones, already tested in other genetic diseases [26], could be relevant in the case of VLCAD missense mutations which do not irreversibly affect the expression of mutated enzyme (R366H, K382Q, V174M, A416T). Accordingly, analysis of the consequences of the mutations at the structural and biochemical level could therefore provide important clues not only to improve patient management, but also for the research of future treatments.
Acknowledgments
This work was supported by a grant from the Association Francaise contre les Myopathies. We thank Toshiyuki Fukao and Ronald Wanders for providing some cell lines.
References
- 1.Aoyama T, Souri M, Ushikubo S, Kamijo T, Yamaguchi S, Kelley RI, Rhead WJ, Uetake K, Tanaka K, Hashimoto T. Purification of human very-long-chain acyl-coenzyme A dehydrogenase and characterization of its deficiency in seven patients. J Clin Invest. 1995;95:2465–73. doi: 10.1172/JCI117947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arnold GL, Van Hove J, Freedenberg D, Strauss A, Longo N, Burton B, Garganta C, Ficicioglu C, Cederbaum S, Harding C, Boles RG, Matern D, Chakraborty P, Feigenbaum A. A Delphi clinical practice protocol for the management of very long chain acyl-CoA dehydrogenase deficiency. Mol Genet Metab. 2009;96:85–90. doi: 10.1016/j.ymgme.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andresen BS, Olpin S, Poorthuis BJ, Scholte HR, Vianey-Saban C, Wanders R, Ijlst L, Morris A, Pourfarzam M, Bartlett K, Baumgartner ER, deKlerk JB, Schroeder LD, Corydon TJ, Lund H, Winter V, Bross P, Bolund L, Gregersen N. Clear correlation of genotype with disease phenotype in very-long-chain acyl-CoA dehydrogenase deficiency. Am J Hum Genet. 1999;64:479–94. doi: 10.1086/302261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liebig M, Schymik I, Mueller M, Wendel U, Mayatepek E, Ruiter J, Strauss AW, Wanders RJ, Spiekerkoetter U. Neonatal screening for very long-chain acyl-coA dehydrogenase deficiency: enzymatic and molecular evaluation of neonates with elevated C14:1-carnitine levels. Pediatrics. 2006;118:1065–9. doi: 10.1542/peds.2006-0666. [DOI] [PubMed] [Google Scholar]
- 5.Gregersen N, Andresen BS, Pedersen CB, Olsen RK, Corydon TJ, Bross P. Mitochondrial fatty acid oxidation defects--remaining challenges. J Inherit Metab Dis. 2008;31:643–57. doi: 10.1007/s10545-008-0990-y. [DOI] [PubMed] [Google Scholar]
- 6.Gregersen N, Andresen BS, Corydon MJ, Corydon TJ, Olsen RK, Bolund P, Bross L. Mutation analysis in mitochondrial fatty acid oxidation defects: Exemplified by acyl-CoA dehydrogenase deficiencies, with special focus on genotype-phenotyperelationship. Hum Mutat. 2001;18:169–89. doi: 10.1002/humu.1174. [DOI] [PubMed] [Google Scholar]
- 7.McAndrew RP, Wang Y, Mohsen AW, He M, Vockley J, Kim JJ. Structural basis for substrate fatty acyl chain specificity: crystal structure of human very-long-chain acyl-CoA dehydrogenase. J Biol Chem. 2008;283:9435–43. doi: 10.1074/jbc.M709135200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Souri M, Aoyama T, Cox GF, Hashimoto T. Catalytic and FAD-binding residues of mitochondrial very long chain acyl-coenzyme A dehydrogenase. J Biol Chem. 1998;273:4227–31. doi: 10.1074/jbc.273.7.4227. [DOI] [PubMed] [Google Scholar]
- 9.Kim JJ, Miura R. Acyl-CoA dehydrogenases and acyl-CoA oxidases. Structural basis for mechanistic similarities and differences. Eur J Biochem. 2004;271:483–93. doi: 10.1046/j.1432-1033.2003.03948.x. [DOI] [PubMed] [Google Scholar]
- 10.Gobin-Limballe S, Djouadi F, Aubey F, Olpin S, Andresen BS, Yamaguchi S, Mandel H, Fukao T, Ruiter JP, Wanders RJ, McAndrew R, Kim JJ, Bastin J. Genetic basis for correction of very-long-chain acyl-coenzyme A dehydrogenase deficiency by bezafibrate in patient fibroblasts: toward a genotype-based therapy. Am J Hum Genet. 2007;81:1133–43. doi: 10.1086/522375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Djouadi F, Bonnefont JP, Thuillier L, Droin V, Khadom N, Munnich A, Bastin J. Correction of fatty acid oxidation in carnitine palmitoyl transferase 2-deficient cultured skin fibroblasts by bezafibrate. Pediatr Res. 2003;54:446–51. doi: 10.1203/01.PDR.0000083001.91588.BB. [DOI] [PubMed] [Google Scholar]
- 12.Manning NJ, Olpin SE, Pollitt RJ, Webley J. A comparison of [9,10–3H]palmitic and [9,10–3H]myristic acids for the detection of defects of fatty acid oxidation in intact cultured fibroblasts. J Inherit Metab Dis. 1990;13:58–68. doi: 10.1007/BF01799333. [DOI] [PubMed] [Google Scholar]
- 13.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 14.The PyMOL reference Manuel. DeLano Scientific LLC; San Carlos, CA, USA: 2004. [Google Scholar]
- 15.Kim JJ, Wang M, Paschke R. Crystal structures of medium-chain acyl-CoA dehydrogenase from pig liver mitochondria with and without substrate. Proc Natl Acad Sci U S A. 1993;90:7523–7. doi: 10.1073/pnas.90.16.7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Battaile KP, Molin-Case J, Paschke R, Wang M, Bennett D, Vockley J, Kim JJ. Crystal structure of rat short chain acyl-CoA dehydrogenase complexed with acetoacetyl-CoA: comparison with other acyl-CoA dehydrogenases. J Biol Chem. 2002;277:12200–7. doi: 10.1074/jbc.M111296200. [DOI] [PubMed] [Google Scholar]
- 17.Pedersen CB, Bross P, Winter VS, Corydon TJ, Bolund L, Bartlett K, Vockley J, Gregersen N. Misfolding, degradation, and aggregation of variant proteins. The molecular pathogenesis of short chain acyl-CoA dehydrogenase (SCAD) deficiency. J Biol Chem. 2003;278:47449–58. doi: 10.1074/jbc.M309514200. [DOI] [PubMed] [Google Scholar]
- 18.Nasser I, Mohsen AW, Jelesarov I, Vockley J, Macheroux P, Ghisla S. Thermal unfolding of medium-chain acyl-CoA dehydrogenase and iso(3)valeryl-CoA dehydrogenase: study of the effect of genetic defects on enzyme stability. Biochim Biophys Acta. 2004;1690:22–32. doi: 10.1016/j.bbadis.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 19.Andresen BS, Bross P, Vianey-Saban C, Divry P, Zabot MT, Roe CR, Nada MA, Byskov A, Kruse TA, Neve S, Kristiansen K, Knudsen I, Corydon MJ, Gregersen N. Cloning and characterization of human very-long-chain acyl-CoA dehydrogenase cDNA, chromosomal assignment of the gene and identification in four patients of nine different mutations within the VLCAD gene. Hum Mol Genet. 1996;5:461–72. doi: 10.1093/hmg/5.4.461. [DOI] [PubMed] [Google Scholar]
- 20.Fukao T, Watanabe H, Orii K, Takahashi Y, Hirano A, Kondo T, Yamaguchi S, Aoyama T, Kondo N. Myopathic form of very-long chain acyl-coa dehydrogenase deficiency: evidence for temperature-sensitive mild mutations in both mutant alleles in a Japanese girl. Pediatr Res. 2001;49:227–31. doi: 10.1203/00006450-200102000-00016. [DOI] [PubMed] [Google Scholar]
- 21.Souri M, Aoyama T, Orii K, Yamaguchi S, Hashimoto T. Mutation analysis of very-long-chain acyl-coenzyme A dehydrogenase (VLCAD) deficiency: identification and characterization of mutant VLCAD cDNAs from four patients. Am J Hum Genet. 1996;58:97–106. [PMC free article] [PubMed] [Google Scholar]
- 22.Takusa Y, Fukao T, Kimura M, Uchiyama A, Abo W, Tsuboi Y, Hirose S, Fujioka H, Kondo N, Yamaguchi S. Identification and characterization of temperature-sensitive mild mutations in three Japanese patients with nonsevere forms of very-long-chain acyl-CoA dehydrogenase deficiency. Mol Genet Metab. 2002;75:227–34. doi: 10.1006/mgme.2002.3297. [DOI] [PubMed] [Google Scholar]
- 23.Goetzman ES, Wang Y, He M, Mohsen AW, Ninness BK, Vockley J. Expression and characterization of mutations in human very long-chain acyl-CoA dehydrogenase using a prokaryotic system. Mol Genet Metab. 2007;91:138–47. doi: 10.1016/j.ymgme.2007.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olsen RK, Andresen BS, Christensen E, Bross P, Skovby F, Gregersen N. Clear relationship between ETF/ETFDH genotype and phenotype in patients with multiple acyl-CoA dehydrogenation deficiency. Hum Mutat. 2003;22:12–23. doi: 10.1002/humu.10226. [DOI] [PubMed] [Google Scholar]
- 25.Toogood HS, van Thiel A, Scrutton NS, Leys D. Stabilization of non-productive conformations underpins rapid electron transfer to electron-transferring flavoprotein. J Biol Chem. 2005;280:30361–6. doi: 10.1074/jbc.M505562200. [DOI] [PubMed] [Google Scholar]
- 26.Chaudhuri TK, Paul S. Protein-misfolding diseases and chaperone-basedtherapeutic approaches. Febs J. 2006;273:1331–49. doi: 10.1111/j.1742-4658.2006.05181.x. [DOI] [PubMed] [Google Scholar]